

Abstract
Malaria is one of the most important infectious diseases in the world, and it has many economic and social impacts on populations, especially in poor countries. Transmission-blocking vaccines (TBVs) are valuable tools for malaria eradication. A study on Anopheles gambiae revealed that polyclonal antibodies to carboxypeptidase B1 of A. gambiae can block sexual parasite development in the mosquito midgut. Hence, it was introduced as a TBV target in regions where A. gambiae is the main malaria vector. However, in Iran and neighboring countries as far as China, the main malaria vector is Anopheles stephensi. Also, the genome of this organism has not been sequenced yet. Therefore, in this study, carboxypeptidase B1 of A. stephensi was characterized by genomic and proteomic approaches. Furthermore, its expression pattern after ingestion of Plasmodium falciparum gametocytes and the effect of anti-CPBAs1 antibodies on sexual parasite development were evaluated. Our results revealed that the cpbAs1 expression level was increased after ingestion of the mature gametocytes of P. falciparum and that anti-CPBAs1 directed antibodies could significantly reduce the mosquito infection rate in the test group compared with the control group. Therefore, according to our findings and with respect to the high similarity of carboxypeptidase enzymes between the two main malaria vectors in Africa (A. gambiae) and Asia (A. stephensi) and the presence of other sympatric vectors, CPBAs1 could be introduced as a TBV candidate in regions where A. stephensi is the main malaria vector, and this will broaden the scope for the potential wider application of CPBAs1 antigen homologs/orthologs.
INTRODUCTION
Malaria is one of the major public health problems in the world, especially in Africa and Asia. In recent years, malaria eradication programs have been managed by WHO, and the research agenda for malaria eradication has focused on tools that can be used in current campaigns. Vaccines that interrupt malaria transmission are one of the emphasized tools (1). Furthermore, vaccines are economical and effective tools for solving public health problems, especially in poor countries. Most of the malaria vaccine development studies have focused on reducing the morbidity and mortality of malaria, particularly with Plasmodium falciparum. According to the malaria eradication research agenda guidelines, one of the criteria that should be considered in malaria vaccine development is the efficacy of a designed vaccine in blocking malaria transmission (1). One kind of vaccine which interrupts malaria transmission is the classical transmission-blocking vaccine (TBV) that targets especially the sexual and mosquito stages of parasite antigens. The majority of the TBV projects have focused on parasite antigens, which have a critical role in sexual parasite development (2–6). However, some recent studies focusing on mosquito-derived antigens such as carboxypeptidase B of Anopheles gambiae (CPBAg1) have revealed that these mosquito cofactors are necessary for parasite development (7–11).
In 2001, Bonnet et al. (12) identified a transcript whose expression was specifically upregulated after ingestion of P. falciparum gametocytes. In 2005, Lavazec et al. (13) determined the full-length sequence of that transcript (the cpbAg1 gene), and the recombinant form of its related protein was characterized.
Later in 2007, the effect of anti-CPB polyclonal antibodies on P. falciparum development was evaluated by Lavazec et al. (11). They found that these antibodies could inhibit parasite sexual development in the mosquito midgut. In consequence, CPBAg1 was introduced as a TBV candidate for areas where A. gambiae plays a critical role in malaria transmission.
The main malaria vector in Africa is A. gambiae. Anopheles stephensi has a central role in malaria transmission as well, and it is predominant in the Indian subcontinent (except for Nepal and Sri Lanka) and also distributed across the Middle East and South Asia in Afghanistan, Bahrain, Bangladesh, China, Egypt, India, Iran, Iraq, Oman, Pakistan, Saudi Arabia, and Thailand. Therefore, we decided to characterize the equivalent gene (cpbAs1) and its related protein (CPBAs1) in A. stephensi and evaluate its competency as a TBV target.
Carboxypeptidases are exopeptidases that remove a single amino acid residue from the C terminus of proteins or peptides. Digestive carboxypeptidases belong to a family of zinc-containing enzymes that, based on their substrate specificity, can be divided into three groups: A, B, and C (13). Group A (CPA) preferentially cleaves the C-terminal hydrophobic residues, group B cleaves basic residues (arginine [Arg] and lysine [Lys]) from the C terminus (14, 15), and group C has specificity for glutamate residues (16). In insects, the activity of carboxypeptidase A or B has been found in the midgut of diverse species of both phytophagous and hematophagous insects.
In hematophagous insects, the activity of carboxypeptidase A is increased significantly after blood feeding, and gene sequences encoding carboxypeptidase A have been reported in different insects (17–19). Furthermore, midgut activity of CPB has been reported in Glossina, Aedes, and Anopheles species (18, 20, 21). In addition, a gene that encodes CPB has been described in Glossina morsitans (22). However, Bown and Gatehouse (16) believe that this gene probably encodes carboxypeptidase C. In this study, the full-length mRNA sequence of the cpbAs1 gene and its related protein (CPBAs1), its expression pattern after P. falciparum gametocyte ingestion, and the effect of anti-CPB directed antibodies on P. falciparum development in A. stephensi midgut have been reported. Notably, CPBAs1 is the second CPB from the medically important insect vectors that had not been characterized previously.
MATERIALS AND METHODS
Primer design.
Because the A. stephensi genome has not been sequenced yet, and according to the recommendations of Scotto-Lavino et al. (23) for 3′ rapid amplification of cDNA ends (RACE) of nonsequenced species, the cpb-1 mRNA sequences of different mosquito vectors, such as A. gambiae (GenBank accession no. AY545988), Culex pipiens (GenBank accession no. XM_001856118), and Aedes aegypti (GenBank accession no. AY590494), were aligned by MEGA4 software. After analysis, five regions were selected for designing the gene-specific primers and AnSt314, AnSt615, AnSt808, R1, and R2 primers were designed (Table 1).
Table 1.
List of the primers that were designed and used in this studya
| Primer name | Sequence |
|---|---|
| Linker | 5′-GAGATTTGAATCTTGCTTCTGGGCCCTCTATTGTCATTGTCTTTTTTTTTTTTTTTTT-3′ |
| Outer | 5′- GAGATTTGAATCTTGCTTCTG-3′ |
| Inner | 5′-GGCCCTCTATTGTCATTGTC-3′ |
| AnSt314 | 5′-CAGGAGTTGCTGAATCGAG-3′ |
| AnSt615 | 5′-GGTATTCATGCCAGAGAATG-3′ |
| AnSt808 | 5′-CAACTTCCCCTTCCAGTG-3′ |
| R1 | 5′-GTCAGCTCGAGCGTGTAC-3′ |
| R2 | 5′-GGTTGATTTATTGCCACTG-3′ |
| AnSt223 | 5′-CCGTAACTTCCCTTTCATG-3′ |
| AnSt297 | 5′-CCGAGAACGAAACCAAAG-3′ |
| AnSt408 | 5′-ACGACTTCCTGCACGCAC-3′ |
| AnSt440 | 5′-CTGCAGCGTTTGGGTGAG-3′ |
| R427 | 5′-TGCAGATCTTCCGCGTTC-3′ |
| R328 | 5′-CGCCTTGTACTGGTCCATC-3′ |
| R74 | 5′-AGCTGTTCCGCATAGTCTTC-3′ |
| As.RT.F | 5′-GAGGATGTACAGGAGTTGCTGAAC-3′ |
| As.RT.R | 5′-CGTCCAGAAGTGTTCAAAGTTCAC-3′ |
| S7F | 5′-TGGAAATGAACTCGGATCTGAAG-3′ |
| S7R | 5′-ACCGGCACGTAGATGATGATC-3′ |
| ClCPBAs-F | 5′-GCTCTCGGATCCGGTTCCTATCATGAATTTGAAGTG-3′ |
| ClCPBAs-R | 5′-CTCAGGCTCGAGCGGCGTCAACGACAACGT-3′ |
Bold letters and underlining are related to the BamHI and XhoI recognition sites in cloning primers.
In the next step, regarding the middle part of the cpbAs1 mRNA sequence (GenBank accession no. GU818728.1), 3′ RACE (AnSt408, AnSt440, AnSt297, and AnSt223) and 5′ RACE (R427, R328, R74, and R68) gene-specific primers were designed (Table 1).
After total sequence determination of cpbAs1 mRNA sequence (GenBank accession no. GU818731.1), real-time PCR primers (As.RT.F and As.RT.R) were designed for cpbAs1 expression analysis (Table 1). Also, for normalization, the S7 gene was selected as an endogenous housekeeping gene (11, 13) and S7F and S7R were designed according to the sequence with GenBank accession no. AF539918 (Table 1). Furthermore, to eliminate the effect of probable genomic DNA contamination, real-time PCR primers were designed based on exon junction regions.
Bioinformatics.
In this study, all primers were designed with Gene Runner software (version 3.05, 1994; Hastings Software Inc.) and their specificity for PCR was checked by nucleotide BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Also, the three-dimensional structure of the proteins was predicted by the SWISS-MODEL web server (http://swissmodel.expasy.org/), and the signal peptide was analyzed by the SignalP 4.0 server (http://www.cbs.dtu.dk/services/SignalP/).
Ethics statement.
This work has been carried out just on mosquito samples collected in the field and on the insectarium strain from the National Insectarium, Pasteur Institute of Iran (PII). After in silico studies, the laboratory phase was started. Eggs and larvae of A. stephensi were collected from the Chabahar district in southeastern Iran, Sistan and Baluchestan province. All necessary permits were obtained for the described field studies as follows.
Before approval by PII, this project was reviewed and cleared by the Ethical Committee of PII. Hence, for mosquito collection from private residences, all residents provided their oral informed consent to have their residences used in the study, and this was coordinated through the local manager of the malaria control program of the Public Health Center in the study areas. Briefly, the principal investigator of the project, N. Dinparast Djadid, presented different sections of the project to the local staff, especially the field collection section. One to 2 weeks before field collection, the local manager of the malaria control program of the Public Health Center visited the study areas and, in a meeting with residents, asked their permission for mosquito collection at the defined date and time. As it is a routine procedure for other activities of the malaria control program in the region, the residents agreed to cooperate in the project. Then, the project was followed by the arrival of the research team at the agreed date and time within the study areas, and mosquito collection was performed. This procedure has been documented in the weekly activity report book of the malaria control program in the study areas and signed by N.D.D., the local manager of the malaria control program, and a representative from the study areas. Except for the private residences, no other collection sites were used.
Also, the rabbits used in this study were immunized in order to produce anti-CPB polyclonal antibodies. The license for work with experimental animal models (mouse and rabbit) was issued by the Animal Unit of PII to A. Raz.
Mosquitoes.
After in silico studies, the laboratory phase was started. Eggs and larvae of A. stephensi were collected from the Chabahar district in southeastern Iran, Sistan and Baluchestan province. Then, they were transferred to the National Insectarium of PII for routine breeding based on standard conditions: specifically, a temperature range of 26 to 28°C, 80% humidity, and a 12-h light/dark cycle (13). Day 5 postemergence mosquitoes were used in this study.
RNA extraction for standard reverse transcription-PCR (RT-PCR).
Total RNA was extracted from the midgut of anesthetized live mosquitoes with the RNeasy RNA extraction kit (Qiagen, Germany) and then treated with DNase I enzyme according to the manufacturer's instruction (Fermentas, USA) to remove probable DNA contamination. In addition, genomic DNA contamination was checked by As.RT.F and R427 primers after each RNA extraction (Table 1).
RT.
Reverse transcription (RT) was performed in a final volume of 20 μl by using the random hexamer, linker, or gene-specific primer (regarding the next reaction) as the primer. The volume of 5 μg of total RNA was adjusted to 14 μl by adding RNase-free distilled water. RNA was left at 75°C for 5 min to remove the secondary structures and immediately was cooled on ice. Then, RT mix (RevertAid Moloney murine leukemia virus [M-MULV], RNasin, deoxynucleoside triphosphate [dNTP] solution, RT buffer, and primer) was added to the cooled RNA, and RT was started by the following program: 10 min at 25°C, 60 min at 42°C, and 10 min at 70°C. All reagents were purchased from Fermentas, USA.
PCRs.
PCRs were performed in a total volume of 25 μl. The reaction mixture contained 400 nM (each) primers, 1 unit of Taq DNA polymerase, 0.2 mM (each) dNTPs, 0.001% gelatin, 2.5 μl of 10× reaction buffer, 1.5 mM MgCl2, and 1 μl of RT reaction product (for RT-PCR) or 100 ng of DNA for PCR as the template. The amplification program was as follows: initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 50 s, and extension at 72°C for 1 min, and an additional final extension at 72°C for 10 min. Then, the PCR products were analyzed by agarose gel electrophoresis.
In the first step, R1 and R2 were used as reverse primers in combination with AnSt314, AnSt584, and AnSt808 to perform PCR on prepared cDNA with random hexamers. With regard to our previous studies on different genes of A. stephensi and their close evolutionary relatedness to similar A. gambiae genes (24, 25), the size of the cpbAs1 PCR products was estimated based on the attachment site of primers on the cpbAg1 mRNA sequence (GenBank accession no. AY545988). Then, only primer combinations whose products were close to the expected sizes were selected for amplification with the Expand High Fidelity PCR system (Roche, Germany). Next, the desired bands were recovered with a gel extraction kit (Qiagen, Germany) and TA cloned in pDrive vector (Qiagen, Germany). For each band, more than five clones were sequenced with an ABI 310 DNA sequencer machine. After that, their contigs were aligned with nucleotide BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), and sequences with maximum similarity to cpbAg1 were selected.
3′ RACE.
Regarding the middle part of the sequence of the cpbAs1 mRNA molecule which was sequenced in the previous step, four primers were designed for 3′ RACE. First, the linker was used for reverse transcription as primer, and then Outer and AnSt223 primers were used for first-round PCR. Afterward, the combination of Inner (as reverse primer) and AnSt297, AnSt408, and AnSt440 (as forward primers) was used for the second-round PCR in three separate reactions to confirm the first round. After the confirmation, the first-round PCR product was TA cloned and sequenced (as mentioned above).
5′ RACE.
To perform 5′ RACE, R427 primer was used (as primer) for the reverse transcription reaction. At the end of the reaction, by using the deoxyribonucleotide terminal transferase enzyme (Fermentas, USA) and ATP (Fermentas, USA), a poly(A) tail was added to the 3′ end of the synthesized cDNA molecules according to the manufacturer's instruction. Then, linker and R328 primers (Table 1) were used for the first-round PCR. In the second-round PCR, cDNA molecules with added poly(A) (as the negative control) and first-round PCR product (1 μl of 1:20-diluted PCR product) were used as the templates for R74 (as reverse primer) and Outer (as forward primer) in two separate reactions. The confirmed PCR product was TA cloned and sequenced for 5′-end sequence determination of the cpbAs1 mRNA molecule.
Cloning, recombinant CPBAs1 expression, purification, and rabbit immunization and endotoxin measurement of the purified protein.
After complete sequence determination of the cpbAs1 mRNA molecule, ClCPBAs-F and ClCPBAs-R cloning primers (Table 1) with BamHI and XhoI restriction sites were designed to amplify the coding sequence of CPBAs1 without its signal peptide. After PCR amplification of cDNA with the Expand High Fidelity PCR system (Roche, Germany) and digestion with BamHI and XhoI, the digested PCR product was cloned into the digested pET-23a vector (Novagen, USA) with the same restriction enzymes. In the next step, the recombinant vector was verified and chemically transformed to Escherichia coli Origami for CPBAs1 in vitro expression.
By using the pET-23a vector, a C-terminal His tag-fused recombinant form of CPBAs1 protein was expressed. After 3 h of isopropyl-β-d-thiogalactopyranoside (IPTG) induction, nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen, Germany) was used for recombinant CPBAs1 purification according to the manufacturer's instructions.
To determine the amount of bacterial endotoxin in purified CPBAs1 recombinant protein, the Limulus amebocyte lysate (LAL) test was performed in the Quality Control Unit of the Recombinant Protein Production Complex of PII with the LAL chromogenic kit (Lonza, USA).
Then, the purified CPBAs1 protein was used for rabbit immunization with Freund's adjuvant (Sigma, Germany). Two rabbits (test and control) were used in this study. Before immunization, blood was collected from the two rabbits for confirmation of the immunization process and blocking assay tests. On day 0, 100 μg of recombinant CPBAs1 in sterile phosphate-buffered saline (PBS) emulsified with complete Freund's adjuvant (Sigma, Germany) was injected subcutaneously into the test rabbit and sterile PBS-adjuvant was injected into the control rabbit with the same protocol on the same day. On day 21, 50 μg of CPBAs1 formulated in incomplete Freund's adjuvant (Sigma, Germany) was injected into the test rabbit as the first booster and sterile PBS-incomplete adjuvant was injected simultaneously into the control rabbit. The immunization protocol on day 21 was repeated on day 42 as the second booster (26). Finally, on day 63, blood was collected from the two rabbits, their sera were separated, and immunization efficiency was analyzed by Western blotting.
Mosquito midgut protein extraction and Western blotting.
Twenty mosquito midguts were separated by dissection and homogenized with a tissue homogenizer and a plastic pestle in 200 μl of 25 mM Tris-HCl, pH 8. Then, the homogenized solution was centrifuged at 18,000 × g at 4°C for 15 min, and the supernatant (midgut protein cocktail) was used for analysis using SDS-PAGE and Western blotting.
This protein cocktail and His-tagged recombinant CPBAs1 were analyzed by 12% SDS-PAGE and then transferred to nitrocellulose (Amersham Hybond ECL) for Western blotting. After saturation in PBS containing 2% bovine serum albumin, the membrane was incubated with anti-CPBAs1 polyclonal rabbit antibodies (dilution, 1:1,000) that were produced in the previous steps. Horseradish peroxidase-conjugated anti-rabbit antibody (dilution, 1:5,000; Santa Cruz Biotechnology, USA) was used as secondary antibody, and its binding was visualized by incubating the membrane with SigmaFast diaminobenzidine (DAB) with metal enhancer (Sigma, Germany).
Biological activity assay.
Because CPBAs1 had been expressed in zymogen form, for mature (active) enzyme production, tryptic cleavage was performed as described by Ventura et al. (27). Briefly, CPBAs1 concentration was adjusted to 1 mg/ml in an activation buffer (50 mM Tris-HCl, 10 mM CaCl2, 150 mM NaCl, pH 8.0). Then, it was treated at a 100:1 (wt/wt) ratio of zymogen to trypsin at 25°C in the activation buffer for 30 min.
Biological activity was assayed by using the synthetic dipeptides hippuryl (Hip)-Arg and Hip-Lys as the CPB-specific substrate and Hip-Phe (phenylalanine) as the carboxypeptidase A-specific substrate as described by Lavazec et al. (13). All reagents were purchased from Sigma (Germany). In brief, 10 ng/μl of activated CPBAs1 in E buffer (100 mM NaCl, 50 mM HEPES, 100 μM ZnCl2, pH 7.2) was added to 20 μl of E buffer containing different concentrations of substrates (depending on type of assay), and then it was incubated at 25°C for 30 min. The reaction was stopped by adding an equal volume of ninhydrin reagent (Merck, Germany). Enzyme activity was determined by measuring the released amino acids using the ninhydrin procedure. The preparation of the ninhydrin reagent and the ninhydrin assay were performed as described by Sun et al. (28). Finally, the absorbance at 570 nm was measured by SmartSpect Plus (Bio-Rad, USA), and 1 unit of enzyme activity was defined as μmol of released amino acids/min.
Km and kcat were determined experimentally by recording the progress of the CPBAs1-catalyzed reaction with the fixed amounts of enzyme and a series of different Hip-Arg concentrations (0.02 to 10 mM); all assays were performed in triplicate. Finally, the CPBAs1 kinetic parameters were deduced from a Lineweaver-Burk plot.
More characterizations were performed through the inhibition assay with 1,10-phenanthroline (a chelating agent) (Sigma, Germany) and arginine analog GEMSA (2-guanidinoethylmercaptosuccinic acid; Santa Cruz Biotechnology, USA) on CPBAs1 activity with 0.1 to 1,000 μM inhibitor concentration. First, inhibitor was incubated with the active form of CPBAs1 at 25°C for 30 min, and enzyme activity was assayed by Hip-Arg as described above. Finally, results were reported as the percentage of activity in test samples relative to the control samples, whose activity was considered 100% (13).
Parasite culture.
According to the previous studies, one of the best gametocyte-producing strains of P. falciparum, the 3D7 strain, was used in this study. In continuous in vitro cultures, the ability to produce gametocytes and their infectiousness seem to drop significantly after 3 months (29). Therefore, for more gametogenesis and for effective mosquito infection, parasites were cultured from freshly thawed samples in a period of less than 3 months.
Parasites were continuously cultured based on the Trager and Jensen method (30) with the exception mentioned above (restarting the cultures from freshly thawed samples in a period of less than 3 months). Also, for efficient mosquito infection, gentamicin sulfate was not added to parasite culture medium (31).
Concisely, parasites were cultured in continuous culture for less than 3 months on human erythrocytes (RBCs) (blood group O+ was obtained from the Blood Transfusion Organization, Tehran, Iran) in RPMI 1640 medium (Gibco, USA) supplemented with 10% pooled human AB+ serum, 25 mM HEPES, 25 mM NaHCO, pH 7.2. The cultures were incubated at 37°C in an atmosphere of 6% CO2, 3% O2, and 91% N2.
Gametocyte culture.
Gametocytes were produced based on the previously described method (31) with minor modifications for improvement. Briefly, before starting the gametocyte production, parasite cultures were synchronized twice (on days −8 and −4) to the ring stage by treatment with 5% d-sorbitol (32). Large tissue culture flasks (75 cm3) were used for large-scale production of gametocytes. At day 0, cultures were set up at 0.7% parasitemia and 6% hematocrit in complete RPMI. Before starting the large-scale production of gametocytes, medium and flasks were warmed to 37°C to prevent the effect of cold shock on cultures. Medium (15 ml complete medium per flask) was replaced at the same time every day. Furthermore, medium replacement was performed in the minimum time and flasks were placed immediately in an incubator. Also, flasks were shaken very gently until high parasitemia was detected (usually until the 5th day). Smears were taken on day 4 to determine the best time for starting the next step. High parasitemia and parasites showing signs of stress (such as triangular ring stages, blurred-looking trophozoites, and schizonts) were the signs of the suitable time. These signs were seen on day 5, and then some changes were applied on parasite cultures. After that, shaking was stopped and medium replacement volume was increased from 15 ml to 22.5 ml. A final concentration of 50 mM N-acetylglucosamine was added to complete medium for at least 5 days to isolate the late-stage gametocytes (stages III to V) and eliminate the parasite asexual stages. N-Acetylglucosamine has selective toxicity for trophozoites and schizonts and kills these stages with no toxic effect on gametocyte development (33). Elimination of asexual stages is very important for quantitative real-time PCR experiments. Smears were taken twice a week to check the parasite stages, probable contamination, and the progress in elimination of asexual stages. Mature gametocytes were seen at day 15. Different stages of gametocytes were distinguished according to the Carter and Miller guidelines (34), and mosquito infection was performed on day 17.
Mosquito feeding.
Female mosquitoes were selected on day 4 postemergence and starved for 12 h before feeding. Artificial blood feeding of female mosquitoes was performed on day 5 postemergence. Three mosquito groups were considered for transmission-blocking assays and quantitative real-time PCR analysis: group 1, the quantitative real-time PCR control group; group 2, the transmission-blocking assay control group; and group 3, the transmission-blocking assay test group. Groups 1 and 2 were compared using quantitative real-time PCR experiments to analyze the effect of mature P. falciparum gametocytes on cpbAs1 expression pattern, and groups 2 and 3 were included in transmission-blocking assays to evaluate the effect of anti-CPBAs1 antibodies on sexual parasite development. After feeding, engorged mosquitoes were maintained inside the insectarium and all mosquito cages were kept in the same condition. Artificial membrane feeding was performed according to the previously described methods (35, 36).
Preparation of blood samples for mosquito feeding.
In a series of transmission-blocking assays on day 17, serum (3 ml [1 ml for each group]) of fresh human O+ blood group was removed after centrifugation at 4,000 × g for 5 min. The remaining erythrocytes (RBCs) were washed three times with an equal amount of heat-inactivated AB+ serum. Then, a thin smear on a glass slide was prepared from the parasite culture with Giemsa stain to evaluate the probable contamination, gametocytemia percentage, and accuracy in elimination of the parasite asexual stage. Afterward, the culture medium was removed by centrifugation at 4,000 × g for 5 min and gametocytemia of washed RBCs for the transmission-blocking assay was adjusted to 0.7% (1 ml of washed RBCs was not infected and was used for feeding the quantitative PCR [Q-PCR] control group). Next, an equal volume of the serum removed from the washed RBCs was replaced by (i) naive rabbit serum before immunization for the Q-PCR and transmission-blocking assay control groups and (ii) immunized rabbit serum against recombinant CPBAs1 for the transmission-blocking assay test group. Rates and intensities of infections in each blocking assay were scored on day 7 after blood feeding, and their significant differences were determined by the chi-square test and analysis of variance (ANOVA), respectively.
Transmission-blocking assay.
Two mosquito groups were included in transmission-blocking assays: (i) the control group that was fed with infected RBCs by mature gametocytes and naive rabbit serum replacement and (ii) the test group that was fed with infected RBCs by mature gametocytes and anti-CPBAs1 directed rabbit antibody replacement. Transmission-blocking assays and quantitative real-time PCR experiments were performed in three independent experiments.
Sampling and RNA extraction for gene expression analysis.
Sampling was carried out at 0 (after finishing the feeding), 1, 3, 6, 8, and 24 h after blood feeding from the control and infected groups. Six mosquitoes were sampled at the mentioned times from the Q-PCR and transmission-blocking assay control groups (total of 36 mosquitoes for each group) in each infection experiment, and their results were compared. Mosquito midguts were separated by dissection and kept at −80°C until RNA extraction. RNA extraction was performed as previously described, and finally extracted RNAs were treated with DNase I enzyme (Fermentas, USA). To reduce deviation during the reverse transcription reaction, RT reactions were performed in triplicate (as described above) and RT products were pooled, aliquoted, and kept at −20°C until use. Also, thawed products were not used (37), and genomic DNA contamination was checked with As.RT.F and R427 (Table 1) primers after each RNA extraction. Furthermore, a negative control was included in reverse transcription reactions to check the genomic DNA contamination in quantitative real-time PCR experiments.
Quantitative real-time PCR.
Quantitative real-time PCR was carried out in triplicate by using the Power SYBR Green PCR Master Mix (Applied Biosystems, United Kingdom) and the Roto-Gene Q instrument (Qiagen, USA). PCRs were performed in 20-μl final volumes containing 900 nM (each) forward and reverse primers, 5 μl of a 1:5 dilution of the pooled RT product, and 10 μl of 2× Master Mix components according to the manufacturer's instructions.
The threshold cycle (2−ΔΔCT) method was selected for quantitative real-time PCR data analysis. Also, for validated ΔΔCT calculation, ΔCT of S7 and cpbAs1 genes was evaluated in different template dilutions according to the guidelines of Livak and Schmittgen (38). Quantitative PCR was performed for three independent infections, and their results were compared by the Wilcoxon test (11).
Nucleotide sequence accession numbers.
In this study, different parts of the cpbAs1 mRNA sequence were determined: the middle part of the cpbAs1 mRNA sequence (GenBank accession no. GU818728.1), the 3′ end of the cpbAs1 mRNA sequence (GenBank accession no. GU818729.1), the 5′ end of the cpbAs1 mRNA sequence (GenBank accession no. GU818730.1), the cpbAs1 total mRNA sequence (GenBank accession no. GU818731.1), and the cpbAs1 genomic sequence (GenBank accession no. GU818732.1).
RESULTS
cpbAs1 mRNA and gene sequence characterization.
The sequencing and analyzing of the first PCR revealed that only the product of AnSt.584 and R1 primers was related to the cpbAs1 gene. This sequence was submitted to GenBank (GenBank accession no. GU818728.1) and used for designing the gene-specific primers for 3′ and 5′ RACE techniques. In the next step, the 3′ end (GenBank accession no. GU818729.1) and the 5′ end (GenBank accession no. GU818730.1) of the cpbAs1 mRNA sequence were determined. Finally, by assembling these sequences (GenBank accession no. GU818728.1, GenBank accession no. GU818729.1, and GenBank accession no. GU818730.1), the cpbAs1 total mRNA sequence was determined (GenBank accession no. GU818731.1). The alignment of cpbAs1 mRNA sequence with cpbAg1 mRNA sequence revealed that they have 83.8% similarity. The full-length cpbAs1 mRNA sequence is 1,436 bp, and its open reading frame is 1,269 bp, encoding a protein composed of 423 amino acids (aa) with a predicted molecular mass of 48.1 kDa (Fig. 1). Moreover, the cpbAs1 gene was amplified with the cloning primers ClCPBAs-F and ClCPBAs-R, sequenced, and submitted to GenBank (GenBank accession no. GU818732.1). Comparison of GenBank sequences with the accession numbers GU818731.1 and GU818732.1 revealed that the cpbAs1 gene has two introns and that its structure is different from that of the cpbAg1 gene, which was reported to have three introns (13).
Fig 1.

Full-length sequence of cpbAs1 cDNA and its predicted amino acid sequence. Nucleotide and amino acid numbers are shown on the left. The solid arrow shows the signal-peptide cleavage site, and the open arrow shows the zymogen activation site. The amino acids that are predicted to include the active site and are involved in zinc binding are shown in boxes, and residues that are involved in substrate binding and cleavage are shown in bold. The aspartic acid residue that is predicted to determine the substrate specificity is underlined. The lowercase letters at the begining and the end of the sequences are related to 3′- and 5′-untranslated regions, respectively.
Structural characterization of CPBAs1.
The CPBAs1 sequence was analyzed with the SignalP 4.0 server, and it was demonstrated that this protein is translated as a preproprotein with a signal sequence at aa 1 to 20 and a prodomain at aa 21 to 115. The presence of a putative signal peptide revealed that its zymogen is secreted and activated after cleavage at the Arg114 position. These features are similar to those of other characterized carboxypeptidases which are translated as preproproteins (39).
Furthermore, CPBAs1 and CPBs of different organisms were aligned by ClustalW for structural analysis (Fig. 2). Interestingly, the critical amino acids in the active site and the pocket binding site of CPBAs1 were completely similar to well-characterized CPB enzymes of other organisms (Fig. 2). According to this alignment, it is predicted that His187, Glu190, and His309 compose the active site of the CPBAs1 enzyme and coordinate the zinc ion. These residues were completely conserved among all aligned CPBs. Also, Glu382 and Arg242, which cover the polarization and cleavage of the accessible carbonyl group of the substrate, were conserved and close to the zinc ion (Fig. 2).
Fig 2.

Alignment of CPBAs1 with 10 CPB enzymes of other organisms. The residues that are involved in zinc and substrate binding are shown in boxes. The GenBank nucleotide sequence accession numbers of the aligned sequences are as follows: carboxypeptidase B of Anopheles stephensi, ADD31639.1; carboxypeptidase B precursor of Anopheles gambiae, AAS99341.1; carboxypeptidase B preproprotein of Homo sapiens, NP_001862; carboxypeptidase B2 of Homo sapiens, AAT97987.1; carboxypeptidase B precursor of Rattus norvegicus, NP_036665.1; carboxypeptidase B precursor of Sus scrofa (porcine), P09955.5; carboxypeptidase B of Aedes aegypti, AAT36734.1; carboxypeptidase B of Aedes aegypti, AAT36733.1; carboxypeptidase B of Aedes polynesiensis, AAT36736.1; zinc carboxypeptidase of Glossina morsitans morsitans, ADD19124.1; Drosophila sechellia, XP_002035702.1; carboxypeptidase B of Culex quinquefasciatus, XP_001856154.1; carboxypeptidase of Eupolyphaga sinensis, AEI58636.1.
In addition, another conserved critical functional domain was the peptide-binding pocket that included Arg189, Tyr310, Phe391, Arg259, Tyr360, and Asp367. According to the work of Titani et al. (14), it seems that Tyr360 and Arg259 interact with the terminal carboxyl group of substrates and that Asp367 determines the enzyme specificity for basic C-terminal residues.
Also, the three-dimensional structures of CPBAs1 and CPBAg1 were predicted and analyzed with the SWISS-MODEL web server. Based on their high similarity in critical domains, their structures and three-dimensional conformations (alpha helices and beta sheets) were very similar, especially in the substrate-binding pocket and the active site (Fig. 3).
Fig 3.
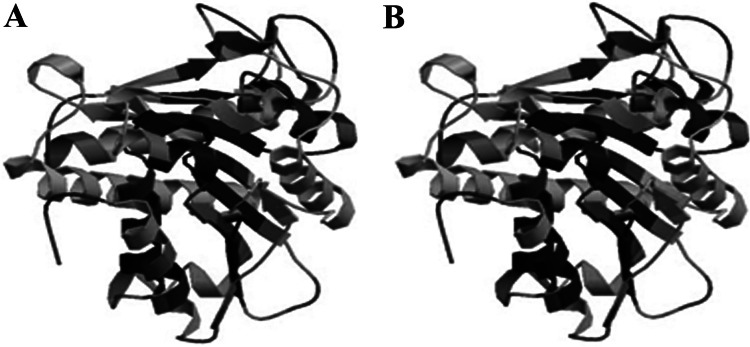
Three-dimensional structures of CPBAs1 (A) and CPBAg1 (B). The model visualization was performed by the SWISS-MODEL web server (http://swissmodel.expasy.org/).
Recombinant CPBAs1 in vitro expression and Western blotting.
In vitro expression of CPBAs1 in E. coli Origami resulted in production of a recombinant protein with about a 48-kDa molecular mass (without signal peptide) (Fig. 4). After purification in the native condition, the amount of endotoxin in the purified CPBAs1 was determined by the Limulus amebocyte lysate test. The endotoxin concentration of the purified CPBAs1 was 720 endotoxin units (EU)/ml. Then, recombinant CPBAs1 was used for rabbit immunization. According to the CPBAs1 purified protein (1.5 mg/ml) and endotoxin (720 EU/ml) concentrations, 48 EU of endotoxin in the first injection and 24 EU in the first and second boosters were injected into the test rabbit in the animal immunization process with the purified CPBAs1. It should be mentioned that our animals were checked by a veterinarian routinely three times per week, but in the case of any injection, animals were under observation for the first 72 h after injection to control any adverse effects such as fever. However, our follow-up showed no sign of fever at all in the examined animals. Western blotting was performed to evaluate the ability of rabbit anti-CPBAs1 polyclonal antibodies to recognize the CPBAs1, which had been extracted from A. stephensi midgut. Notably, two natural forms of CPBAs1 were detected by rabbit polyclonal antibodies (Fig. 4). The two forms, with molecular masses of about 48 kDa and 37 kDa, were related to zymogen and mature (activated) forms of CPBAs1, respectively.
Fig 4.

SDS-PAGE and Western blotting of CPBAs1. (A) SDS-PAGE analysis of recombinant CPBAs1 that was expressed in E. coli Origami after purification with Ni-NTA agarose. (B) Western blotting of midgut-extracted protein cocktail (lane 1) and purified recombinant CPBAs1 (lane 2). To perform Western blotting, rabbit anti-CPBAs1 polyclonal antibody and horseradish peroxidase-conjugated anti-rabbit antibody were used as first and second antibodies, respectively.
Biological activity assay.
The specific activity of recombinant CPBAs1 was determined against Hip-Arg and Hip-Lys (as the CPB-specific substrates) and Hip-Phe (as the carboxypeptidase A-specific substrate). CPBAs1 specific activity against Hip-Arg, Hip-Lys, and Hip-Phe was 15,200 ± 6, 2,757.4 ± 4, and 0 unit/mg, respectively (1 unit of enzyme is defined as the amount of enzyme that produces 1 μmol of product/min).
Consequently, our results showed that recombinant CPBAs1 has enzymatic activity against Hip-Arg and Hip-Lys but not against Hip-Phe. In addition, CPBAs1 activity against Hip-Arg was approximately 5.5-fold greater than that against Hip-Lys, indicating its preference for arginine residue cleavage.
The effect of CPB inhibitors on CPBAs1 activity showed that CPB activity was inhibited by GEMSA, which is an Arg analog and active site-directed inhibitor (Fig. 5). Furthermore, the general zinc protease inhibitor 1,10-phenanthroline as a chelating agent inhibited CPBAs1 activity, probably due to the chelation of zinc ion cofactor (Fig. 5). These results confirmed that cpbAs1 encodes an active CPB, and they are consistent with other CPB reports (13, 40–42). The CPBAs1 kinetic parameters were determined by the progress of an enzyme-catalyzed reaction using fixed amounts of CPBAs1 and a series of different concentrations of Hip-Arg as the substrate according to the standard Michaelis-Menten method. The Km was calculated by a Lineweaver-Burk plot as 2.22 mM with a specificity constant (kcat/Km) of 5,488 mM−1 s−1.
Fig 5.
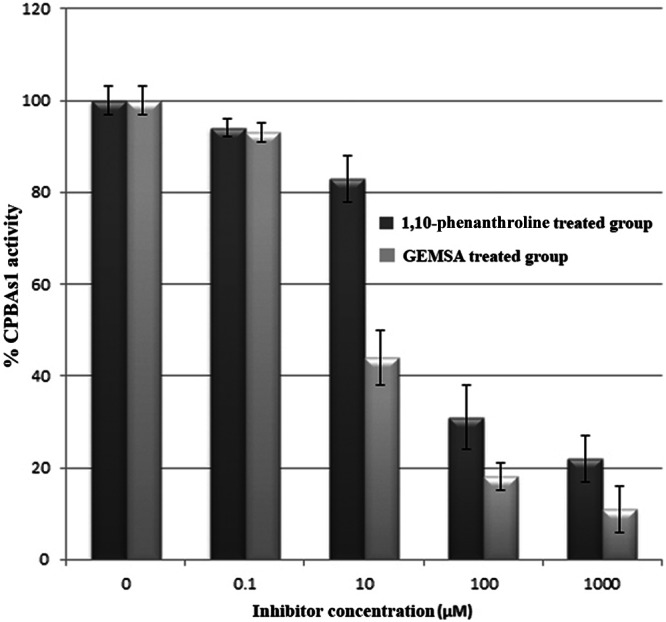
The effect of GEMSA (2-guanidinoethylmercaptosuccinic acid) and 1,10-phenanthroline on CPBAs1 activity. The activity of the recombinant CPBAs1 was assayed after incubation with the zinc-carboxypeptidase inhibitor 1,10-phenanthroline and GEMSA (the active site-directed inhibitor), using Hyp-Arg as the substrate. The activity of 100% corresponds to 15,125 and 14,840 units/mg for 1,10-phenanthroline and GEMSA experiments, respectively.
Quantitative real-time PCR experiments.
The expression pattern of cpbAs1 was analyzed by quantitative real-time PCR after blood feeding in different time frames in control and infected groups in three independent infection experiments. Our results revealed that cpbAs1 expression is downregulated in response to blood feeding in the control group until 24 h post-blood meal (PBM). However, the expression pattern of cpbAs1 in the infected group was completely different. The cpbAs1 expression was increased after 6 h PBM in comparison to the control group with 2.2-, 4.5-, and 2.18-fold increases at 6, 8, and 24 h PBM, respectively (Fig. 6). Comparing the quantitative real-time PCR results of the two groups by the Wilcoxon test showed that differences in expression levels of cpbAs1 at 6, 8, and 24 h PBM are statistically significant.
Fig 6.

Expression pattern analysis of cpbAs1 gene in A. stephensi midgut after ingestion of mature gametocytes. The expression pattern of the cpbAs1 gene was analyzed in different time frames after blood feeding on day 5 postemergence. The S7 ribosomal protein gene was used as the housekeeping gene for normalizing the data. Asterisks indicate statistically significant differences compared to the noninfected blood-fed group, based on the P value from the Wilcoxon test (<0.05). Bars indicate standard deviations from the three independent experiments.
Transmission-blocking assays.
To evaluate the effect of anti-CPBAs1 antibodies on P. falciparum sexual development in the midgut of A. stephensi, we used a rabbit serum directed against recombinant CPBAs1 in the test group in comparison to naive rabbit serum (the same rabbit serum before immunization), which was used in the control group. Our results on day 7 postinfection showed that the addition of anti-CPBAs1 rabbit serum to infected blood with P. falciparum mature gametocytes can inhibit sexual parasite development and reduce the infection rate to 2.6% in the test group in comparison to 55.6% in the control group (Fig. 7). These results were consistently obtained in three independent infection experiments. Furthermore, the infected mosquitoes in the test group that had fed on anti-CPBAs1 antibodies harbored fewer oocysts than did the infected mosquitoes in the control group that had fed on the same infected blood, with the exception of naive rabbit serum addition (Fig. 7).
Fig 7.
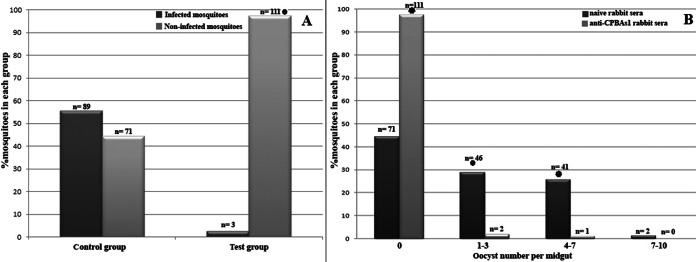
Evaluating the effect of anti-CPBAs1 antibodies on sexual P. falciparum development. The frequency of infected mosquitoes (A) and intensity of infection (B) were evaluated on day 7 postinfection after midgut dissection. The proportion of the infected mosquitoes was determined by mean value from each series of three independent infections. Asterisks indicate statistically significant differences at the 95% confidence level, based on the P value from the chi-square test (A) and ANOVA (B). Numbers above the columns indicate the sample size.
DISCUSSION
The mechanism of CPB in parasite development has not been elucidated yet. The P. falciparum genome project revealed that there is not a coding gene for CPB in its genome (43, 44). Therefore, it seems that a part of the parasite nutritional requirements is provided by mosquito carboxypeptidases. The findings of Lavazec and Bourgouin (44) support this hypothesis. They found that supplementing the P. falciparum-infected blood with Arg and Lys increases the efficiency of parasite development in A. gambiae and the mosquito infection rate. The outcome of the current study is the first report of molecular characterization of CPBAs1 and the second report of CPB characterization in insect vector species. Similarities of 83.8% and 89.6% between the mRNA and protein sequences of CPB in A. stephensi and A. gambiae, respectively, point out that their structure and function are the same. Moreover, our complementary experiments on biological activity assays support this hypothesis. Comparing the three-dimensional structure of CPBAs1 with that of CPBAg1 revealed that these two enzymes have similar conformations especially in the signal peptide, prodomains, and active moiety (Fig. 3).
Biological activity assays revealed that CPBAs1 has a preference for Arg and Lys residue cleavage and has no activity against Hip-Phe as the carboxypeptidase A-specific substrate. Moreover, its specificity for Arg is higher than that for Lys residues as reported for other CPBs (13, 45). The recombinant CPBAs1 Km is about 8.5-fold greater than the amounts which have been reported for the CPB enzyme of vertebrates by using the same substrates (13, 40–42). Furthermore, the CPBAs1 alignment with CPB enzymes of other organisms (Fig. 2) disclosed that CPBAs1 in critical points, such as the active site (His187, Glu190, and His309), the substrate-binding pocket (Arg189, Tyr310, Phe391, Arg259, and Tyr360), and the residue that determines the enzyme specificity (Asp367), is completely similar to other CPBs, especially CPBAg1 (13) (Fig. 2).
Expression pattern analysis of cpbAs1 in different time frames showed that the presence of sexual stages of P. falciparum triggers its upregulation in the mosquito midgut lumen. Because the serum was related to the same rabbit (before and after immunization with recombinant CPBAs1), it seems that this upregulation is related to the presence of P. falciparum gametocytes in the mosquito midgut. This increase at 6, 8, and 24 h PBM was statistically significant (by the Wilcoxon test) compared to the control group, and the highest increase was seen at 8 h PBM, which corresponds to the transformation of zygotes to ookinetes in the mosquito lumen (36, 46). Also, having a high level of expression at 24 h PBM may be related to the ookinete presence, since this time corresponds to the peak of ookinete abundance in the midgut (36, 46).
Moreover, transmission-blocking assays in three independent infections demonstrated that anti-CPBAs1 antibody could reduce significantly the infectivity of P. falciparum parasite in the A. stephensi midgut.
Briefly, a TBV candidate should have the following specifications: (i) antibodies directed against the TBV candidate should be able to restrict the Plasmodium parasite development in the mosquito, (ii) it should have very low antigenic diversity, and (iii) it should have good antigenicity for the human immune system and the ability to induce high titers of antibody. Therefore, a parasite or vector molecule that possesses these features can be introduced as a vaccine candidate that interrupts malaria transmission. As mentioned before, most of the research has focused on parasite-derived targets, and in a few studies, mosquito-based targets have been introduced. Our data revealed that CPBAs1 has the essential criteria for a TBV candidate in the Middle East region, where A. stephensi is the main malaria vector. In addition, CPBAs1 is a mosquito-derived antigen, which is not under the pressure of the human immune system.
To sum up, with regard to the high similarity of carboxypeptidase enzymes among different insect species, especially between the two main malaria vectors in Africa (A. gambiae) and Asia (A. stephensi), it seems that CPB can be considered a universal TBV candidate for countries in which malaria is endemic, after further investigations. This hypothesis may be supported by the fact that A. stephensi is sympatric with other prevalent major malaria vector species (mostly species complexes) from the Middle East up to China, including A. culicifacies, A. fluviatilis, A. superpictus, A. pulcherrimus, A. minimus, A. hyrcanus, A. annularis, and A. sundaicus. Accordingly, with respect to the transmission dynamics, the presence of those primary and secondary vectors, and the evolutionary relatedness of Anopheles species, the outcome of the current study will somehow extend a broader scope for the potential wider application of CPBAs1 or its homologs (or orthologs) due to its possible cross-reactivity as a TBV in those sympatric vector species when anti-CPB antibodies are ingested.
ACKNOWLEDGMENTS
We thank A. Raeisi and his colleagues in the Malaria Control Program, Center for Diseases Management and Control, Iran, and Zahedan University of Medical Sciences for their kind assistance in national and provincial coordination. Also, our gratitude goes to Catherine Bourgouin for reviewing the manuscript and her comments. We appreciate the Tehran Blood Transfusion Organization for providing the blood and serum required for malaria parasite culture and the Research and Production Complex of PII (Dr. Azizi and his colleagues) for assistance in performing the LAL test.
This project has been funded by PII through grant 364 to Navid Dinparast Djadid. The Education Office of PII has also provided partial funding to Abbasali Raz for his Ph.D. thesis.
Footnotes
Published ahead of print 8 April 2013
REFERENCES
- 1. malERA Consultative Group on Vaccines 2011. A research agenda for malaria eradication: vaccines. PLoS Med. 8:e1000398 doi:10.1371/journal.pmed.1000398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duffy PE, Kaslow DC. 1997. A novel malaria protein, Pfs28, and Pfs25 are genetically linked and synergistic as falciparum malaria transmission-blocking vaccines. Infect. Immun. 65:1109–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hisaeda H, Yasutomo K. 2002. Development of malaria vaccines that block transmission of parasites by mosquito vectors. J. Med. Invest. 49:118–123 [PubMed] [Google Scholar]
- 4. Kaslow DC. 1997. Transmission-blocking vaccines: uses and current status of development. Int. J. Parasitol. 27:183–189 [DOI] [PubMed] [Google Scholar]
- 5. Kaslow DC. 2002. Transmission-blocking vaccines. Chem. Immunol. 80:287–307 [DOI] [PubMed] [Google Scholar]
- 6. Tsuboi T, Tachibana M, Kaneko O, Torii M. 2003. Transmission-blocking vaccine of vivax malaria. Parasitol. Int. 52:1–11 [DOI] [PubMed] [Google Scholar]
- 7. Dinglasan RR, Fields I, Shahabuddin M, Azad AF, Sacci JB., Jr 2003. Monoclonal antibody MG96 completely blocks Plasmodium yoelii development in Anopheles stephensi. Infect. Immun. 71:6995–7001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lal AA, Patterson PS, Sacci JB, Vaughan JA, Paul C, Collins WE, Wirtz RA, Azad AF. 2001. Anti-mosquito midgut antibodies block development of Plasmodium falciparum and Plasmodium vivax in multiple species of Anopheles mosquitoes and reduce vector fecundity and survivorship. Proc. Natl. Acad. Sci. U. S. A. 98:5228–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shahabuddin M, Lemos FJ, Kaslow DC, Jacobs-Lorena M. 1996. Antibody-mediated inhibition of Aedes aegypti midgut trypsins blocks sporogonic development of Plasmodium gallinaceum. Infect. Immun. 64:739–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Srikrishnaraj KA, Ramasamy R, Ramasamy MS. 1995. Antibodies to Anopheles midgut reduce vector competence for Plasmodium vivax malaria. Med. Vet. Entomol. 9:353–357 [DOI] [PubMed] [Google Scholar]
- 11. Lavazec C, Boudin C, Lacroix R, Bonnet S, Diop A, Thiberge S, Boisson B, Tahar R, Bourgouin C. 2007. Carboxypeptidases B of Anopheles gambiae as targets for a Plasmodium falciparum transmission-blocking vaccine. Infect. Immun. 75:1635–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonnet S, Prevot G, Jacques JC, Boudin C, Bourgouin C. 2001. Transcripts of the malaria vector Anopheles gambiae that are differentially regulated in the midgut upon exposure to invasive stages of Plasmodium falciparum. Cell. Microbiol. 3:449–458 [DOI] [PubMed] [Google Scholar]
- 13. Lavazec C, Bonnet S, Thiery I, Boisson B, Bourgouin C. 2005. cpbAg1 encodes an active carboxypeptidase B expressed in the midgut of Anopheles gambiae. Insect Mol. Biol. 14:163–174 [DOI] [PubMed] [Google Scholar]
- 14. Titani K, Ericsson LH, Walsh KA, Neurath H. 1975. Amino-acid sequence of bovine carboxypeptidase B. Proc. Natl. Acad. Sci. U. S. A. 72:1666–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gardell SJ, Craik CS, Clauser E, Goldsmith EJ, Stewart CB, Graf M, Rutter WJ. 1988. A novel rat carboxypeptidase, CPA2: characterization, molecular cloning, and evolutionary implications on substrate specificity in the carboxypeptidase gene family. J. Biol. Chem. 263:17828–17836 [PubMed] [Google Scholar]
- 16. Bown DP, Gatehouse JA. 2004. Characterization of a digestive carboxypeptidase from the insect pest corn earworm (Helicoverpa armigera) with novel specificity towards C-terminal glutamate residues. Eur. J. Biochem. 271:2000–2011 [DOI] [PubMed] [Google Scholar]
- 17. Edwards MJ, Lemos FJ, Donnelly-Doman M, Jacobs-Lorena M. 1997. Rapid induction by a blood meal of a carboxypeptidase gene in the gut of the mosquito Anopheles gambiae. Insect Biochem. Mol. Biol. 27:1063–1072 [DOI] [PubMed] [Google Scholar]
- 18. Noriega FG, Edgar KA, Bechet R, Wells MA. 2002. Midgut exopeptidase activities in Aedes aegypti are induced by blood feeding. J. Insect Physiol. 48:205–212 [DOI] [PubMed] [Google Scholar]
- 19. Ramos A, Mahowald A, Jacobs-Lorena M. 1993. Gut-specific genes from the black fly Simulium vittatum encoding trypsin-like and carboxypeptidase-like proteins. Insect Mol. Biol. 1:149–163 [DOI] [PubMed] [Google Scholar]
- 20. Gooding RH. 1977. Digestive processes of haematophagous insects. XIV. Haemolytic activity in the midgut of Glossina morsitans morstians Westwood (Diptera: Glossinidae). Can. J. Zool. 55:1899–1905 [DOI] [PubMed] [Google Scholar]
- 21. Moskalyk LA. 1998. Carboxypeptidase B in Anopheles gambiae (Diptera: Culicidae): effects of abdominal distention and blood ingestion. J. Med. Entomol. 35:216–221 [DOI] [PubMed] [Google Scholar]
- 22. Yan J, Cheng Q, Li CB, Aksoy S. 2002. Molecular characterization of three gut genes from Glossina morsitans morsitans: cathepsin B, zinc-metalloprotease and zinc-carboxypeptidase. Insect Mol. Biol. 11:57–65 [DOI] [PubMed] [Google Scholar]
- 23. Scotto-Lavino E, Du G, Frohman MA. 2006. 3′ end cDNA amplification using classic RACE. Nat. Protoc. 1:2742–2745 [DOI] [PubMed] [Google Scholar]
- 24. Djadid ND, Barjesteh H, Raeisi A, Hassanzahi A, Zakeri S. 2006. Identification, sequence analysis, and comparative study on GSTe2 insecticide resistance gene in three main world malaria vectors: Anopheles stephensi, Anopheles culicifacies, and Anopheles fluviatilis. J. Med. Entomol. 43:1171–1177 [DOI] [PubMed] [Google Scholar]
- 25. Djadid ND, Gholizadeh S, Aghajari M, Zehi AH, Raeisi A, Zakeri S. 2006. Genetic analysis of rDNA-ITS2 and RAPD loci in field populations of the malaria vector, Anopheles stephensi (Diptera: Culicidae): implications for the control program in Iran. Acta Trop. 97:65–74 [DOI] [PubMed] [Google Scholar]
- 26. Jackson LR, Fox JG. 1995. Institutional policies and guidelines on adjuvants and antibody production. ILAR J. 37:141–152 [DOI] [PubMed] [Google Scholar]
- 27. Ventura S, Villegas V, Sterner J, Larson J, Vendrell J, Hershberger CL, Aviles FX. 1999. Mapping the pro-region of carboxypeptidase B by protein engineering. Cloning, overexpression, and mutagenesis of the porcine proenzyme. J. Biol. Chem. 274:19925–19933 [DOI] [PubMed] [Google Scholar]
- 28. Sun S-W, Lin Y-C, Weng Y-M, Chen M-J. 2006. Efficiency improvements on ninhydrin method for amino acid quantification. J. Food Compos. Anal. 19:112–117 [Google Scholar]
- 29. Jensen JB. 1979. Observations on gametogenesis in Plasmodium falciparum from continuous culture. J. Protozool. 26:129–132 [DOI] [PubMed] [Google Scholar]
- 30. Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675 [DOI] [PubMed] [Google Scholar]
- 31. Hyde JE. (ed). 1993. Methods in molecular biology, vol 21 Protocols in molecular parasitology. Humana Press Inc, New York, NY [Google Scholar]
- 32. Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418–420 [PubMed] [Google Scholar]
- 33. Fivelman QL, McRobert L, Sharp S, Taylor CJ, Saeed M, Swales CA, Sutherland CJ, Baker DA. 2007. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol. Biochem. Parasitol. 154:119–123 [DOI] [PubMed] [Google Scholar]
- 34. Carter R, Miller LH. 1979. Evidence for environmental modulation of gametocytogenesis in Plasmodium falciparum in continuous culture. Bull. World Health Organ. 57(Suppl 1):37–52 [PMC free article] [PubMed] [Google Scholar]
- 35. Xi Z, Das S, Garver L, Dimopoulos G. 2007. Protocol for Plasmodium falciparum infections in mosquitoes and infection phenotype determination. J. Vis. Exp. 2007(5):222 doi:10.3791/222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tchuinkam T, Mulder B, Dechering K, Stoffels H, Verhave JP, Cot M, Carnevale P, Meuwissen JH, Robert V. 1993. Experimental infections of Anopheles gambiae with Plasmodium falciparum of naturally infected gametocyte carriers in Cameroon: factors influencing the infectivity to mosquitoes. Trop. Med. Parasitol. 44:271–276 [PubMed] [Google Scholar]
- 37. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622 [DOI] [PubMed] [Google Scholar]
- 38. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 39. Clauser E, Gardell SJ, Craik CS, MacDonald RJ, Rutter WJ. 1988. Structural characterization of the rat carboxypeptidase A1 and B genes. Comparative analysis of the rat carboxypeptidase gene family. J. Biol. Chem. 263:17837–17845 [PubMed] [Google Scholar]
- 40. Alter GM, Leussing DL, Neurath H, Vallee BL. 1977. Kinetic properties of carboxypeptidase B in solutions and crystals. Biochemistry 16:3663–3668 [DOI] [PubMed] [Google Scholar]
- 41. Bradley G, Naude RJ, Muramoto K, Yamauchi F, Oelofsen W. 1996. Ostrich (Struthio camelus) carboxypeptidase B: purification, kinetic properties and characterization of the pancreatic enzyme. Int. J. Biochem. Cell Biol. 28:521–529 [DOI] [PubMed] [Google Scholar]
- 42. Marinkovic DV, Marinkovic JN, Erdos EG, Robinson CJ. 1977. Purification of carboxypeptidase B from human pancreas. Biochem. J. 163:253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allary M, Schrevel J, Florent I. 2002. Properties, stage-dependent expression and localization of Plasmodium falciparum M1 family zinc-aminopeptidase. Parasitology 125:1–10 [DOI] [PubMed] [Google Scholar]
- 44. Lavazec C, Bourgouin C. 2008. Mosquito-based transmission blocking vaccines for interrupting Plasmodium development. Microbes Infect. 10:845–849 [DOI] [PubMed] [Google Scholar]
- 45. Tan AK, Eaton DL. 1995. Activation and characterization of procarboxypeptidase B from human plasma. Biochemistry 34:5811–5816 [DOI] [PubMed] [Google Scholar]
- 46. Vaughan JA, Noden BH, Beier JC. 1992. Population dynamics of Plasmodium falciparum sporogony in laboratory-infected Anopheles gambiae. J. Parasitol. 78:716–724 [PubMed] [Google Scholar]