

Abstract
Persistent colonization of the human stomach with Helicobacter pylori is a risk factor for gastric adenocarcinoma, and H. pylori-induced carcinogenesis is dependent on the actions of a bacterial oncoprotein known as CagA. Epidemiological studies have shown that high dietary salt intake is also a risk factor for gastric cancer. To investigate the effects of a high-salt diet, we infected Mongolian gerbils with a wild-type (WT) cagA+ H. pylori strain or an isogenic cagA mutant strain and maintained the animals on a regular diet or a high-salt diet. At 4 months postinfection, gastric adenocarcinoma was detected in 100% of the WT-infected/high-salt-diet animals, 58% of WT-infected/regular-diet animals, and none of the animals infected with the cagA mutant strain (P < 0.0001). Among animals infected with the WT strain, those fed a high-salt diet had more severe gastric inflammation, higher gastric pH, increased parietal cell loss, increased gastric expression of interleukin 1β (IL-1β), and decreased gastric expression of hepcidin and hydrogen potassium ATPase (H,K-ATPase) compared to those on a regular diet. Previous studies have detected upregulation of CagA synthesis in response to increased salt concentrations in the bacterial culture medium, and, concordant with the in vitro results, we detected increased cagA transcription in vivo in animals fed a high-salt diet compared to those on a regular diet. Animals infected with the cagA mutant strain had low levels of gastric inflammation and did not develop hypochlorhydria. These results indicate that a high-salt diet potentiates the carcinogenic effects of cagA+ H. pylori strains.
INTRODUCTION
Helicobacter pylori is a Gram-negative bacterium that is present in half of the world's population and persistently colonizes the human stomach despite a robust immune response (1–3). Although most H. pylori-infected persons remain asymptomatic, the presence of this organism in the stomach increases the risk of gastric adenocarcinoma (4, 5), and H. pylori has been classified as a class I carcinogen (6). The clinical outcomes of H. pylori infection are determined by a variety of factors, including host genetics, environmental factors (including diet), and variation among H. pylori strains in expression of virulence determinants (4, 5, 7).
There is a high degree of genetic diversity among clinical isolates of H. pylori (8, 9). One of the strain-specific genetic features associated with adverse clinical outcome is a 40-kb region of chromosomal DNA known as the cag pathogenicity island (PAI). The cag PAI encodes a “bacterial oncoprotein” known as CagA and a type IV secretion system (T4SS) that delivers CagA into host cells (10–12). Upon translocation into host cells, CagA interacts with a variety of host cell target molecules, resulting in pleiotropic effects that include cytoskeletal rearrangements, activation of NFκB, alteration of tight junctions, and perturbation of iron trafficking (10, 13–16).
The expression of cagA is regulated in response to variations in several environmental conditions, including iron concentration, pH, and salt concentration (17–20). The molecular mechanisms by which cagA is regulated are not yet completely understood, but the ferric uptake regulator Fur is known to modulate cagA expression (21). In response to elevated salt concentrations, cagA expression is upregulated in some H. pylori strains but not in others (20). In an analysis of clinical strains of H. pylori from Colombian patients, the capacity of strains to upregulate cagA expression in response to high-salt conditions was dependent on the presence of two copies of a TAATGA motif in the cagA promoter region (22). Strain-specific variation in cagA sequences (15, 23), levels of basal cagA expression (24), and regulation of cagA expression in response to environmental conditions (22) all may influence the extent of CagA-mediated cellular alterations caused by individual H. pylori strains.
Epidemiological studies have shown that H. pylori infection and high dietary salt intake increase the risk of gastric cancer in human subjects (25–30). Several studies have evaluated the effects of a high-salt diet on H. pylori infection and gastric cancer in animal models (31–37). One study reported that high dietary salt consumption increased the incidence of gastric cancer in a chemical-induced carcinogenesis model (33), and another study reported that H. pylori infection and a high-salt diet could independently induce atrophic gastritis and intestinal metaplasia in Mongolian gerbils (34). Other studies reported that high salt intake could modulate H. pylori colonization of the stomach or alter Th2 responses, but overall disease outcomes were unaffected by the combination of H. pylori infection and increased dietary salt intake (35–37). Mouse-adapted H. pylori Sydney strain-1 (SS1) was used in many of those previous studies. It was recently reported that this strain has a nonfunctional cag pathogenicity island (PAI) (38, 39); therefore, experiments with this strain do not allow assessment of cag PAI-dependent effects.
The goal of the current study was to analyze the effect of a high-salt diet on H. pylori-induced gastric carcinogenesis in a Mongolian gerbil model and test the hypothesis that elevated dietary salt intake would be associated with enhanced disease progression in this model. We utilized H. pylori strain 7.13, which possesses a functional cag PAI, causes adenocarcinoma in Mongolian gerbils (18, 40, 41), and upregulates CagA expression in vitro in response to high-salt conditions (22). Animals were maintained on either a regular diet alone or a diet supplemented with an additional 8% sodium chloride (34–37). Our results indicate that H. pylori-infected animals maintained on a high-salt diet exhibit increased gastric inflammation and a higher rate of gastric adenocarcinoma in comparison to infected animals maintained on a regular diet. The high-salt diet exacerbates gastric disease caused by the WT cagA+ strain but has no detectable effect on gastric histology in animals infected with an isogenic cagA mutant strain. We show that the development of gastric adenocarcinoma is associated with hypochlorhydria. Finally, cagA transcription is increased in vivo in animals fed a high-salt diet compared to animals fed a regular diet, similar to the effects of high-salt conditions on cagA transcription that have been observed in vitro (20, 22).
MATERIALS AND METHODS
Bacterial strains and culture conditions.
H. pylori strain 7.13 was used as the parental wild-type (WT) strain for these experiments. An isogenic cagA mutant strain, distinct from similar mutants used in previous studies (14, 41), was constructed by insertion of a kanamycin resistance cassette (aphA) in a unique NdeI site corresponding to nucleotide 1067 in cagA from strain 7.13, using previously described methodology (42). H. pylori strains were grown on tryptic soy agar plates containing 5% sheep blood or in brucella broth (Thermo-Fisher) supplemented with 5% fetal bovine serum at 37°C either in ambient air containing 5% CO2 or in a microaerobic atmosphere generated by a BD GasPak EZ Campy container system. For selection of isogenic cagA mutants, blood agar plates were supplemented with kanamycin (40 μg/ml). Insertional inactivation of cagA was confirmed by PCR and Western blot analyses.
In vivo challenge of Mongolian gerbils.
All procedures were approved by the Institutional Animal Care and Use Committee of Vanderbilt University. A single cohort of 108 male Mongolian gerbils, ages 6 to 8 weeks, each weighing 41 to 50 g, was purchased from Charles River Laboratories. This cohort of gerbils was used for all experiments reported in this study. The gerbils were maintained on either a regular diet (Purina 5001 diet; Purina, LLC) (0.75% sodium chloride) or a high-salt modification of the Purina 5001 diet supplemented with an additional 8% sodium chloride (BioServ, LLC). The only difference between the regular diet and the high-salt diet was the addition of 8% sodium chloride, resulting in a final sodium chloride concentration of 8.75%. Animals were maintained on their respective diets for 1 week prior to orogastric challenge with H. pylori and throughout the remainder of the study. Bacteria were grown in brucella broth containing 10% (vol/vol) fetal bovine serum (FBS) overnight under microaerobic conditions (BD GasPak EZ Campy container system) prior to challenge. Gerbils (19 to 20 animals per group) were subjected to orogastrical gavage with H. pylori WT strain 7.13 or the isogenic cagA mutant strain in two doses spaced 24 to 48 h apart (each 1.5 × 109 CFU in 500 μl of fresh brucella broth) when the animals were 8 to 10 weeks old. Animals on a high-salt diet and animals on a regular diet were infected with aliquots of bacteria from the same cultures. Uninfected control animals (3 to 5 per group) were maintained on a regular diet or a high-salt diet. Gerbils were euthanized at 16 weeks postinoculation, and the stomach was excised. The gastric contents were gently removed, and the gastric pH was measured by touching pHydrion pH paper (Micro Essential Laboratory) to the antral portion of the glandular stomach. Subsequently, the stomach was gently washed with phosphate-buffered saline (PBS) and cut into sections extending from the esophagogastric squamocolumnar junction through the proximal duodenum, and these were used for histological examination, analysis of bacterial burden, and RNA extraction. For bacterial burden analyses, stomach sections were homogenized with a tissue tearor (Biospec Products). For RNA extraction, stomach sections were homogenized with a gentleMACS Dissociator and M-tubes (Miltenyi Biotech).
Evaluation of bacterial burden in the gerbil stomach.
Gerbil gastric tissue was isolated, weighed, homogenized in sterile brucella broth, and plated in serial dilutions onto Trypticase soy agar plates containing 5% sheep blood (Hemostat Laboratories), vancomycin (Sigma-Aldrich) (20 μg/ml), nalidixic acid (Sigma-Aldrich) (10 μg/ml), bacitracin (Sigma-Aldrich) (30 μg/ml), and amphotericin B (Sigma-Aldrich) (2 μg/ml) to select for H. pylori growth. These plates were incubated at 37°C in a microaerobic chamber (BD GasPak EZ Campy container system) for 5 days. The bacterial burden was calculated by determining the number of CFU present per gram of tissue.
Histological examination of gerbil gastric tissue.
Slices of stomach from the forestomach to the pylorus along the lesser curvature were fixed in 10% neutral buffered formalin solution (Fisher Scientific), paraffin embedded, sectioned into 5-μm-thick sections, and stained with hematoxylin and eosin. Two sections of each tissue block were prepared, yielding multiple fields for histologic analysis of each animal. Indices of inflammation and the presence of dysplasia and adenocarcinoma were evaluated in a blinded fashion by a pathologist (M. K. Washington). Severity of gastric inflammation was evaluated on a 12-point scale, which graded acute and chronic inflammation in the corpus or the antrum based on a score of 0 to 3 for each of these parameters (0 = no detectable inflammation and 3 = high prevalence of immune cells) (43). Acute inflammation was defined by the presence of polymorphonuclear leukocytes, and chronic inflammation was defined by the presence of mononuclear cell infiltration that was independent of lymphoid follicles. Dysplasia was characterized by the presence of irregular, angulated, and, occasionally, cystically dilated glands with enlarged overlapping hyperchromatic nuclei. Gastric adenocarcinoma was characterized by irregular, angulated, cystically dilated glands with occasional cribriform architecture in the submucosa and muscularis propria, spreading laterally to the surface mucosal component.
Immunohistochemistry detection of H,K-ATPase.
Gastric tissue was treated with citrate buffer (pH 6.0) at 105°C for 20 min with 10 min of cooling prior to blocking with mouse immunoglobulin (Vector Laboratories) for 60 min. After quenching with 0.03% hydrogen peroxide containing sodium azide and blocking with serum-free protein, primary antibody to mouse hydrogen potassium ATPase (H,K-ATPase; Abcam) was added and the reaction mixture was incubated for 60 min. Detection of the primary antibody was performed with an EnVision+ labeled polymer system and chromogen 3,3′-diaminobenzidine tetrahydrochloride (DAB) reagent (Dako-Agilent) before analysis by light microscopy was performed. The sections were scored in a blinded fashion by a pathologist on a scale of 0 to 3. A score of “0” indicates normal parietal cell distribution (no parietal cell loss), “1” indicates patchy distribution and mild loss of parietal cells, “2” indicates moderate loss of parietal cells, and “3” indicates a complete or nearly complete loss of parietal cells, as determined in multiple fields.
Real-time RT-PCR expression analyses.
Gastric tissue from WT-infected, cagA mutant-infected, and uninfected control animals was placed in RNAlater (Ambion) and stored at −20°C until total RNA was subsequently extracted by using a TRIzol isolation protocol (Invitrogen) with some modifications. Gastric tissue was homogenized in 1 ml of TRIzol reagent and subjected to two chloroform extractions. RNA was precipitated with isopropanol and washed with 70% ethanol before further purification with an RNeasy Minikit (Qiagen). RNA was treated with RNase inhibitor (Applied Biosystems) and subjected to DNA-free DNase treatment (Ambion) before synthesis of cDNA using a High Capacity cDNA reverse transcription kit (Applied Biosystems). For real-time reverse transcription-PCR (RT-PCR) of host gene expression, we employed a relative gene expression method utilizing glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a normalizer and tissue from uninfected rodents as a calibrator. RNA extracts from the stomachs of 5 to 7 individual H. pylori-infected rodents were analyzed, along with RNA from uninfected control animals (maintained under the same diet conditions) pooled into a single control. For analysis of cagA, cagA transcript signals were normalized to 16S rRNA. The ΔCT of cagA was calculated as the difference between the cycle threshold (CT) of cagA and the cycle threshold of 16S rRNA (normalizer). Levels of cagA expression are indicated as relative units, calculated as 2−ΔΔCT, where ΔΔCT is equal to the difference between the ΔCT of cagA of each experimental sample and the ΔCT of cagA of the calibrator sample (H. pylori grown in vitro). The in vitro reference control was a pool of three biological replicates of RNA derived from bacteria grown overnight in brucella broth. All cDNA samples were analyzed in duplicate or triplicate along with controls in which no reverse transcriptase was added. For analyses of eukaryotic gene expression, we excluded culture-negative animals. Real-time PCR was performed using a StepOne Plus Real-Time PCR machine (Applied Biosystems), with SYBR green as the fluorochrome. Primers are shown in Table S1 in the supplemental material.
Statistical analyses.
Bacterial colonization densities were normalized by log transformation prior to analysis using an unpaired Student's t test. Histology scores of different groups of animals were compared using the Mann-Whitney U-test. Colonization efficiency and rates of dysplasia or adenocarcinoma in different groups of animals were compared by Fisher's exact test. The correlation between gastric pH and inflammation scores was evaluated by the Pearson correlation coefficient. Gene expression results were analyzed by unpaired Student's t test and analysis of variance (ANOVA). All statistical analyses were performed with a GraphPad Prism Software package.
RESULTS
Colonization of the stomach with WT and cagA mutant strains.
Mongolian gerbils were infected with WT H. pylori 7.13 or an isogenic cagA mutant strain and were maintained on either a regular diet or a high-salt diet (19 to 20 animals per group). At 16 weeks postinfection, animals were euthanized and H. pylori colonization of the stomach was assessed as described in Materials and Methods. When comparing animals infected with the WT strain that were maintained on a regular diet to WT-infected animals on a high-salt diet, there was no significant difference in bacterial burden (2.7 × 107 ± 6 × 106 and 1.2 × 107 ± 2.8 × 106 bacteria per gram of stomach tissue, respectively, P = 0.488) (Fig. 1). Among animals fed a high-salt diet, the WT strain colonized more efficiently than did the cagA mutant strain (100% of animals colonized with the WT strain and 60% of animals colonized with the cagA mutant strain, respectively, P < 0.0001). If noncolonized animals are excluded from the analysis, there was no significant difference between the WT strain and the cagA mutant in colonization efficiency or bacterial burden.
Fig 1.

H. pylori colonization of the gerbil stomach. Gerbils were infected with WT H. pylori or an isogenic cagA mutant strain and maintained on either a regular diet or a high-salt diet. At 16 weeks postinfection, the stomach was removed, homogenized, and plated onto bacteriological culture medium. The bacterial burden was calculated by determining the number of CFU per gram of tissue. The dotted line indicates the limit of detection. Data represent the means ± standard errors of the means (SEM) for each group (n = 19 to 20 animals per group).
High dietary salt intake exacerbates H. pylori-induced inflammation.
To analyze the effect of a high-salt diet on H. pylori-induced gastric pathology, we analyzed gastric tissue from the H. pylori-infected gerbils. Representative sections of the gastric antrum are shown in Fig. 2 for uninfected animals (Fig. 2A and B), WT-infected animals (Fig. 2C and D), and cagA mutant-infected animals (Fig. 2E and F). Sections of gastric tissue from animals in each group were scored for severity of total gastric inflammation on a 12-point scale, which evaluated acute and chronic inflammation in both the corpus and the antrum. Uninfected animals exhibited no detectable inflammation, regardless of diet. WT-infected animals maintained on a high-salt diet exhibited significantly higher total gastric inflammation scores than their regular-diet counterparts (P = 0.0058; Fig. 2G). Similarly, when inflammation was analyzed in discrete regions of the stomach (corpus and antrum) and classified with respect to either chronic or acute inflammatory processes, WT-infected animals maintained on a high-salt diet had increased acute inflammation in the antrum (P = 0.0346), increased chronic inflammation in the antrum (P = 0.0372), increased acute inflammation in the corpus (P = 0.0077), and increased chronic inflammation in the corpus (P = 0.0477) compared to WT-infected animals maintained on a regular diet (see Fig. S1 in the supplemental material). Consistent with previous reports (18, 41, 44), animals infected with the cagA mutant strain and fed a regular diet exhibited only trace levels of inflammation (Fig. 2G). We did not detect any increased inflammation in cagA mutant-infected animals that were fed a high-salt diet compared to those that were fed a regular diet (Fig. 2G). These data demonstrate that high dietary salt intake significantly increases the severity of gastric inflammation caused by a WT cagA+ H. pylori strain but does not have any detectable effect on inflammation in animals infected with a cagA mutant strain.
Fig 2.
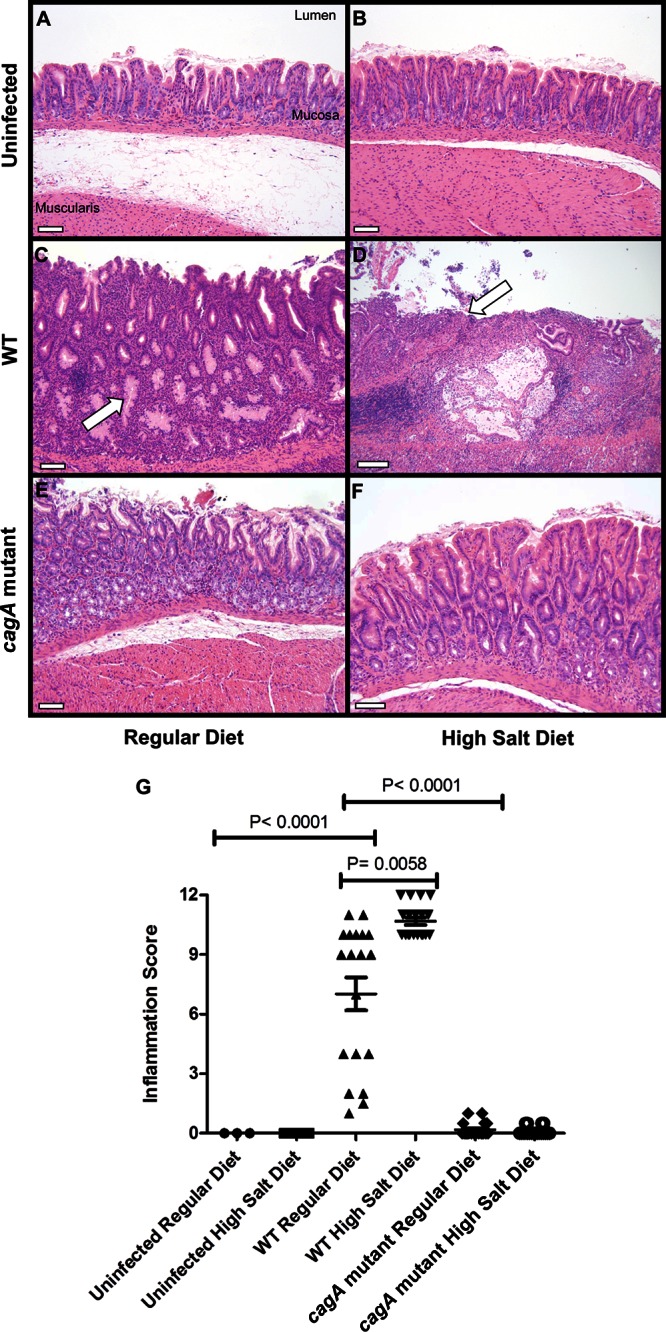
Analysis of gastric inflammation. Gerbils were infected with WT H. pylori or an isogenic cagA mutant strain and maintained on either a regular diet or a high-salt diet. As controls, uninfected gerbils were maintained on either a regular diet or a high-salt diet. At 16 weeks postinfection, gastric tissue was collected and sections were stained with hematoxylin and eosin. (A to F) Representative sections of the gastric antrum are shown. Panel C demonstrates dysplasia (arrow), and panel D demonstrates ulceration (arrow). Magnification bars indicate 100 μm. (G) Representative micrographs of gastric tissue were scored for total inflammation on a scale of 0 to 12 (sum of chronic [0 to 3] and acute [0 to 3] inflammation in both the corpus and antrum of the gerbil stomach). Animals infected with WT H. pylori and maintained on a high-salt diet had significantly higher inflammation scores than WT-infected animals maintained on a regular diet (P = 0.0058). In addition, WT-infected animals maintained on a regular diet had significantly higher inflammation scores than uninfected animals or cagA mutant-infected animals maintained on a regular diet (P < 0.0001 and P < 0.0001). Horizontal bars indicate mean total inflammation ± SEM. Statistical analyses were performed using the Mann-Whitney U-test.
High dietary salt intake results in increased dysplasia and invasive gastric adenocarcinoma.
Invasive gastric adenocarcinoma was observed more frequently in WT-infected gerbils maintained on a high-salt diet than in WT-infected gerbils maintained on a regular diet (100% versus 58%) (P < 0.0001) (Fig. 3). The gastric adenocarcinomas were characterized by the presence of irregularly shaped glandular structures comprised of tall mucin-producing cells, which invaded through the muscularis mucosa (Fig. 3A and B). The morphology of the tumors in animals fed a high-salt diet was similar to that of the tumors in animals fed a regular diet, but the tumors in animals fed a high-salt diet tended to be larger and more deeply invasive. Gastric dysplasia, characterized by noninvasive dilated glands with pseudostratified nuclei (Fig. 2C), was also detected more commonly in WT-infected gerbils maintained on a high-salt diet than in those maintained on a regular chow diet (Fig. 3C). The total gastric inflammation score (scale of 0 to 12) was higher in animals with gastric dysplasia than in animals without dysplastic lesions (Fig. 3D) (P < 0.0001), which suggests that inflammation may contribute to the development of dysplasia. Dysplastic lesions and adenocarcinomas were not detected in either uninfected animals or cagA mutant-infected animals, which is consistent with the results of several previous studies (4, 41). These data demonstrate that a high-salt diet significantly increases the incidence of premalignant and malignant lesions in animals infected with cagA+ H. pylori but not in animals infected with a cagA mutant or uninfected animals.
Fig 3.

Analysis of gastric dysplasia and adenocarcinoma. Gerbils were infected with WT H. pylori or an isogenic cagA mutant strain and maintained on either a regular diet or a high-salt diet. As controls, uninfected gerbils were maintained on either a regular diet or a high-salt diet. Animals were euthanized at 16 weeks postinfection. (A and B) Representative micrographs of gastric tissue derived from WT-infected animals maintained on a high-salt diet (arrows indicate tumors). Magnification bars indicate 100 μm. (C) Micrographs were evaluated for dysplastic lesions and invasive adenocarcinoma. Only animals infected with the WT strain exhibited these abnormalities. Bars indicate the percentage of animals within each group exhibiting dysplasia or gastric adenocarcinoma. Gastric cancer and dysplasia were detected significantly more frequently in the WT-infected high-salt diet group than in WT-infected regular-diet counterparts (P < 0.001). (D) Analysis of gastric inflammation in animals with gastric dysplasia. WT-infected animals maintained on a regular diet and WT-infected animals maintained on a high-salt diet were analyzed. The total gastric inflammation score (scale of 0 to 12) was higher in WT-infected animals with gastric dysplasia than in WT-infected animals without dysplastic lesions. Horizontal bars indicate mean inflammation score ± SEM for each group. Statistical analyses were performed using Fisher's exact test (C) and Mann-Whitney U analysis (D).
High dietary salt intake exacerbates H. pylori-induced hypochlorhydria.
The development of gastric cancer in humans is often preceded by atrophic gastritis and associated hypochlorhydria (5, 7, 45). To evaluate the possible development of hypochlorhydria in H. pylori-infected gerbils, the gastric pH of each animal was measured at the time of necropsy. As shown in Fig. 4A, all uninfected animals had a gastric pH of 3 regardless of dietary salt intake. All cagA mutant-infected animals had a gastric pH of 3 to 4, and there was no significant difference between the regular-diet and high-salt-diet groups with respect to gastric pH. The mean gastric pH of WT-infected animals maintained on a regular diet was 4.6 ± 1.3 compared to a mean pH of 6.1 ± 0.5 in WT-infected animals maintained on a high-salt diet (P = 0.0027). Two animals in the WT-infected high-salt diet group exhibited a gastric pH of 7. These results suggest that CagA contributes to H. pylori-induced gastric hypochlorhydria and indicate that H. pylori-induced hypochlorhydria is exacerbated by high dietary salt intake.
Fig 4.
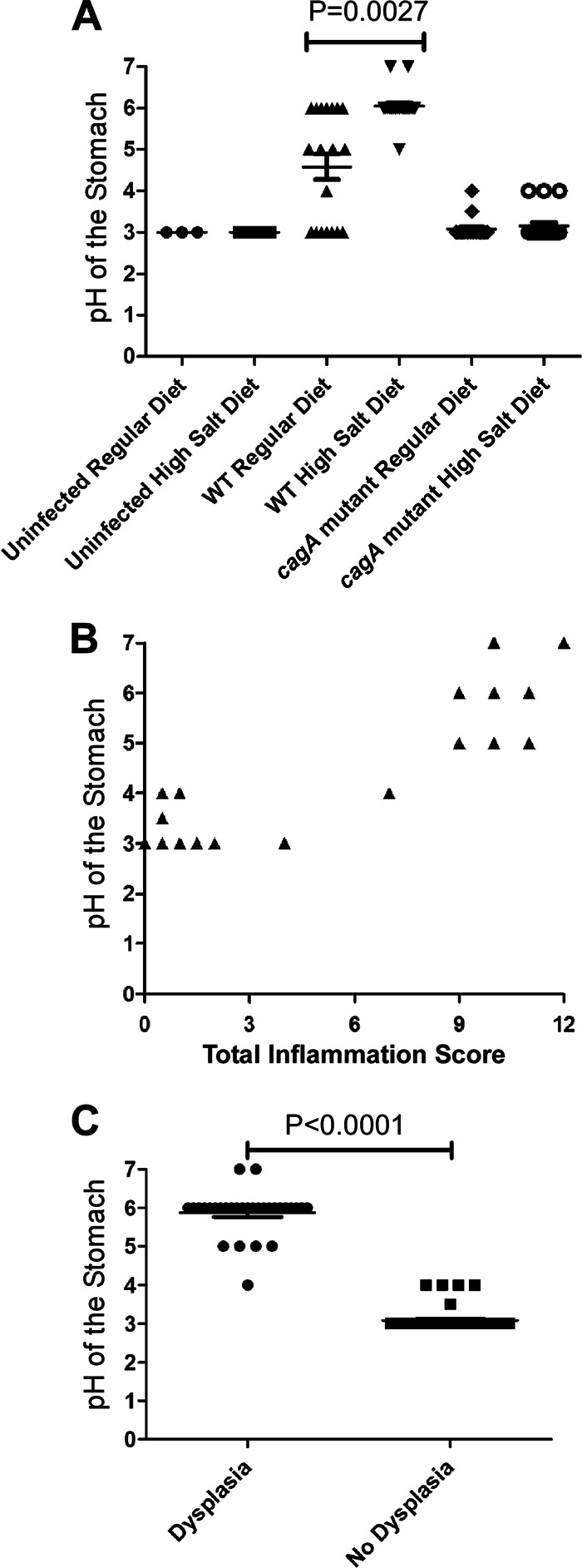
Analysis of gastric pH. Gerbils were infected with WT H. pylori or an isogenic cagA mutant strain and maintained on either a regular diet or a high-salt diet. As controls, uninfected gerbils were maintained on either a regular diet or a high-salt diet. Animals were euthanized at 16 weeks postinfection, and gastric pH was measured at the time of necropsy. Horizontal bars in panels A and C indicate mean pH ± SEM. (A) The highest gastric pH values were detected in animals infected with the WT strain and maintained on a high-salt diet and were significantly higher than in animals infected with WT H. pylori and maintained on a regular diet (P = 0.0027, Mann-Whitney U analysis). (B) The gastric histology of all animals in the study (including uninfected, WT-infected, and cagA mutant strain-infected animals maintained on either high-salt or regular diets) was scored for total inflammation on a scale of 0 to 12 (sum of chronic [0 to 3] and acute [0 to 3] inflammation in both the corpus and antrum). Each triangle represents 3 to 5 animals. Gastric pH values were positively correlated with severity of gastric inflammation (R = 0.9410 and P < 0.0001), based on Pearson correlation coefficient (assuming Gaussian populations). (C) In an analysis of WT-infected animals maintained on a regular diet and WT animals maintained on a high-salt diet, the gastric pH was higher in animals with dysplastic lesions than in animals without dysplastic lesions (P < 0.0001, Mann Whitney U-test).
Analysis of the hematoxylin-and-eosin-stained gastric tissue suggested that there were reductions in parietal cell numbers in hypochlorhydric animals compared to animals with acidic gastric pH and that there was greater parietal cell loss in WT-infected animals fed a high-salt diet than in WT-infected animals fed a regular diet (or other groups). No thinning of the gastric mucosa was detected in these animals (data not shown); instead, reactive hyperplasia (characterized by increased thickness of the foveolar epithelial compartment with loss of mucin) was present. To more rigorously analyze parietal cell numbers and distribution, we stained gastric sections of representative animals with an anti-H,K-ATPase antibody. Uninfected animals had a thick band-like zone of parietal cells in the gastric corpus (Fig. 5A), with a few H,K-ATPase-positive cells in deep antral glands. In comparison to the uninfected animals, the majority of the infected animals exhibited parietal cell loss (Fig. 5B and C). The most extensive parietal cell loss, corresponding to loss of staining in the gastric body, was detected in WT-infected animals on a high-salt diet (P = 0.0285; Fig. 5D).
Fig 5.

Immunohistochemical analysis of gastric H,K-ATPase in tissue from the gastric body of H. pylori-infected gerbils. (A) Parietal cells in an uninfected control stomach. (B) A WT-infected animal maintained on a regular diet exhibits patchy distribution and moderate loss of parietal cells. (C) A WT-infected animal maintained on a high-salt diet exhibits extensive loss of parietal cells. (D) Parietal cell loss was scored in two groups of infected animals (5 animals per group; bars indicate means ± SEM). A score of “0” indicates no parietal cell loss; normal distribution in the corpus and the antrum, “1” indicates patchy distribution and mild loss of parietal cells, “2” indicates moderate loss of parietal cells, and “3” indicates a complete or near complete loss of parietal cells within the gastric tissue. Animals infected with WT H. pylori and maintained on a high-salt diet exhibited a significantly increased loss of parietal cells compared to WT-infected animals on a regular diet (P = 0.0463, Mann-Whitney U analysis).
Hypochlorhydria is associated with increased gastric inflammation and dysplasia.
We next analyzed if there was a correlation between gastric pH and the severity of gastric inflammation. In a combined analysis of all animals included in this study (including uninfected animals, WT-infected animals, and cagA mutant-infected animals on both high-salt and regular diets), there was a significant correlation between gastric pH and severity of gastric inflammation (Fig. 4B) (P < 0.0001). There was also a significant correlation when the analysis was limited to WT-infected animals on a regular diet (P < 0.0001). Animals with dysplasia had a mean gastric pH of 6.0 ± 0.7, whereas animals lacking dysplasia had a mean gastric pH of 3.4 ± 0.4 (P < 0.0001) (Fig. 4C). Taken together, these results demonstrate that hypochlorhydria occurs more commonly in H. pylori-infected animals with severe gastric inflammation and dysplasia than in H. pylori-infected animals lacking these features.
Increased expression of IL-1β in response to cagA+ H. pylori and a high-salt diet.
To further analyze the gastric mucosal inflammatory response, we analyzed a set of host factors relevant for innate immune responses and T-cell recruitment. The levels of expression of several proinflammatory cytokines (interleukin 1β [IL-1β], IL-6, IL-17, and gamma interferon [IFN-γ]), anti-inflammatory cytokines (IL-10), chemokines (KC, CCL12), and inducible nitric oxide synthase (iNOS) in the gastric tissue were measured by real-time RT-PCR. Gerbils infected with WT H. pylori exhibited increased expression of all of these immune modulators (P = 0.001) (Fig. 6A; see also Fig. S2 in the supplemental material). Genes corresponding to proinflammatory T cell cytokines representing Th1 and Th17 responses, IFN-γ and IL-17, were among the most highly upregulated genes, and their expression in gerbils maintained on a high-salt diet was similar to that in gerbils maintained on a regular diet (Fig. 6A). Expression levels of IL-1β (a proinflammatory cytokine, produced by monocytes, macrophages, and dendritic cells) and iNOS (which is induced in response to the presence of proinflammatory cytokines in the same cell types) were significantly increased in WT-infected animals maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet (P < 0.01 and P = 0.006) (Fig. 6A; see also Fig. S2 in the supplemental material). Gerbils infected with the cagA mutant strain did not exhibit any increase in gastric IL-1β compared to uninfected animals (Fig. 6A).
Fig 6.

Real-time RT-PCR analysis of the host immune response, modulators of gastric acid production, and H. pylori cagA in the gerbil stomach. For panels A and B, RNA extracts from the stomachs of 5 to 7 individual H. pylori-infected rodents were analyzed, along with RNA from uninfected control animals (same diet conditions) pooled into a single control. For panel C (analysis of cagA transcription), gastric RNA extracts were analyzed along with RNA derived from H. pylori grown in modified brucella broth alone (regular medium) or supplemented with 0.5% sodium chloride (high-salt medium). The results represent mean values based on analyses of 5 to 7 animals per group. (A) IL-1β, IL-17, and IFN-γ were induced by WT infection. IL-1β transcript abundance was significantly increased in WT-infected animals maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet. (B) Hepcidin, gastrin, and H,K-ATPase are downregulated in the WT-infected high-salt-diet animals compared to the WT-infected regular-diet animals. For panels A and B, transcript abundance was normalized to GAPDH, and relative units were calculated as described in Materials and Methods. (C) cagA gene expression is elevated in WT-infected animals maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet (P < 0.05). Similarly, cagA transcription was increased in bacteria grown in vitro in high-salt medium compared to bacteria grown in regular medium. For panel C, bacterial transcript levels were normalized to the corresponding 16S rRNA levels in each sample. Relative units were calculated as described in Materials and Methods.
We hypothesized not only that the hypochlorhydria observed in some H. pylori-infected animals might be attributable to a reduction in parietal cell number but also that increased expression of IL-1β might lead to decreased gastric acid secretion (46). IL-1β is known to negatively regulate hepcidin, gastrin, and H,K-ATPase, which in turn regulate or mediate gastric acid secretion (46–48). Therefore, we analyzed the transcript abundance of these three molecules. Expression of all three targets was diminished in WT-infected animals maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet (Fig. 6B).
Expression of cagA is enhanced in gerbils maintained on a high-salt diet.
Since previous in vitro studies showed that production of the oncogenic effector molecule CagA by H. pylori is positively regulated in response to elevated concentrations of sodium chloride (20, 22), we hypothesized that a similar upregulation of cagA expression might also occur in vivo in response to increased dietary salt intake. To test this hypothesis, we quantified bacterial expression of cagA in the gerbil stomach, as described in Materials and Methods. A significant increase in cagA expression was observed in WT-infected animals maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet (P < 0.05) (Fig. 6C), a result that mimicked the upregulation of cagA expression that occurs when H. pylori is cultured in vitro in the presence of high-salt conditions (20). We also observed that expression of cagA was increased in vivo compared to the expression seen in vitro, which is consistent with previously reported results (49).
DISCUSSION
In this report, we show in a Mongolian gerbil model that a high dietary salt intake enhances H. pylori-induced carcinogenesis. The observed effects of a high-salt diet on H. pylori-induced gastric cancer in the gerbil model correlate well with human epidemiologic data, which have repeatedly shown increased rates of gastric cancer in persons who consume a high-salt diet (25–30). The high-salt diet used in this study (8.75% sodium chloride) approximates the concentration of sodium chloride in some foods consumed by humans. For example, dried fish is often preserved in 3 to 20% salt, pickled foods contain up to 25% salt, and soy sauce contains 19% salt (35). In contrast to human diets, which vary considerably from day to day, the gerbils in this study were fed a high-salt diet with no variation over a prolonged time period.
In the current study, we observed that among gerbils infected with the WT strain and fed a regular diet, gastric cancer was present at 4 months postinfection in 60% of the animals. This incidence of gastric cancer in the gerbil model is similar or slightly higher than that reported in several previous studies (18, 40, 41). Gastric cancer developed in animals that were infected with a WT cagA+ H. pylori strain but not in animals infected with an isogenic cagA mutant strain. Although we did not analyze a complemented mutant strain in the current study, the failure of a cagA mutant strain to cause gastric cancer is concordant with the results of several other studies (18, 41). Collectively, these results in animal models of H. pylori infection, combined with evidence from transgenic animal experiments (50), cell culture experiments (16), and human epidemiologic studies (51), provide evidence that CagA has an important role in gastric cancer pathogenesis.
The administration of a high-salt diet to uninfected animals did not stimulate the development of gastric cancer, which leads us to conclude that the effects of a high-salt diet on gastric cancer are dependent on the presence of H. pylori infection. Notably, we observed that a high-salt diet led to an increased incidence of gastric carcinoma in animals infected with a WT cagA+ H. pylori strain but not in animals infected with a cagA mutant strain. This provides evidence that the effects of a high-salt diet on gastric cancer pathogenesis are relevant mainly in the context of infection with cagA+ H. pylori strains. We are unaware of any studies that have examined the relationships among a high-salt diet, infection with cagA+ H. pylori strains, and gastric cancer in human populations, but in several parts of the world that have high rates of gastric cancer, there is a high prevalence of cagA+ strains and a large proportion of the population consumes a high-salt diet.
Several previous studies have analyzed the effect of a high-salt diet (typically 8% added salt, similar to the current study) on H. pylori-induced gastric pathology in animal models but have not reached a consistent conclusion (31–37). Notably, the rodent studies in many of these reports were carried out using H. pylori Sydney Strain 1 (SS1), which harbors an inactive cag PAI (38, 39). We propose that an effect of a high-salt diet on H. pylori-induced gastric cancer may be more readily detectable in experiments that employ a cagA+ strain and a gerbil model.
In addition to a high-salt diet, a low-iron diet was recently shown to augment H. pylori-induced carcinogenesis in the gerbil model (18). Low-iron conditions lead to alterations in the expression of many H. pylori genes, including upregulation of cagA expression (17), and also stimulate assembly of pili associated with the cag T4SS when H. pylori is in contact with gastric epithelial cells (18). Potentially, there are related features of these two distinct dietary interventions that lead to increased H. pylori virulence or host disease progression. For example, iron absorption may be impaired in the setting of hypochlorhydria (52–54).
Previous studies have reported that exposure of H. pylori to high-salt conditions in vitro leads to alterations in the expression of multiple H. pylori genes, including cagA (22). In the current study, we observed that cagA gene expression was elevated in the stomachs of WT-infected animals maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet (when normalized based on comparison to 16S rRNA). This upregulation of cagA transcription in response to a high-salt diet mimics the upregulation of cagA gene expression that is observed in vitro in response to high-salt conditions. In agreement with a previous report (49), we also observed that cagA transcripts were more abundant in the gastric tissue samples than in bacteria grown in laboratory medium. We propose that an upregulation of cagA gene expression in response to the high-salt diet is an important feature of the mechanism by which a high-salt diet enhances carcinogenesis.
We observed that animals infected with the WT strain and fed a high-salt diet had significantly higher levels of gastric inflammation than WT-infected animals on a regular diet. To elucidate a potential immunologic basis for this difference, we analyzed the expression of several cytokines, chemokines, and immune modulatory molecules that regulate inflammation. This analysis was limited in scope because reagents and sequences are not readily available for immunologic studies in gerbils. Nevertheless, we were able to demonstrate that the relative levels of expression of IFN-γ, IL-17, IL-1β, IL-6, IL-10, KC, iNOS, and CCL12 were increased in WT H. pylori-infected animals compared to uninfected animals. Interestingly, both IL-1β transcription and iNOS transcription were significantly elevated in WT-infected gerbils maintained on a high-salt diet compared to the regular-diet counterparts, which suggests that these factors may contribute to the increased inflammation that accompanies a high-salt diet.
Since atrophic gastritis and hypochlorhydria commonly precede the development of gastric cancer in humans (45), we analyzed the gastric pH of animals at the time of necropsy. As expected, uninfected animals had an acidic gastric pH. WT-infected animals maintained on a high-salt diet had markedly increased gastric pH compared to their regular diet WT-infected counterparts. Animals infected with the cagA mutant exhibited only minor alterations in gastric pH, and among these animals, a high-salt diet did not have any detectable effects on gastric pH. Therefore, CagA contributes to the development of hypochlorhydria, and a high-salt diet exacerbates the development of hypochlorhydria in animals infected with a WT strain. We observed that the increases in pH were significantly correlated with increased inflammation, which suggests that inflammation contributes to this process. Reductions in parietal cell number were detected in the hypochlorhydric animals, which likely accounts at least in part for the observed hypochlorhydria. In addition, hypochlorhydria could be due to perturbations in parietal cell function. We detected increased expression of IL-1β (a known inhibitor of gastric acid production) in WT-infected animals on a high-salt diet compared to WT-infected animals on a regular diet. We also analyzed the expression of several other host factors that regulate or mediate gastric acid production (gastrin, hepcidin, and H,K-ATPase). Hepcidin is an antimicrobial peptide involved in iron metabolism homeostasis and is a regulator of gastric acid secretion (47). Gastrin is a regulator of gastric acid secretion that is negatively regulated by IL-1-β through the activation of NFκB (55). H,K-ATPase is the parietal cell enzyme that mediates acid secretion and is also transcriptionally repressed by NFκB (56). Animals infected with WT H. pylori and maintained on a high-salt diet had decreased hepcidin, gastrin, and H,K-ATPase transcripts compared to animals infected with WT H. pylori and maintained on a regular diet. We propose that these alterations all contribute to the observed hypochlorhydria.
In summary, the results of this study reveal that increased dietary salt consumption markedly alters the outcome of infection with a cagA+ H. pylori strain. In the simplest model, high salt concentrations stimulate increased expression of cagA, and the actions of CagA lead to inflammation, hypochlorhydria, and enhanced carcinogenesis. An equally plausible model proposes that high salt concentrations lead to altered expression of multiple H. pylori genes (including cagA) (22) as well as to alterations in the host and that this constellation of alterations stimulates enhanced carcinogenesis. Regardless of which mechanism is operative, the current results compare favorably with a large body of epidemiologic evidence indicating that a high-salt diet is a risk factor for gastric cancer in humans (25–30). Potentially, reductions in dietary salt intake could lead to a reduction in the risk of H. pylori-associated gastric adenocarcinoma in populations who have a high risk for this malignancy.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH AI068009 (T.L.C.), CA116087 (T.L.C.), F32 AI102568 (J.A.G.), R01 DK58587 (R.M.P.), R01 CA77955 (R.M.P.), and the Department of Veterans Affairs (T.L.C. and H.M.S.A.). This work was also supported by the Vanderbilt Ingram Cancer Center, and Core Services performed through Vanderbilt University Medical Center's Digestive Disease Research Center were supported by NIH grant P30DK058404.
We thank Judy Romero-Gallo and Jenny Noto for helpful advice regarding the gerbil model.
Footnotes
Published ahead of print 8 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01271-12.
REFERENCES
- 1. Cover T, Blaser M. 2009. Helicobacter pylori in health and disease. Gastroenterology 136:1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atherton J, Blaser M. 2009. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest. 119:2475–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Algood H, Cover T. 2006. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin. Microbiol. Rev. 19:597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wroblewski L, Peek R, Jr, Wilson K. 2010. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev. 23:713–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fox J, Wang T. 2007. Inflammation, atrophy, and gastric cancer. J. Clin. Invest. 117:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. IARC 1994. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 61:1–241 [PMC free article] [PubMed] [Google Scholar]
- 7. Peek R, Jr, Blaser M. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2:28–37 [DOI] [PubMed] [Google Scholar]
- 8. Blaser M, Berg D. 2001. Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Invest. 107:767–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suerbaum S, Josenhans C. 2007. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 5:441–452 [DOI] [PubMed] [Google Scholar]
- 10. Tegtmeyer N, Wessler S, Backert S. 2011. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 278:1190–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fischer W. 2011. Assembly and molecular mode of action of the Helicobacter pylori Cag type IV secretion apparatus. FEBS J. 278:1203–1212 [DOI] [PubMed] [Google Scholar]
- 12. Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287:1497–1500 [DOI] [PubMed] [Google Scholar]
- 13. Bourzac K, Guillemin K. 2005. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol. 7:911–919 [DOI] [PubMed] [Google Scholar]
- 14. Tan S, Noto J, Romero-Gallo J, Peek R, Jr, Amieva M. 2011. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog. 7:e1002050 doi:10.1371/journal.ppat.1002050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hatakeyama M. 2011. Anthropological and clinical implications for the structural diversity of the Helicobacter pylori CagA oncoprotein. Cancer Sci. 102:36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hatakeyama M. 2004. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 4:688–694 [DOI] [PubMed] [Google Scholar]
- 17. Merrell DS, Thompson LJ, Kim CC, Mitchell H, Tompkins LS, Lee A, Falkow S. 2003. Growth phase dependent response of Helicobacter pylori to iron starvation. Infect. Immun. 71:6510–6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Noto J, Gaddy J, Lee J, Piazuelo M, Friedman D, Colvin D, Romero-Gallo J, Suarez G, Loh J, Slaughter J, Tan S, Morgan D, Wilson K, Bravo L, Correa P, Cover T, Amieva M, Peek R., Jr 2013. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J. Clin. Invest. 123:479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Merrell D, Goodrich M, Otto G, Tompkins L, Falkow S. 2003. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71:3529–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Loh J, Torres V, Cover T. 2007. Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res. 67:4709–4715 [DOI] [PubMed] [Google Scholar]
- 21. Pich O, Carpenter B, Gilbreath J, Merrell D. 2012. Detailed analysis of Helicobacter pylori Fur-regulated promoters reveals a Fur box core sequence and novel Fur-regulated genes. Mol. Microbiol. 84:921–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loh J, Friedman D, Piazuelo M, Bravo L, Wilson K, Peek R, Jr, Correa P, Cover T. 2012. Analysis of Helicobacter pylori cagA promoter elements required for salt-induced upregulation of CagA expression. Infect. Immun. 80:3094–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duncan S, Valk P, Shaffer C, Bordenstein S, Cover T. 2012. J-Western forms of Helicobacter pylori cagA constitute a distinct phylogenetic group with a widespread geographic distribution. J. Bacteriol. 194:1593–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loh J, Shaffer C, Piazuelo M, Bravo L, McClain M, Correa P, Cover T. 2011. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epidemiol. Biomarkers Prev. 20:2237–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee S, Kang D, Shim K, Choe J, Hong W, Choi H. 2003. Effect of diet and Helicobacter pylori infection to the risk of early gastric cancer. J. Epidemiol. 13:162–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsugane S, Sasazuki S. 2007. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 10:75–83 [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Terry P, Yan H. 2009. Review of salt consumption and stomach cancer risk: epidemiological and biological evidence. World J. Gastroenterol. 15:2204–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. D'Elia L, Rossi G, Ippolito R, Cappuccio F, Strazzullo P. 2012. Habitual salt intake and risk of gastric cancer: a meta-analysis of prospective studies. Clin. Nutr. 31:489–498 [DOI] [PubMed] [Google Scholar]
- 29. Brenner H, Rothenbacher D, Arndt V. 2009. Epidemiology of stomach cancer. Methods Mol. Biol. 472:467–477 [DOI] [PubMed] [Google Scholar]
- 30. Krejs G. 2010. Gastric cancer: epidemiology and risk factors. Dig. Dis. Sci. 28:600–603 [DOI] [PubMed] [Google Scholar]
- 31. Toyoda T, Tsukamoto T, Hirano N, Mizoshita T, Kato S, Takasu S, Ban H, Tatematsu M. 2008. Synergistic upregulation of inducible nitric oxide synthase and cyclooxygenase-2 in gastric mucosa of Mongolian gerbils by a high-salt diet and Helicobacter pylori infection. Histol. Histopathol. 23:593–599 [DOI] [PubMed] [Google Scholar]
- 32. Gamboa-Dominguez A, Ubbelohde T, Saqui-Salces M, Romano-Mazzoti L, Cervantes M, Domínguez-Fonseca C, de la Luz Estreber M, Ruíz-Palacios GM. 2007. Salt and stress synergize H. pylori-induced gastric lesions, cell proliferation, and p21 expression in Mongolian gerbils. Dig. Dis. Sci. 52:1517–1526 [DOI] [PubMed] [Google Scholar]
- 33. Kato S, Tsukamoto T, Mizoshita T, Tanaka H, Kumagai T, Ota H, Katsuyama T, Asaka M, Tatematsu M. 2006. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int. J. Cancer 119:1558–1566 [DOI] [PubMed] [Google Scholar]
- 34. Bergin I, Sheppard B, Fox J. 2003. Helicobacter pylori infection and high dietary salt independently induce atrophic gastritis and intestinal metaplasia in commercially available outbred Mongolian gerbils. Dig. Dis. Sci. 48:475–485 [DOI] [PubMed] [Google Scholar]
- 35. Fox J, Dangler C, Taylor N, King A, Koh T, Wang T. 1999. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 59:4823–4828 [PubMed] [Google Scholar]
- 36. Fox J, Rogers A, Ihrig M, Taylor N, Whary M, Dockray G, Varro A, Wang T. 2003. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 63:942–950 [PubMed] [Google Scholar]
- 37. Rogers A, Taylor N, Whary M, Stefanich E, Wang T, Fox J. 2005. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 65:10709–10715 [DOI] [PubMed] [Google Scholar]
- 38. Crabtree J, Ferrero R, Kusters J. 2002. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter 7:139–140 (Author's reply, 7:140–141.) [DOI] [PubMed] [Google Scholar]
- 39. Philpott D, Belaid D, Troubadour P, Thiberge J, Tankovic J, Labigne A, Ferrero R. 2002. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylori isolates. Cell Microbiol. 4:285–296 [DOI] [PubMed] [Google Scholar]
- 40. Franco A, Israel D, Washington M, Krishna U, Fox J, Rogers A, Neish A, Collier-Hyams L, Perez-Perez G, Hatakeyama M, Whitehead R, Gaus K, O'Brien D, Romero-Gallo J, Peek R., Jr 2005. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 102:10646–10651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Franco A, Johnston E, Krishna U, Yamaoka Y, Israel D, Nagy T, Wroblewski L, Piazuelo MB, Correa P, Peek RM., Jr 2008. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 68:379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tummuru M, Cover T, Blaser M. 1994. Mutation of the cytotoxin-associated cagA gene does not affect the vacuolating cytotoxin activity of Helicobacter pylori. Infect. Immun. 62:2609–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romero-Gallo J, Harris E, Krishna U, Washington M, Perez-Perez G, Peek R., Jr 2008. Effect of Helicobacter pylori eradication on gastric carcinogenesis. Lab. Invest. 88:328–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rieder G, Merchant J, Haas R. 2005. Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128:1229–1242 [DOI] [PubMed] [Google Scholar]
- 45. Correa P. 1992. Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52:6735–6740 [PubMed] [Google Scholar]
- 46. El-Omar E. 2001. The importance of interleukin 1 beta in Helicobacter pylori associated disease. Gut 48:743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schwarz P, Kubler J, Strnad P, Muller K, Barth T, Gerloff A, Feick P, Peyssonnaux C, Vaulont S, Adler G, Kulaksiz H. 2012. Hepcidin is localised in gastric parietal cells, regulates acid secretion and is induced by Helicobacter pylori infection. Gut 61:193–201 [DOI] [PubMed] [Google Scholar]
- 48. Schubert M. 2011. Gastric secretion. Curr. Opin. Gastroenterol. 27:536–542 [DOI] [PubMed] [Google Scholar]
- 49. Scott D, Marcus E, Wen Y, Oh J, Sachs G. 2007. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc. Natl. Acad. Sci. U. S. A. 104:7235–7240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, Yamada G, Azuma T, Hatakeyama M. 2008. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. U. S. A. 105:1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blaser M, Perez-Perez G, Kleanthous H, Cover T, Peek R, Chyou P, Stemmermann G, Nomura A. 1995. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 55:2111–2115 [PubMed] [Google Scholar]
- 52. Krieg L, Milstein O, Krebs P, Xia Y, Beutler B, Du X. 2011. Mutation of the gastric hydrogen-potassium ATPase alpha subunit causes iron-deficiency anemia in mice. Blood 118:6418–6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miret S, Simpson R, McKie A. 2003. Physiology and molecular biology of dietary iron absorption. Annu. Rev. Nutr. 23:283–301 [DOI] [PubMed] [Google Scholar]
- 54. Annibale B, Capurso G, Delle Fave G. 2003. The stomach and iron deficiency anaemia: a forgotten link. Dig. Liver Dis. 35:288–295 [DOI] [PubMed] [Google Scholar]
- 55. Datta De D, Bhattacharjya S, Maitra M, Datta A, Choudhury A, Dhali G, Roychoudhury S. 2011. IL1B induced Smad 7 negatively regulates gastrin expression. PLoS One 6:e14775 doi:10.1371/journal.pone.0014775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smolka A, Backert S. 2012. How Helicobacter pylori infection controls gastric acid secretion. J. Gastroenterol. 47:609–618 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.