

Abstract
OspZ is an effector protein of the type III secretion system in Shigella spp. that downregulates the human inflammatory response during bacterial infection. The ospZ gene is located on the large virulence plasmid of Shigella. Many genes on this plasmid are transcriptionally repressed by the nucleoid structuring protein H-NS and derepressed by VirB, a DNA-binding protein that displays homology to the plasmid partitioning proteins ParB and SopB. In this study, we characterized the ospZ promoter and investigated its regulation by H-NS and VirB in Shigella flexneri. We show that H-NS represses and VirB partially derepresses the ospZ promoter. H-NS-mediated repression requires sequences located between −731 and −412 relative to the beginning of the ospZ gene. Notably, the VirB-dependent derepression of ospZ requires the same VirB binding sites as are required for the VirB-dependent derepression of the divergent icsP gene. These sites are centered 425 bp upstream of the ospZ gene but over 1 kb upstream of the icsP transcription start site. Although these VirB binding sites lie closer to ospZ than icsP, the VirB-dependent increase in ospZ promoter activity is lower than that observed at the icsP promoter. This indicates that the proximity of VirB binding sites to Shigella promoters does not necessarily correlate with the level of VirB-dependent derepression. These findings have implications for virulence gene regulation in Shigella and other pathogens that control gene expression using mechanisms of transcriptional repression and derepression.
INTRODUCTION
Shigella species are Gram-negative, nonmotile rods that are causative agents of bacillary dysentery. The pathogenesis of Shigella spp. is primarily attributed to a large virulence plasmid (∼230 kb) that carries many of the genes required for colonization of humans and primates, including invasion of the colonic epithelium and actin-based motility within the host cell cytoplasm. The precise regulation of virulence genes is central to Shigella pathogenicity. This regulation relies heavily upon the antagonistic relationship between the nucleoid structuring protein H-NS, which functions as a transcriptional repressor, and one of two transcription factors encoded by the large virulence plasmid, VirF and VirB (1), which function to counteract H-NS-mediated repression (2, 3) in a process called derepression (reviewed in reference 4).
OspZ is encoded by the large virulence plasmid, is secreted by the type III secretion system (5, 6), and is homologous to NleE of enteropathogenic Escherichia coli (7). NleE and full-length OspZ block nuclear translocation of the p65 subunit of NF-κB, thereby downregulating the interleukin-8 (IL-8) response during bacterial infection (8). Full-length OspZ is therefore a member of a growing family of bacterial proteins that cause the suppression of NF-κB-regulated genes, thereby subverting innate immune signaling within the host (9–11). ospZ of Shigella flexneri serotype 6 encodes full-length OspZ, but the protein in S. flexneri serotype 2a has a 42-amino-acid truncation at the C terminus. The truncated form of OspZ in Shigella flexneri 2a is unable to inhibit p65 nuclear translocation (8). Instead of reducing polymorphonuclear leukocyte (PMN) migration, an activity predicted for the full-length OspZ protein, the truncated form of OspZ has been shown to stimulate PMN migration across polarized T84 intestinal epithelial cells (7). Although the activity of both forms of the OspZ protein has been characterized and implicated in the virulence of Shigella species (7, 8), the promoter region of ospZ in Shigella species remains largely uncharacterized.
The ospZ promoter region lies within an unusually long intergenic region on the large Shigella virulence plasmid (depicted in Fig. 1A). This intergenic region also contains the promoter of the divergent gene icsP. IcsP is an outer membrane protease that cleaves IcsA, the actin tail assembly protein, from the surface of Shigella and functions to regulate the actin-based motility of Shigella within epithelial cells (12, 13). Like many virulence genes in Shigella, icsP is transcriptionally repressed by H-NS and derepressed by VirB (14). This mechanism of gene regulation has been termed “silencing and anti-silencing” (4). Two VirB binding sites, located over 1 kb upstream of the icsP transcription start site (TSS), are essential for the VirB-dependent regulation of the icsP promoter (15). These VirB binding sites are centered only 425 bp upstream of the ospZ gene (Fig. 1A).
Fig 1.
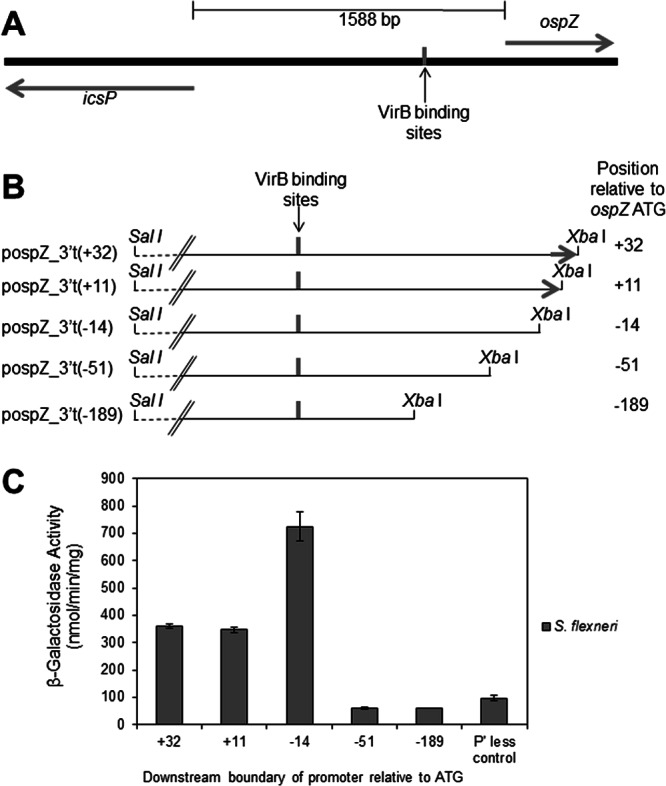
Analysis and characterization of the ospZ promoter. (A) Overview of the intergenic region between icsP and ospZ. VirB binding sites identified by Castellanos et al. (15) are indicated and centered 425 bp and 1,160 bp upstream of the ospZ and icsP translation start sites, respectively. (B) Schematic of the 3′-truncated ospZ promoter fragments found in the pospZ_3′t series. For each construct, the position of the 3′ truncation relative to the ospZ ATG is given in parentheses. The 5′ end of the ospZ gene (denoted by the arrow) is found only in pospZ_3′t(+32) and pospZ_3′t(+11). (C) Activity of the pospZ_3′t series in wild-type S. flexneri. pMIC21 serves as the promoterless (P' less) control. Assays were run in triplicate, and the means and standard deviations are shown.
As a starting point for this work, we chose to characterize the ospZ TSS and its associated promoter elements. Since ospZ lies divergent to icsP on the Shigella virulence plasmid, we tested our hypothesis that the ospZ promoter, like that of icsP, is regulated by VirB and H-NS. Having established the involvement of H-NS and VirB as regulatory proteins, we identified DNA sequences necessary for the regulation of the ospZ promoter (PospZ). The two major goals of this study were to improve our understanding of the transcriptional regulation of ospZ, a gene implicated in the pathogenicity of Shigella species, and to further examine how H-NS-mediated repression and VirB-dependent derepression of virulence genes are orchestrated on the Shigella virulence plasmid.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. E. coli was grown on LB agar (LB broth containing 1.5% [wt/vol] agar), and S. flexneri strains were grown on Trypticase soy agar (TSA; Trypticase soy broth [TSB] containing 1.5% [wt/vol] agar). To ensure that Shigella strains maintained the virulence plasmid, Congo red binding was tested on TSA plates containing 0.01% (wt/vol) Congo red. E. coli and S. flexneri strains were grown at 37°C with aeration in Luria-Bertani (LB) broth for all assays. Where appropriate, antibiotics were added at the following final concentrations: ampicillin, 100 µg/ml, and chloramphenicol, 25 µg/ml.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| S. flexneri | ||
| 2457T | S. flexneri serotype 2a | 41 |
| AWY3 | 2457T virB::Tn5 | 14 |
| E. coli | ||
| MC4100 | E. coli strain K-12 with araD and lacZ deletion | 42 |
| MC4100 hns | MC4100 hns::Kn. The first 37 amino acids of H-NS are expressed, giving rise to a trans-dominant negative effect over other H-NS-like proteins in the cell. | 43 |
| Plasmids | ||
| pHJW20 | PicsP-lacZ reporter plasmid derived from pACYC184. Carries 1,259 bp upstream and 48 bp downstream of the icsP gene on a PstI-XbaI DNA fragment. Restriction with SalI and XbaI completely removes icsP promoter and gene sequences. | 15 |
| pDB05 | pHJW20 carrying 1,613 bp of wild-type sequence upstream and 32 bp downstream of the ospZ translation start site in the SalI and XbaI sites. Also known as pospZ_3′t(+32). | This work |
| pospZ_3′t(+11) | pHJW20 carrying 1,613 bp of wild-type sequence upstream and 11 bp downstream of the ospZ translation start site in the SalI and XbaI sites | This work |
| pospZ_3′t(−14) | pHJW20 carrying wild-type sequences located between 1613 bp and 14 bp upstream of the ospZ translation start site in the SalI and XbaI sites | This work |
| pospZ_3′t(−51) | pHJW20 carrying wild-type sequences located between 1613 bp and 51 bp upstream of the ospZ translation start site in the SalI and XbaI sites | This work |
| pospZ_3′t(−189) | pHJW20 carrying wild-type sequences located between 1613 bp and 189 bp upstream of the ospZ translation start site in the SalI and XbaI sites | This work |
| pAFW04 | Identical to pHJW20, but the lambda oop terminator is cloned immediately upstream of the icsP promoter fragment to prevent possible transcriptional read-through into the promoter region. Restriction with SalI and XbaI completely removes icsP promoter sequences but leaves the lambda oop terminator. | This work |
| pospZ_5′t(−1613) | pAFW04 carrying 1,613 bp of wild-type sequence upstream and 32 bp downstream of the ospZ translation start site on a SalI-XbaI DNA fragment. Identical to pDB05, but the lambda oop terminator is cloned immediately upstream of the ospZ promoter fragment to prevent possible transcriptional read-through into the promoter region. | This work |
| pospZ_5′t(−1203) | pAFW04 carrying 1,203 bp of wild-type sequence upstream and 32 bp downstream of the ospZ translation start site on a SalI-XbaI DNA fragment | This work |
| pospZ_5′t(−731) | pAFW04 carrying 731 bp of wild-type sequence upstream and 32 bp downstream of the ospZ translation start site on a SalI-XbaI DNA fragment | This work |
| pospZ_5′t(−495) | pAFW04 carrying 495 bp of wild-type sequence upstream and 32 bp downstream of the ospZ translation start site on a SalI-XbaI DNA fragment | This work |
| pospZ_5′t(−412) | pAFW04 carrying 412 bp of wild-type sequence upstream and 32 bp downstream of the ospZ translation start site on a SalI-XbaI DNA fragment | This work |
| pospZ_Δ45 | pAFW04 with a 45-bp deletion between −270 and −225 relative to the ospZ translation start site | This work |
| pMAP07 | pAFW04 lacking all promoter sequences | This work |
| pMIC16 | pBluescript II KS(+) carrying 809 bp upstream and 331 bp downstream of the ospZ translation start site with 14-bp substitutions in the VirB binding sites | 15 |
| pDB15 | pospZ_5′t(−1613) carrying 14-bp substitutions in the VirB binding sites, so that the original sequence ATTTCAGtATGAAAT was altered to GCCCAGCtCGACCCGa | This work |
| pMIC21 | pHJW20 lacking all promoter sequences | 15 |
| pBAD-virB | pATM324 derived from pBAD18; Ampr | 44 |
| pBAD18 | Arabinose-inducible pBAD expression vector, pBR ori; Ampr | 45 |
Uppercase letters represent sites found to be essential for VirB-dependent regulation of the icsP promoter (16) or the corresponding mutated sites. Note that the original sites are organized as a near-perfect inverted repeat, separated by a single nucleotide (lowercase).
Plasmid construction.
The ospZ promoter region was PCR amplified from the large virulence plasmid of S. flexneri serotype 2a strain 2457T using primers W152 and W153. (Primers used in this study are listed in Table 2.) The resulting fragment was cut with the restriction endonucleases SalI and XbaI and cloned into pHJW20 (15) in order to replace the icsP promoter region. The resulting plasmid, pDB05, carries the entire ospZ promoter (1,613 bp upstream of the ospZ translation start site) and the first 32 bp of the ospZ coding region, cloned upstream of a translation stop site in each reading frame and a promoterless lacZ gene, so that expression of lacZ is directly regulated by the ospZ promoter.
Table 2.
Oligonucleotide primers used in this study
| Primer | DNA sequencea |
|---|---|
| W73 | CAATAAAATGGTTGGTTGAAGGTCGTG |
| W152 | CCGAGTCGACCAAGTACAAAGAATTTTAATTTCATCG |
| W153 | CCGATCTAGAACGTTCTTAATATTCTTGATGGGAC |
| W154 | CCGATCTAGAAAACCAGAACCTCGCTTAGGCC |
| W160 | TTTGCGCTCCTTCAACTGGGCA |
| W180 | CCGAGTCGACCCTCCTGTATTTGACGTAACGTG |
| W181 | CCGAGTCGACTAATGTGGTTGTCCGATTAAGGAC |
| W201 | TTACTTCTAGACCTAAGTGGAATGTCTCCACGG |
| W202 | TTACTTCTAGACTCAAATATAAACATTACCATGAAC |
| W203 | TTACTTCTAGAGGACTAATCATTTTAATCTCTATACTC |
| W291 | CCGAGTCGACGGATAAGCTATTGTCCTTATTCCAC |
| W336 | ATTTTGTCGACGAAGTATTCAATCAAACAATTACACC |
| W355 | GTGGGTAGGATCAATAAAGACGTTTTATATGTGTCTGC |
| W356 | GTAATATGGACACAGGCCTAAGCG |
| W357 | CCTCGCTTAGGCCTGTGTCCATATTACGTGGGTAGGATCAATAAAGACGTTTTATATGTGTCTGC |
| M13 | GAGCGGATAACAATTTCACACAGG |
Underlined sequences represent unique restriction sites introduced for cloning purposes.
The 3′ promoter truncations of ospZ were PCR amplified from pDB05 using forward primer W152 in combination with reverse primer W203, W202, W201, or W154. PCR products were digested with SalI and XbaI and then ligated to pDB05 also digested with SalI and XbaI. This created a series of PospZ-lacZ fusion plasmids with the same 5′ end but different 3′ ends: +11, −14, −51, and −189 relative to the translation start site of the ospZ gene, respectively. These plasmids and pDB05 are described as the pospZ_3′t series in this work.
The 5′ promoter truncations of ospZ were isolated or PCR amplified from pDB05 using forward primers W152, W180, W181, W291, and W336 in combination with reverse primer W153. PCR products were digested with SalI and XbaI and were ligated to pAFW04, which had also been digested with SalI and XbaI. This created a series of PospZ-lacZ fusion plasmids with 5′ boundaries of −1613, −1203, −731, −495, and −412 relative to the ospZ translation start site. The 3′ end of each promoter fragment was the same, +32 relative to the ospZ translation start site. These plasmids are described as the pospZ_5′t series in this work.
To create pDB15, the plasmid pMIC16 (15) was digested with restriction enzymes BglII and PstI to remove a 197-bp fragment containing the mutated VirB binding sites. The resulting fragment was subsequently cloned into pospZ_5′t(−1613) to replace the 197-bp BglII PstI restriction fragment containing the wild-type VirB binding sites.
To create pospZ_Δ45, a three-step PCR was used. In step one, two DNA fragments upstream and downstream of the region to be deleted were amplified from pospZ_5′t(−1613) DNA using the following primer pairs: W73-W357 and W356-W160. In step two, the upstream fragment was further amplified using W73 and W355 (W355 is identical to W357 except that its 5′ end is complementary to sequences downstream of the region to be deleted). In step three, the fragments generated in steps one and two were mixed and amplified using W73 and W160. The resulting DNA was then cut with BglII and XbaI and used to replace the existing BglII and XbaI fragment present in pospZ_5′(−1613).
All constructs generated in this study were confirmed by DNA sequencing.
β-Galactosidase assays.
β-Galactosidase activity was determined using the Miller protocol (16). Overnight cell cultures were back-diluted 1:100 and grown for 5 h at 37°C before being assayed, because prior experiments have shown that both icsP and ospZ promoter activities are maximal after 5 h (14). In experiments in which virB was induced from the pBAD18 derivative, Shigella and E. coli strains were grown for 3 h and 5 h, respectively, at either 30 or 37°C prior to induction with 0.2% (wt/vol) l-arabinose. Cells were then grown at either 30 or 37°C for an additional 2 h before being assayed.
Primer extension analysis.
The ospZ TSS was identified through RNA extraction and primer extension analysis as described recently (17) using a protocol adapted from that of Aiba et al. (18). Total cellular RNA was extracted using the hot-phenol method from 109 cells harvested from early-stationary-phase cultures. Samples were digested with DNase I (Invitrogen) for 1 h at 37°C in DNase I buffer (Ambion), and total RNA integrity was verified by formaldehyde gel electrophoresis and ethidium bromide staining. Primers W153 and W160 were 5′ end labeled with [γ-32P]ATP using T4 polynucleotide kinase (Promega). One picomole of 32P-labeled primer and 5 μg of total RNA were dissolved in 30 μl of hybridization buffer (18). The annealing reaction mixture was heated at 50°C for 5 min, incubated at 75°C for 15 min, and maintained at 45°C for a total of 3 h. Samples were ethanol precipitated and cDNA was generated using Superscript II reverse transcriptase (Invitrogen) at 37°C for 50 min. Reactions were aborted by heating samples to 70°C for 10 min, and RNA was removed by digestion with 10 mg/ml of RNase A (Sigma) for 30 min at 37°C. Samples were ethanol precipitated and dissolved in 5 μl of loading dye (95% [vol/vol] formamide, 20 mM EDTA, 0.05% [wt/vol] bromophenol blue, 0.05% [wt/vol] xylene cyanol; Sequenase, version 2.0, DNA sequencing kit [Affymetrix]) prior to separation by electrophoresis on a 6% (vol/vol) glycerol-tolerant polyacrylamide gel (PAGE) containing 7 M urea. PAGE gels were transferred to Whatman paper and vacuum dried. Dried gels were exposed to a phosphor screen overnight and visualized using a Typhoon 9410 variable-mode imager (Amersham). A sequencing ladder generated from pBluescript II KS(+) (Stratagene) and an M13 reverse primer with the Sequenase, version 2.0, DNA sequencing kit (Affymetrix) was routinely used to determine the sizes of primer extension products.
RESULTS
Analysis of the ospZ promoter region.
To identify the location of associated ospZ promoter sequences, a series of 3′ truncations of putative promoter sequences were created. Promoter fragments were generated with the same upstream boundary (−1613 relative to the beginning of the ospZ gene) but different downstream boundaries ranging from + 32 to −189 relative to the beginning of the ospZ gene (Fig. 1B). Each promoter fragment was then cloned upstream of a promoterless lacZ gene in a reporter plasmid, and β-galactosidase activity was measured in a wild-type S. flexneri 2a strain. Our data show that the three longest constructs display β-galactosidase activity above baseline, but the two shortest constructs do not (Fig. 1C). This indicates that under the conditions tested, the only active promoter(s) lie(s) within 50 nucleotides (nt) of the 5′ end of the ospZ gene. The β-galactosidase activity generated by cells carrying pospZ_3′t(−14) was routinely 2-fold higher than that generated by cells carrying plasmids with longer promoter fragments. Although the underlying reason for this remains unknown, it is possible that the unique 3′ junction between the ospZ sequence and the lacZ gene increases mRNA stability or translation efficiency of lacZ in this construct.
Transcription start site mapping.
To precisely map the TSS of the ospZ promoter, we next performed primer extension analysis on mRNA isolated from the wild-type S. flexneri 2a strain or the same strain carrying the PospZ-lacZ fusion plasmid, pDB05. Two primers were used for our analyses: W153 is complementary to the beginning of the ospZ transcript, a sequence that is present in both the ospZ gene and the PospZ-lacZ fusion plasmid, and W160 is complementary to the beginning of the lacZ transcript, a sequence present only in the PospZ-lacZ fusion plasmid. Regardless of whether the ospZ promoter was carried by the large virulence plasmid or the low-copy-number PospZ-lacZ reporter, a single primer extension product was observed when primer W153 was used (Fig. 2A). This indicates that a single TSS is associated with the ospZ gene and that this TSS is used regardless of whether the transcript is generated from the native ospZ gene or the PospZ-lacZ fusion plasmid. When primer W160 was used for primer extension analysis of mRNA isolated from cells carrying the PospZ-lacZ fusion plasmid, a larger product was detected. This was expected because primer W160 binds downstream of W153. Even so, the size of this product mapped exactly to the position of the TSS identified by primer W153. The identification of a single transcription start site 28 nt upstream of the ospZ translation start site by primer extension analyses is consistent with our previous findings (Fig. 1C), which predicted that the ospZ TSS lies within 50 nucleotides of the start of the ospZ gene. Using this information, the potential −10 and −35 promoter elements of the ospZ promoter were identified (Fig. 2B) and were found to display a 5/6 and 4/6 match to each consensus sequence, respectively.
Fig 2.

Identification of the transcription start site of the ospZ gene. (A) Primer extension analysis of ospZ transcripts generated by the wild type (2457T) or the wild type carrying the plasmid pDB05. The primer W153 binds within ospZ, and the primer W160 binds within lacZ. Arrows indicate identified transcription start sites, which map to the same nucleotide position. (B) Sequence of the ospZ promoter region and beginning of the gene. The ospZ transcription start site identified by this work is indicated (bold type and designated TSS), and associated potential −10 and −35 elements are shown (bold and underlined). The gene sequence is italicized, and encoded amino acids are given. The sequence bearing strong sequence similarity to the 3′ end of IS3 (92% identity over 185 nt) is highlighted in gray (GenBank number AL391753.1 [32]).
VirB upregulates the ospZ promoter.
Our previous studies of the genetic locus containing icsP and ospZ have demonstrated that two VirB binding sites, organized as an inverted repeat and located over 1 kb upstream of the icsP TSS, are required for the VirB-dependent regulation of the icsP promoter (15). These VirB binding sites are located closer to the beginning of the divergent ospZ gene than the icsP gene (Fig. 1A). Therefore, we next chose to determine whether or not these VirB binding sites influence the activity of the ospZ promoter. To do this, β-galactosidase assays were performed in wild-type Shigella and a virB mutant background carrying either the full-length ospZ promoter [pospZ_5′t(−1613)] or a derivative that contained base pair substitutions throughout the VirB binding sites (pDB15). Our data show that the activity of the ospZ promoter carrying wild-type VirB binding sites is significantly higher in wild-type Shigella than in the virB mutant and that this increase is completely dependent on the previously identified VirB binding sites (15) (Fig. 3). Notably, complementation of the virB mutant with a plasmid carrying an inducible copy of virB rescued the VirB-dependent regulation of the ospZ promoter (Fig. 3B) at both 30 and 37°C, but this effect was observed only in virB mutants carrying constructs bearing wild-type VirB binding sites (and not in those cells carrying pDB15). Taken together, these results indicate that ospZ promoter activity is dependent upon VirB for its activity and that this dependency requires the two VirB binding sites, which were previously characterized as regulating the icsP promoter (15).
Fig 3.

Activity of the PospZ-lacZ fusions in wild-type Shigella and a virB mutant. (A) β-Galactosidase activity resulting from cells carrying either the wild-type full-length promoter (pospZ-5′t(−1613), the full-length promoter containing mutated VirB binding sites (pDB15), or a promoterless control (pMAP07) grown at 37°C. Assays were run in triplicate, and the means and standard deviations are shown.*, P < 0.01. (B) Fold increase in ospZ promoter activity in cells grown with (pBAD-virB) or without (pBAD18) induction of virB. In each case, β-galactosidase activity from the promoterless control in each strain background was subtracted from those generated by cells carrying PospZ-lacZ fusions. The resulting data were normalized to activities obtained in the virB mutant strain carrying pDB15 (construct with the mutated VirB binding sites) when these cells were grown at either 30 or 37°C. Assays were run in triplicate, and the means and standard deviations are shown.
H-NS represses the ospZ promoter at 37°C.
The nucleoid structuring protein H-NS plays a major role in silencing the transcription of virulence genes on the Shigella virulence plasmid (19–22). At the nonpermissive temperature of 30°C, many genes on the virulence plasmid are repressed by H-NS (23). Upon a switch to 37°C, a complex regulatory cascade is triggered, which results in the production of VirF and subsequently VirB (3, 24, 25). Although some virulence genes are derepressed solely by the increase in temperature (23), others are derepressed at 37°C by either VirF or VirB, which function to relieve the H-NS-mediated repression of these virulence gene promoters on the large virulence plasmid (3, 14, 21, 26).
Our previous studies at the icsP and ospZ locus have revealed that H-NS is able to repress the icsP promoter at 37°C (14). Therefore, we hypothesized that H-NS would also repress transcription of the divergent gene ospZ at 37°C. To test this hypothesis and to identify regions of DNA sequences required for H-NS-mediated regulation, a series of 5′-truncated PospZ-lacZ fusion plasmids were constructed (named the pospZ_5′t series) and introduced into the wild-type E. coli strain MC4100 and an hns mutant derivative. β-Galactosidase levels were then measured after the transformed cells were grown at 37°C. These E. coli strains were used to avoid interference arising from the H-NS-dependent regulation of virB in Shigella (27) and the notorious genetic instability of Shigella hns mutants (A. T. Maurelli, personal communication). A similar strategy has been used by us (14) and others to study H-NS-dependent regulation of Shigella promoters (21).
Consistent with our hypothesis, the activity of the longest construct (−1613) was 6-fold lower in the wild-type background than in the hns mutant background (Fig. 4A). Similar expression levels were observed in wild-type cells carrying the promoters truncated to −1203 and −731 relative to the ospZ gene, indicating that these promoters were also repressed by H-NS. As we successively removed sequences from the 5′ end of the ospZ promoter starting at −731, promoter activity was seen to significantly increase in the wild-type background in a stepwise manner, until it was equivalent to that observed in the hns mutant background. These observations are consistent with the removal of sequences required for H-NS-mediated repression. Based on these analyses, sequences between −495 and −412 allow partial repression of the ospZ promoter by H-NS, but sequences between −731 and −495 are needed for full repression of the ospZ promoter by H-NS.
Fig 4.

Activity of the ospZ promoter truncation series (pospZ_5′t series) in wild-type E. coli and an hns mutant (A) and wild-type S. flexneri 2457T and a virB mutant derivative (B). pMAP07 serves as the promoterless control. Assays were run in triplicate, and each assay was repeated three times. Means and standard deviations of representative data are shown. Note that the activities of pDB05 and pospZ_5′t(−1613) were found to be identical to each other (data not shown).
VirB production in a wild-type Shigella background at 30°C or in an E. coli background at 37°C is sufficient to partially derepress the ospZ promoter.
The virB gene is maximally transcribed at 37°C (28). While VirB production is normally very low at 30°C (29), it has been shown that VirB-regulated genes can overcome H-NS-mediated repression if virB expression is induced from an inducible promoter at this temperature (28, 30). To test if VirB production at 30°C can overcome the H-NS-mediated repression of ospZ in a wild-type Shigella background, wild-type Shigella strains carrying a PospZ-lacZ reporter and either a virB inducible plasmid or an empty plasmid control (pBAD18) were grown under inducing conditions. Our data show that VirB production in a wild-type Shigella background at 30°C leads to a 1.74-fold increase in ospZ promoter activity, but this increase is not observed when the identified VirB binding sites are mutated in the reporter constructs (Fig. 3B). Interestingly, when virB was expressed from an inducible plasmid in wild-type cells grown at 37°C, ospZ promoter activity was seen to increase further (2.32-fold versus 1.35-fold), even though VirB is naturally produced at 37°C in wild-type cells. Because additional VirB protein can further elevate ospZ promoter activity, the VirB-dependent effect on the ospZ promoter observed in wild-type cells does not appear to be saturating under the conditions used in our assay.
At the icsP promoter the VirB protein has no effect in the absence of H-NS, indicating that VirB functions solely to antagonize H-NS-mediated repression (14). Our data show that VirB increases ospZ promoter activity (Fig. 3) and that H-NS is required for full repression of the ospZ promoter (Fig. 4A). We next wanted to test whether VirB functions to relieve H-NS-mediated repression or whether it functions similarly to an activator, increasing the activity of the ospZ promoter even in the absence of H-NS. To test this, a pBAD expression vector carrying virB or an empty plasmid control (pBAD18) was introduced into the E. coli strain MC4100 or an hns mutant derivative carrying either the full-length ospZ promoter fragment [pospZ_5′t(−1613)] or a promoterless lacZ reporter, and β-galactosidase activities were measured 2 h postinduction. Our data show that in wild-type E. coli, ospZ promoter activity increased 2-fold after virB expression (Fig. 5), consistent with our previous results obtained in a Shigella background (Fig. 3). In contrast, in the MC4100 hns mutant, promoter activity was not affected by expression of virB (Fig. 5). These data demonstrate that VirB functions to partially overcome H-NS-mediated repression of the ospZ promoter. Furthermore, since levels of derepression of the ospZ promoter following exogenous expression of virB in an E. coli background (Fig. 5) were similar to those observed in a wild-type Shigella background, this strongly suggests that VirB is the only Shigella-specific protein required for derepression of the full-length ospZ promoter in our assays.
Fig 5.
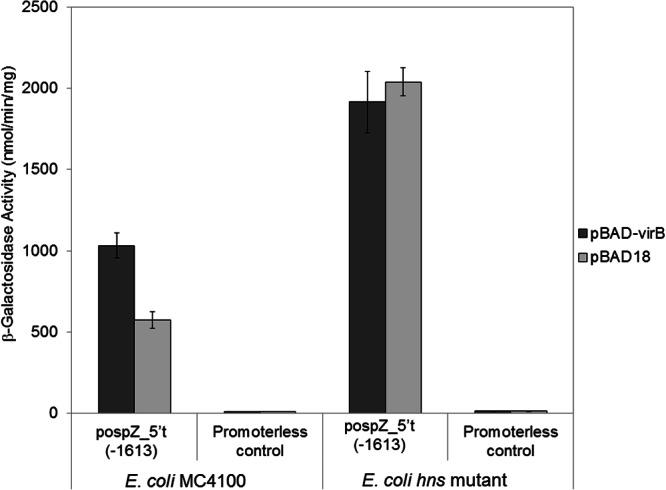
Activity of the ospZ promoter (pospZ_5′t(−1613) in a wild-type E. coli strain and an hns mutant derivative after induction of virB from an l-arabinose-inducible plasmid. pMAP07 serves as the promoterless control. Assays were run in triplicate, and the means and standard deviations are shown. Note that the activities of pDB05 and pospZ_5′t(−1613) were found to be identical to each other (data not shown).
Activity of the PospZ truncation series in an S. flexneri background.
To further characterize the VirB-dependent regulation of the ospZ promoter, we next measured the activity of our 5′ promoter truncation series in a wild-type Shigella background or a virB mutant derivative (Fig. 4B). The pattern of promoter activity observed in the virB mutant was very similar to that seen in the wild-type E. coli strain (Fig. 4A). Both of these strains express H-NS but do not express virB. These data therefore validate our use of E. coli strains in experiments to test the involvement of H-NS in the regulation of the ospZ promoter (Fig. 4A). As expected, all promoter truncations, except −412, which lacks the required VirB binding sites, displayed a VirB-dependent increase in promoter activity. This supports our finding that the VirB binding sites located at position −425 relative to the beginning of the ospZ gene are required for the VirB-dependent regulation of the ospZ promoter. Surprisingly, the promoter truncated to −731 displayed a 4.5-fold increase in VirB-dependent activity, whereas all other VirB-regulated promoters in this series displayed a 2-fold increase. The reason for this remains unclear, but this observation appears to be an artifact inherent to the pospZ_5′t(−731) construct because first, only a 2-fold increase is seen in promoter activity when measured in the context of the entire intergenic region, and second, ospZ expression was previously reported to increase 2- to 3-fold in response to temperature (5).
Comparison of the ospZ and icsP genetic locus in other Shigella strains and species.
Finally, to assess whether our characterization of the ospZ promoter was relevant to other Shigella strains and species, DNA sequences of ospZ promoter regions were collected and compared to the entire icsP and ospZ intergenic region taken from Shigella flexneri serotype 2a, type strain 2457T (Table 3). Our analysis revealed that most sequenced strains show complete sequence identity to the VirB binding sites, promoter elements, and most of the intergenic region in 2457T, despite the occasional point mutation (no more than 4 in any sequence). These findings are consistent with our hypothesis that VirB regulates ospZ genes and icsP genes in these Shigella strains and species as well. The only sequences displaying a significant deviation from those found in 2457T were found in S. dysenteriae serotype 2 [(10)4279] and S. boydii serotype 4 (Sb227, YP_406371). The available sequences of these strains indicate that a 45-bp deletion exists between 270 and 225 bp upstream of the ospZ gene (Table 3). To test the impact that this 45-bp deletion has on ospZ promoter activity, a 45-bp deletion identical to that found in S. dysenteriae serotype 2 and S. boydii serotype 4 was created in our PospZ-lacZ reporter plasmid (pospZ_Δ45), and promoter activity was measured in S. flexneri serotype 2a strain 2457T and a virB mutant derivative. Although the deletion slightly lowered overall promoter activity, ospZ promoter activity was still 2.5-fold higher in the wild-type S. flexneri strain than in the virB mutant (Table 4). These data strongly suggest that the 45-bp deletion found in S. dysenteriae serotype 2 and S. boydii serotype 4 does not interfere with VirB-dependent regulation of the ospZ promoter.
Table 3.
Analysis of DNA sequences found at the ospZ and icsP locus in Shigella spp.a
| Species, serotype, and strain name | GenBank accession no., NCBI reference sequence no., or source of strain | OspZ product (full-length, 230 aa) and coordinates of gene on published sequence | Identity to ospZ and icsP intergenic region taken from 2457T (%) | Description of sequence change in intergenic region |
|---|---|---|---|---|
| S. boydii, Sb227 | CP000037 | Full length (8); complement (123143–123835) | 97.0 | 45-bp deletion between −270 and −225 relative to ospZ gene, and 3 single point substitutions |
| S. boydii CDC3083-94 | NC_010660.1 | Full length; complement (206053–206745) | 99.7 | 2 single point substitutions |
| S. dysenteriae serotype 1, Sd197 | CP000035.1 | Full length (7); complement (179127–179819) | 99.7 | 4 single point substitutions |
| S. dysenteriae serotype 2, (10)4279 | This work | Full length | 97 | 45-bp deletion between −270 and −225 relative to ospZ gene, and 3 single point substitutions |
| S. flexneri serotype 2a, 2104 | RRB strain | 42-aa deletion, truncated to amino acid 188 (8) | 100 | NA |
| S. flexneri serotype 6, 0106164 | RRB strain | Full length (8) | 99.7 | Single point substitution |
| S. flexneri serotype 2a, 2457T | This work | 42-aa deletion, truncated to amino acid 188 | Reference sequence | NA |
| S. flexneri serotype 2a, pCP301 | AF386526.1 | 42-aa deletion, truncated to amino acid 188; complement (218144–218711) | 100 | NA |
| S. flexneri serotype 5a, M90T pWR501 | AF348706.1 | 42-aa deletion, truncated to amino acid 188; complement (218378–218941) | 99.9 | 2 single point substitutions |
| S. flexneri serotype 5a, M90T pWR100 | AL391753.1 | 42-aa deletion, truncated to amino acid 188; complement (212330–212896) | 99.9 | 2 single point substitutions |
| S. sonnei, Ss046 | NC_007385.1 | Full length (8); complement (210796–211488) | 99.7 | 2 single point substitutions and 1 insertion |
aa, amino acid(s); NA, not applicable.
Table 4.
Effect of 45-bp deletion between −270 and −225 relative to the ospZ translation start site
| Reporter plasmid | Avg β-galactosidase activity (nmol/min/mg) ± SD |
Ratio of activity (WT vs. virB mutant) | |
|---|---|---|---|
| WTa (2457T) | virB mutant (AWY3) | ||
| pospZ_5′t(−1613) | 232 ± 5b | 85 ± 2 | 2.72 |
| pospZ_Δ45 | 180 ± 6b | 68 ± 3 | 2.65 |
| Promoterless control (pMAP07) | 19 ± 2 | 19 ± 1 | 0.97 |
WT, wild type.
P < 0.05.
DISCUSSION
The ospZ gene, formerly called orf212 (5, 7), is carried by the large virulence plasmid in all four Shigella species. This gene is located upstream of and divergent from the icsP gene in all Shigella strains and species analyzed (coordinates provided in Table 3), even though this gene is not annotated on the published sequences of the large virulence plasmids found in Shigella flexneri serotypes 2a and 5a (pCP301 [AF386526.1] and pWR501 [AF348706.1], respectively).
The OspZ protein is homologous to NleE, which is encoded by attaching and effacing pathogens Citrobacter, enterohemorrhagic E. coli (EHEC), and enteropathogenic E. coli (EPEC) (7, 8), in which it has been shown to block translocation of the p65 subunit of the transcription factor NF-κB to the host cell nucleus, resulting in NF-κB inhibition, and decreased IL-8 expression in EPEC-infected intestinal epithelial cells (8). The OspZ/NleE protein encoded by these bacteria is 224 to 230 amino acids long. In contrast, most S. flexneri strains produce a C-terminally truncated protein that is only 188 amino acids long (the only exception is S. flexneri serotype 6 [8] [Table 3]). The truncated form of OspZ has also been implicated in Shigella virulence because it is required for effective transepithelial migration of PMNs in vitro (7). During infection, the transepithelial migration of PMNs disrupts the integrity of the epithelial layer and facilitates the passage of Shigella to the basolateral surface of the colonic epithelium, the primary site of epithelial cell invasion.
In this study, we characterized the promoter region of the ospZ gene in S. flexneri serotype 2a. Our initial analysis of the icsP and ospZ intergenic region revealed a 219-bp open reading frame immediately upstream of the ospZ gene that terminates 1 nucleotide prior to the beginning of the ospZ gene. Although the location and juxtaposition of this ORF to ospZ raised the possibility of a bicistronic operon, this hypothesis was not supported by our 3′ truncation analysis or the transcription start site mapping, which placed the only active promoter(s) in this region within this 219-bp ORF. Upon closer inspection, the 219-bp ORF was found to be similar to ORF III of insertion sequence 3 (IS3) (31); a partial IS3 sequence was found in this genetic locus previously (32). Interestingly, the putative −35 element pinpointed by our studies was found to lie within this partial IS3 element (described in reference 32), demonstrating the composite nature of the ospZ promoter and implicating insertion sequences in the evolution of this virulence gene promoter (Fig. 2B). Based on our analyses of the ospZ promoter region, we conclude that the 219-bp ORF does not encode protein, the ospZ message is monocistronic, and it is transcribed from a single transcription start site located 28 nt upstream of the ospZ gene.
Type III effector proteins are typically tightly regulated (6, 33), so it seemed unlikely that ospZ would be constitutively expressed. Based on our studies, the ospZ promoter is repressed by H-NS and partially derepressed by VirB. Two VirB binding sites, organized as an inverted repeat and located 397 bp upstream of the ospZ TSS, increase the activity of the ospZ promoter 2-fold. Removal of the two VirB binding sites by site-directed mutagenesis or through 5′ truncation analysis of the ospZ promoter region resulted in a complete loss of VirB-dependent regulation. These findings are consistent with those of Le Gall et al., who reported a modest increase in ospZ (orf212) expression in response to a shift from 30 to 37°C (5). Since VirB is maximally produced at 37°C and not at 30°C (28), it seems likely that VirB was responsible for the effect reported by Le Gall et al. Even though a 2-fold VirB-dependent increase in ospZ promoter activity appears to be a modest level of regulation, the observed increase in promoter activity is reproducible and statistically significant (Fig. 3 and 4). Furthermore, this increase is completely dependent upon the VirB protein and the two VirB binding sites (Fig. 3). If ospZ mRNA is long-lived, a 2-fold change in ospZ promoter activity at 37°C could considerably increase OspZ production in Shigella. To our knowledge, the half-life of ospZ mRNA has not yet been determined. Alternatively, it is possible that ospZ expression may be further upregulated by another, as-yet-uncharacterized factor. MxiE, a transcription factor that upregulates many genes encoding type III effector proteins (5), does not appear to be involved, based on our studies (data not shown).
The VirB binding sites implicated in the regulation of the ospZ promoter were previously found to be necessary for the VirB-dependent regulation of the divergent icsP promoter (15). We therefore conclude that the VirB binding sites that lie within the intergenic region between icsP and ospZ on the S. flexneri 2a virulence plasmid are involved in the bidirectional regulation of these genes. To our knowledge, this is only the second example of divergent genes or operons on the Shigella virulence plasmid being regulated by the same VirB binding sites. The first example was observed in the 31-kb invasion locus of the virulence plasmid, between the divergent genes icsB and ipgD (3, 34, 35). At this locus, the intergenic region containing the VirB binding sites is much shorter, placing the VirB binding sites only 114 bp upstream of PicsB and 105 bp upstream of PipgD (34, 35). At the icsP and ospZ genetic locus, the VirB binding sites are located unusually far upstream of the promoters they regulate: centered 397 bp upstream of the ospZ TSS and 1,137 bp upstream of the icsP TSS. This clearly demonstrates that VirB breaks some of the paradigms associated with bacterial transcription factors like the cAMP receptor protein (CRP; also known as the catabolite activator protein [CAP]), which typically bind within a 200-bp window of the genes they regulate (36). Clearly, novel mechanisms of gene regulation are at play on the Shigella virulence plasmid. Furthermore, although the VirB binding sites lie much closer to PospZ than PicsP (Fig. 1A), their effect on PospZ is much less dramatic than at PicsP: VirB upregulates the ospZ promoter approximately 2-fold, while it increases activity of the icsP promoter over 12-fold (15). These findings demonstrate that the proximity of VirB binding sites to Shigella promoters does not necessarily correlate with the level of VirB-dependent regulation. This is an important consideration for others studying virulence gene regulation in Shigella and mechanisms of transcriptional repression and derepression in other bacterial pathogens.
VirB is an unusual regulator of transcription that displays 39% identity with the P1 plasmid partitioning protein ParB, a DNA-binding protein involved in segregation of plasmid prophage during cell division. Despite this structural similarity, there is no evidence to suggest that VirB functionally contributes to the maintenance or segregation of the virulence plasmids found in Shigella today (32, 37). Instead, this protein appears to function solely to upregulate the transcription of virulence gene expression in Shigella species. Rather than functioning as an activator of transcription, VirB functions to counter transcriptional repression mediated by the nucleoid structuring protein H-NS (4, 14, 35). In this work, we demonstrate that VirB functions to partially relieve repression mediated either directly or indirectly by H-NS and that VirB has no impact on ospZ promoter activity in an hns mutant. These data are consistent with our findings (14) and those of others (21) that indicate that VirB functions solely to relieve H-NS-mediated transcriptional repression of genes that are encoded by the Shigella virulence plasmid.
To further understand the molecular interplay between H-NS and VirB at the ospZ promoter, we chose to characterize the sequences within the icsP and ospZ intergenic region required for H-NS-mediated repression. Sequences between −412 and −731 (relative to the ATG) were found to be required for full H-NS-mediated repression, but a partial loss of repression was observed as we truncated through the promoter to position −495 (Fig. 4A; compare −731, −495, and −412). The observed increase in promoter activity upon removal of sequences located between −731 and −412 was unlikely to be caused by transcription from a second cryptic promoter, because removal of the ospZ promoter elements identified in this study (potential −10 element and TSS) resulted in a complete loss of activity in the −412 deletion construct (data not shown). Although the H-NS high-affinity binding site 5′-TCGATAAATT-3′ has been reported to facilitate the cooperative binding of H-NS to DNA (38, 39), a complete match to the high-affinity binding sequence was not discovered between −731 and −412. The data presented in Fig. 4A are consistent with two regions being required for the H-NS-mediated repression of the ospZ promoter: one between −731 and −495 and the other between −495 and −412 (relative to the ATG). Alternatively, it is possible that the entire region located between −731 and −412, which is characteristically AT rich (66%, compared to the entire intergenic region at 63%), binds H-NS uniformly, thereby leading to a gradual loss of repression when 5′ sequences are eliminated in our truncation series. Future work will establish which of these scenarios is more likely.
The relative spacing between DNA sites occupied by VirB and H-NS is likely to be important if we are to understand the molecular interplay between VirB and H-NS on the virulence plasmid. Our work at the icsP and ospZ genetic locus reveals that sequences required for the H-NS-mediated repression and VirB-dependent derepression of the ospZ promoter lie adjacent to one another. Sequences located between −412 to −731 relative to the beginning of the ospZ gene are required for full H-NS-mediated repression, and an inverted repeat, displaying a 6/7 and 7/7 match to the reported VirB binding sites (34, 35) and centered at position −425, is required for VirB-dependent derepression. At the icsB and ipgD locus, the VirB binding sites (an inverted repeat) are also located adjacent to the region that binds H-NS (35). Furthermore, a recent study that sought to relieve H-NS-mediated repression of the E. coli proU promoter by transplanting VirB binding sites into different locations within the promoter demonstrated that antagonism of H-NS by VirB occurred optimally when the VirB binding sites were positioned immediately upstream of and adjacent to the upstream regulatory element (URE), one of two regions in this promoter region known to bind H-NS (40). Our findings at the icsP and ospZ locus therefore provide another example of a genetic locus where VirB and H-NS binding sites lie adjacent to one another. Although the importance of the juxtapositioning of these sites is not fully understood at this time, it appears to be an essential feature of promoters that are repressed by H-NS and transcriptionally derepressed by VirB. It is possible that when VirB binds next to regions occupied by H-NS, it destabilizes or displaces H-NS from the DNA. Currently, the molecular details that lead to transcriptional derepression require greater elucidation. This will be a major focus of future research in our laboratory.
Finally, we examined the ospZ promoter of other Shigella strains and species and compared them to that of the Shigella flexneri serotype 2a type strain, 2457T. Regardless of the strain examined, our analysis revealed complete sequence identity of the VirB binding sites, promoter elements, and most of the intergenic regions. The only exceptions were found in S. dysenteriae serotype 2 [(10)4279] and S. boydii serotype 4 (Sb227, YP_406371), which have a 45-bp deletion downstream of the VirB binding sites (−425) and the region required for H-NS-mediated repression (−731 to −412). Although neither of these strains is currently in our possession, an identical 45-bp deletion did not affect the VirB-dependent regulation of promoter activity (Table 4) when created in our PospZ-lacZ reporter (pospZ_Δ45). Taken together, these data strongly suggest that regardless of the Shigella strain background, the ospZ gene is upregulated by VirB.
In summary, this study has characterized the ospZ promoter and demonstrated that this gene, which is implicated in the pathogenicity of Shigella, is repressed by the nucleoid structuring protein H-NS and derepressed by the virulence gene regulator VirB. The DNA sequences needed for this regulation have been identified and found to lie adjacent to one another. As the scientific community continues to develop an interest in the dynamic role that nucleoid associated proteins like H-NS play in shaping the genome and influencing gene expression, it will become important to understand how events that remodel H-NS-DNA complexes are orchestrated and which molecular mechanisms are at play. The mapping of regions required for repression and derepression obtained from this study will provide a framework for future studies seeking to elucidate the molecular mechanism of transcriptional repression and derepression. Since transcriptional repression and derepression can be considered as the molecular switch that controls virulence in many important pathogens, including Shigella, we believe that these findings will be met with general interest.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) grant P20 RR-016464 from the IDeA Networks of Biomedical Research Excellence Program of the National Center for Research Resources and by NIH grant R15 AI090573-01. E.L.H. is supported by an Australian Research Council Future Fellowship. J.S.P. is supported by an Australian Postgraduate Award.
We sincerely thank the anonymous reviewers for their comments and helpful suggestions.
Footnotes
Published ahead of print 29 March 2013
REFERENCES
- 1. Adler B, Sasakawa C, Tobe T, Makino S, Komatsu K, Yoshikawa M. 1989. A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol. Microbiol. 3:627–635 [DOI] [PubMed] [Google Scholar]
- 2. Dorman CJ, Porter ME. 1998. The Shigella virulence gene regulatory cascade: a paradigm of bacterial gene control mechanisms. Mol. Microbiol. 29:677–684 [DOI] [PubMed] [Google Scholar]
- 3. Porter ME, Dorman CJ. 1997. Differential regulation of the plasmid-encoded genes in the Shigella flexneri virulence regulon. Mol. Gen. Genet. 256:93–103 [DOI] [PubMed] [Google Scholar]
- 4. Stoebel DM, Free A, Dorman CJ. 2008. Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology 154:2533–2545 [DOI] [PubMed] [Google Scholar]
- 5. Le Gall T, Mavris M, Martino MC, Bernardini ML, Denamur E, Parsot C. 2005. Analysis of virulence plasmid gene expression defines three classes of effectors in the type III secretion system of Shigella flexneri. Microbiology 151:951–962 [DOI] [PubMed] [Google Scholar]
- 6. Parsot C. 2009. Shigella type III secretion effectors: how, where, when, for what purposes? Curr. Opin. Microbiol. 12:110–116 [DOI] [PubMed] [Google Scholar]
- 7. Zurawski DV, Mumy KL, Badea L, Prentice JA, Hartland EL, McCormick BA, Maurelli AT. 2008. The NleE/OspZ family of effector proteins is required for polymorphonuclear transepithelial migration, a characteristic shared by enteropathogenic Escherichia coli and Shigella flexneri infections. Infect. Immun. 76:369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, Wagstaff KM, Dunstone MA, Sloan J, Whisstock JC, Kaper JB, Robins-Browne RM, Jans DA, Frankel G, Phillips AD, Coulson BS, Hartland EL. 2010. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 6:e1000898 doi:10.1371/journal.ppat.1000898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou JM, Shao F. 2007. The phosphothreonine lyase activity of a bacterial type III effector family. Science 315:1000–1003 [DOI] [PubMed] [Google Scholar]
- 10. Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ. 2007. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat. Immunol. 8:47–56 [DOI] [PubMed] [Google Scholar]
- 11. Zurawski DV, Mumy KL, Faherty CS, McCormick BA, Maurelli AT. 2009. Shigella flexneri type III secretion system effectors OspB and OspF target the nucleus to downregulate the host inflammatory response via interactions with retinoblastoma protein. Mol. Microbiol. 71:350–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Egile C, d'Hauteville H, Parsot C, Sansonetti PJ. 1997. SopA, the outer membrane protease responsible for polar localization of IcsA in Shigella flexneri. Mol. Microbiol. 23:1063–1073 [DOI] [PubMed] [Google Scholar]
- 13. Shere KD, Sallustio S, Manessis A, D'Aversa TG, Goldberg MB. 1997. Disruption of IcsP, the major Shigella protease that cleaves IcsA, accelerates actin-based motility. Mol. Microbiol. 25:451–462 [DOI] [PubMed] [Google Scholar]
- 14. Wing HJ, Yan AW, Goldman SR, Goldberg MB. 2004. Regulation of IcsP, the outer membrane protease of the Shigella actin tail assembly protein IcsA, by virulence plasmid regulators VirF and VirB. J. Bacteriol. 186:699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castellanos MI, Harrison DJ, Smith JM, Labahn SK, Levy KM, Wing HJ. 2009. VirB alleviates H-NS repression of the icsP promoter in Shigella flexneri from sites over 1 kb upstream of the transcription start site. J. Bacteriol. 191:4047–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 17. Hensley CT, Kamneva OK, Levy KM, Labahn SK, Africa LA, Wing HJ. 2011. Two promoters and two translation start sites control the expression of the Shigella flexneri outer membrane protease IcsP. Arch. Microbiol. 193:263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aiba H, Adhya S, de Crombrugghe B. 1981. Evidence for two functional gal promoters in intact Escherichia coli cells. J. Biol. Chem. 256:11905–11910 [PubMed] [Google Scholar]
- 19. Prosseda G, Falconi M, Nicoletti M, Casalino M, Micheli G, Colonna B. 2002. Histone-like proteins and the Shigella invasivity regulon. Res. Microbiol. 153:461–468 [DOI] [PubMed] [Google Scholar]
- 20. Porter ME, Dorman CJ. 1994. A role for H-NS in the thermo-osmotic regulation of virulence gene expression in Shigella flexneri. J. Bacteriol. 176:4187–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beloin C, Dorman CJ. 2003. An extended role for the nucleoid structuring protein H-NS in the virulence gene regulatory cascade of Shigella flexneri. Mol. Microbiol. 47:825–838 [DOI] [PubMed] [Google Scholar]
- 22. Dorman CJ. 2009. Global regulators and environmental adaptation in Gram-negative pathogens. Clin. Microbiol. Infect. 15(Suppl 1):47–50 [DOI] [PubMed] [Google Scholar]
- 23. Falconi M, Colonna B, Prosseda G, Micheli G, Gualerzi CO. 1998. Thermoregulation of Shigella and Escherichia coli EIEC pathogenicity. A temperature-dependent structural transition of DNA modulates accessibility of virF promoter to transcriptional repressor H-NS. EMBO J. 17:7033–7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broach WH, Egan N, Wing HJ, Payne SM, Murphy ER. 2012. VirF-independent regulation of Shigella virB transcription is mediated by the small RNA RyhB. PLoS One 7:e38592 doi:10.1371/journal.pone.0038592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dorman CJ, McKenna S, Beloin C. 2001. Regulation of virulence gene expression in Shigella flexneri, a facultative intracellular pathogen. Int. J. Med. Microbiol. 291:89–96 [DOI] [PubMed] [Google Scholar]
- 26. Berlutti F, Casalino M, Zagaglia C, Fradiani PA, Visca P, Nicoletti M. 1998. Expression of the virulence plasmid-carried apyrase gene (apy) of enteroinvasive Escherichia coli and Shigella flexneri is under the control of H-NS and the VirF and VirB regulatory cascade. Infect. Immun. 66:4957–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tobe T, Yoshikawa M, Mizuno T, Sasakawa C. 1993. Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by virF and repression by H-NS. J. Bacteriol. 175:6142–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tobe T, Nagai S, Okada N, Adler B, Yoshikawa M, Sasakawa C. 1991. Temperature-regulated expression of invasion genes in Shigella flexneri is controlled through the transcriptional activation of the virB gene on the large plasmid. Mol. Microbiol. 5:887–893 [DOI] [PubMed] [Google Scholar]
- 29. Mitobe J, Morita-Ishihara T, Ishihama A, Watanabe H. 2008. Involvement of RNA-binding protein Hfq in the post-transcriptional regulation of invE gene expression in Shigella sonnei. J. Biol. Chem. 283:5738–5747 [DOI] [PubMed] [Google Scholar]
- 30. Kane KA, Dorman CJ. 2012. VirB-mediated positive feedback control of the virulence gene regulatory cascade of Shigella flexneri. J. Bacteriol. 194:5264–5273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Timmerman KP, Tu CP. 1985. Complete sequence of IS3. Nucleic Acids Res. 13:2127–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buchrieser C, Glaser P, Rusniok C, Nedjari H, D'Hauteville H, Kunst F, Sansonetti P, Parsot C. 2000. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38:760–771 [DOI] [PubMed] [Google Scholar]
- 33. Bongrand C, Sansonetti PJ, Parsot C. 2012. Characterization of the promoter, MxiE box and 5′ UTR of genes controlled by the activity of the type III secretion apparatus in Shigella flexneri. PLoS One 7:e32862 doi:10.1371/journal.pone.0032862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taniya T, Mitobe J, Nakayama S, Mingshan Q, Okuda K, Watanabe H. 2003. Determination of the InvE binding site required for expression of IpaB of the Shigella sonnei virulence plasmid: involvement of a ParB boxA-like sequence. J. Bacteriol. 185:5158–5165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turner EC, Dorman CJ. 2007. H-NS antagonism in Shigella flexneri by VirB, a virulence gene transcription regulator that is closely related to plasmid partition factors. J. Bacteriol. 189:3403–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Collado-Vides J, Magasanik B, Gralla JD. 1991. Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev. 55:371–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sergueev K, Dabrazhynetskaya A, Austin S. 2005. Plasmid partition system of the P1par family from the pWR100 virulence plasmid of Shigella flexneri. J. Bacteriol. 187:3369–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lang B, Blot N, Bouffartigues E, Buckle M, Geertz M, Gualerzi CO, Mavathur R, Muskhelishvili G, Pon CL, Rimsky S, Stella S, Babu MM, Travers A. 2007. High-affinity DNA binding sites for H-NS provide a molecular basis for selective silencing within proteobacterial genomes. Nucleic Acids Res. 35:6330–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bouffartigues E, Buckle M, Badaut C, Travers A, Rimsky S. 2007. H-NS cooperative binding to high-affinity sites in a regulatory element results in transcriptional silencing. Nat. Struct. Mol. Biol. 14:441–448 [DOI] [PubMed] [Google Scholar]
- 40. Kane KA, Dorman CJ. 2011. Rational design of an artificial genetic switch: co-option of the H-NS-repressed proU operon by the VirB virulence master regulator. J. Bacteriol. 193:5950–5960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Formal SB, Dammin GJ, LaBrec EH, Schneider H. 1958. Experimental Shigella infections: characteristics of a fatal infection produced in guinea pigs. J. Bacteriol. 75:604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pogliano JA, Beckwith J. 1994. SecD and SecF facilitate protein export in Escherichia coli. EMBO J. 13:554–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamada H, Yoshida T, Tanaka K, Sasakawa C, Mizuno T. 1991. Molecular analysis of the Escherichia coli hns gene encoding a DNA-binding protein, which preferentially recognizes curved DNA sequences. Mol. Gen. Genet. 230:332–336 [DOI] [PubMed] [Google Scholar]
- 44. Schuch R, Sandlin RC, Maurelli AT. 1999. A system for identifying post-invasion functions of invasion genes: requirements for the Mxi-Spa type III secretion pathway of Shigella flexneri in intercellular dissemination. Mol. Microbiol. 34:675–689 [DOI] [PubMed] [Google Scholar]
- 45. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]