

Abstract
RNA polymerase (RNAP) is an extensively studied multisubunit enzyme required for transcription of DNA into RNA, yet the δ subunit of RNAP remains an enigmatic protein whose physiological roles have not been fully elucidated. Here, we identify a novel, so far unrecognized function of δ from Bacillus subtilis. We demonstrate that δ affects the regulation of RNAP by the concentration of the initiating nucleoside triphosphate ([iNTP]), an important mechanism crucial for rapid changes in gene expression in response to environmental changes. Consequently, we demonstrate that δ is essential for cell survival when facing a competing strain in a changing environment. Hence, although δ is not essential per se, it is vital for the cell's ability to rapidly adapt and survive in nature. Finally, we show that two other proteins, GreA and YdeB, previously implicated to affect regulation of RNAP by [iNTP] in other organisms, do not have this function in B. subtilis.
INTRODUCTION
RNA polymerase (RNAP) is the key enzyme responsible for transcription of DNA into RNA. Bacterial RNAP core enzyme consists of several subunits: the α dimer that holds together β and β′, which form the catalytic center, and the ω subunit that binds to β′. This core enzyme, α2ββ′ω, is capable of elongating but not initiating transcription. To initiate transcription, the core enzyme must associate with a σ subunit that allows the holoenzyme to recognize specific sequences in the DNA, i.e., promoters. Typically, several different σ subunits are present in the cell and direct the expression of different subsets of genes (1, 2).
While the α2ββ′ω composition is conserved across the bacterial kingdom, Gram-positive Firmicutes contain an additional subunit, δ, which is encoded by the rpoE gene in the model bacterium Bacillus subtilis. The δ subunit was first reported as an endogenous protein present in RNAP from phage SP01-infected Bacillus subtilis cells, which was required for its accurate middle gene transcription (3, 4). The rpoE gene specifies a protein of 173 amino acids (aa) with a molecular mass of ∼20.5 kDa. The protein is highly acidic (pI, 3.6) (5). As determined by circular dichroism (CD) spectroscopy, it consists of two domains: (i) the N-terminal domain (NTD), which is structured; and (ii) the C-terminal domain, which is unstructured and whose amino acid composition—stretches of glutamic and aspartic acid residues—makes it virtually a polyanion (6). The structure of the ordered N-terminal domain was recently solved based on a truncated construct consisting of the N-terminal domain containing a His tag. The NTD contains four α-helices and an antiparallel β-sheet (7). Delta binds to RNAP in vivo (8), but the binding site is unknown.
The in vitro effects of δ on transcription were previously examined in detail in the B. subtilis system. δ was reported to destabilize complexes between RNAP and DNA in vitro, thus increasing RNAP's specificity for good consensus promoter sequences (9, 10). Despite this inhibitory effect, δ was shown to stimulate transcription on some templates in vitro, possibly by enhancing RNAP recycling (11). However, in spite of these effects in vitro, no clear physiological role for delta has been identified. The protein is not essential, and B. subtilis mutants without δ display only a mild phenotype consisting of a prolonged lag phase of stationary-phase cells diluted into fresh medium (12).
Here, using B. subtilis as a model organism, we show that δ is a crucial component of the transcription machinery that allows the cell to survive in nature. We demonstrate that δ is important for the regulation of RNAP by the concentration of the transcription initiating NTP ([iNTP]). Various types of promoter regulation by [iNTP] were described previously (13–17). Relevant to this study is the regulation in which the [iNTP] affects the stability of the open complex, a significant intermediate formed during transcription initiation. Briefly, an increasing iNTP concentration stimulates transcription by stabilizing the open complex at promoters that form relatively unstable open complexes ([iNTP]-sensitive promoters); a decreasing iNTP concentration has the opposite effect (18, 19). Promoters that form stable open complexes are not affected by this type of regulation ([iNTP]-insensitive promoters). This type of [iNTP] regulation is pivotal for rapid changes in gene expression when the intracellular NTP concentrations change—typically in response to changes in nutrient availability (20–23). We demonstrate that a mutant strain lacking δ displays a decreased ability to rapidly alter the activity of [iNTP]-sensitive promoters. This likely contributes to the decrease in fitness of the cell, as it is unable to survive in a changing environment under the pressure of a competing strain. Furthermore, we show that two other proteins, GreA and YdeB, whose homologs in other bacterial species were previously implicated to affect regulation of RNAP by [iNTP], do not affect this process in B. subtilis.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Strains and plasmids are listed in Table 1. Primers are listed in Table S1 in the supplemental material. Competent Escherichia coli cells—strain DH5α, used for cloning, or strain BL21 (DE3), used for overexpression of proteins—were prepared according to the method of Hanahan (30). Competent B. subtilis cells were prepared as described previously (31). All PCRs were done using the Expand High Fidelity system (Roche). All constructs were verified by sequencing.
Table 1.
List of strains and plasmids
| Strain | Relevant characteristicsa | Source |
|---|---|---|
| B. subtilis | ||
| MH5636 | rpoC-10×His | 24 |
| MO1099 | trpC2 pheA1 amyE::MLS | 25 |
| RLG6943 | MO1099 amyE::Cm, rrnBp2 (−77/+50)-lacZ | 20 |
| RLG7553 | MO1099 amyE::Cm, rrnBp2 (−38/+1)-lacZ | 20 |
| RLG7554 | MO1099 amyE::Cm, rrnBp1 (−39/+1)-lacZ | 20 |
| RLG7555 | MO1099 amyE::Cm, Pveg (−38/−1,+1G)-lacZ | 20 |
| LK34 | MO1099 amyE::Cm, promoterless lacZ | 20 |
| LK293 | MO1099 KO rpoE::Kan, amyE::Cm, rrnBp2 (−38/+1)-lacZ | This study |
| LK633 | MO1099 KO rpoE::Kan, MLS | This study |
| LK637 | MH5636 rpoC-His10, rpoE::Kan, MLS | This study |
| LK642 | MO1099 KO rpoE::Kan, amyE::Cm, rrnBp1 (−39/+1)-lacZ | This study |
| LK643 | MO1099 KO rpoE::Kan, amyE::Cm, Pveg (−38/−1,+1G)-lacZ | This study |
| LK831 | MO1099 KO rpoE::Kan, amyE::Cm, PrpoE-rpoE | This study |
| LK866 | MO1099 amyE::Spc, Pxyl-rpoE-6×His | This study |
| LK1173 | MO1099 amyE::Spc, Pxyl-6×His-ydeB | This study |
| E. coli | ||
| RLG770 | pRLG770 | 26 |
| RLG7023 | BL21/pFL31/Bsu_rpoE | 6 |
| RLG 7019 | pSG1729 | 27 |
| RLG7396 | pDG 3661, rrnBp1 (−39/+1)-lacZ | 20 |
| RLG7397 | pDG 3661, rrnBp2 (−38/+1)-lacZ | 20 |
| RLG7557 | pDG 3661, Pveg (−38/−1/+1G)-lacZ | 20 |
| LK1 | pRLG770 with Pveg (−38/+1,+1G) | 20 |
| LK4 | pRLG770 with Pveg-10DBP1 | 28 |
| LK7 | pRLG770 with rrnBp1(−39/+1) | 28 |
| LK9 | pRLG770 with rrnBp1-10Dveg | 28 |
| LK14 | pRLG770 with rrnAp1 (−39/+1) | 28 |
| LK22 | BL21 pCD2/Bsu_sigA | 29 |
| LK140 | pRLG770 with rrnBp2 (−38/+2) | 20 |
| LK371 | BL21 pGEX1/Bsu_greA | This study |
| LK626 | DH5α pGEX-5X-3/KO rpoE::Kan | This study |
| LK645 | DH5α pSG1729 with rpoE-6×His | This study |
| LK1093 | DH5α pDG3661 with PrpoE-rpoE | This study |
| LK1169 | DH5α pSG1729 with 6×His-ydeB | This study |
MLS, macrolide-lincosamide-streptogramin B resistance; Spc, spectinomycin.
The B. subtilis ΔrpoE knockout (KO) strain was prepared by a double crossover by replacing the rpoE gene with a kanamycin (Kan) resistance gene. The kanamycin resistance gene, including its promoter region, was excised by EcoRI and SalI from the pDK vector obtained from the Bacillus Genetic Stock Center. Homology regions (∼600 bp from both upstream and downstream of the rpoE gene) were amplified by PCR with two pairs of primers (210, containing BamHI, plus 211, containing EcoRI; and 212, containing XhoI, plus 213, containing NotI). The rpoE knockout construct was sequentially assembled in plasmid pGEX-5X-3, where it was cloned into the polycloning region. The cloning was performed in the following order: (i) rpoE upstream homology region, (ii) kanamycin resistance gene, and (iii) rpoE downstream homology region. The resulting construct (LK626) was transformed into competent B. subtilis MO1099. The transformants that appeared on LB plates supplemented with kanamycin (5 μg/ml) were analyzed by PCR and Western blotting, and one clone (LK633) was selected for further experiments. Subsequently, the LK633 strain was transformed with pDG3661 plasmids containing transcriptional fusions to lacZ of the following core promoters: (i) rrnBp1 (RLG7396), (ii) rrnBp2 (RLG7397), and (iii) Pveg+1G (RLG7557) (20), yielding strains LK642, LK293, and LK643, respectively. The transformants were selected on LB plates with chloramphenicol (Cm; 5 μg/ml). The mode and site of integration were verified as described previously (25), as well as by PCR and sequencing.
The strain for purification of His-tagged RNAP without δ was prepared by transforming strain MH5636 with construct LK626, yielding strain LK637.
For rpoE complementation studies, we first created a strain in which the rpoE gene was under the control of a xylose-inducible promoter. We titrated the amount of xylose that resulted in approximately the wild-type (wt) level of δ in the cells as judged by Western blotting performed on cell extracts from exponential phase. However, the native promoter for rpoE varies its activity during the bacterial growth, with a spike at the end of exponential phase (12). This variation was impossible to achieve with the Pxyl promoter, and the strain in which the rpoE expression was driven by Pxyl did not perform well in the competition experiments (data not shown). Hence, we created a strain in which the rpoE gene was ectopically integrated at the amyE site and was transcribed from its own promoter. To prepare this strain, we first PCR amplified the rpoE region, including its promoter, by primers 483 and 484 and cloned this fragment via EcoRI and HindIII into pDG3661. The integration was verified as described above. The construct LK1093 was transformed into the ΔrpoE strain LK633, yielding strain LK831.
For pulldown experiments with δ, we prepared strain LK866, where δ was His tagged. The plasmid construct was prepared using the integration vector pSG1729 (27), into which the rpoE gene (amplified with primers 194 and 235) was inserted, coding for a C-terminal 6×His tag. The insertion was done via KpnI and EcoRI. The gene was under a xylose-inducible promoter. The resulting construct (LK645) was transformed into B. subtilis MO1099, yielding strain LK866, and the integration was verified.
For pulldown experiments of YdeB, we prepared strain LK1173. The ydeB gene (amplified with primers 931 and 963) contained an N-terminal 6×His tag and was cloned into pSG1729. The gene insertion was done via KpnI and BamHI, and the gene was under a xylose-inducible promoter. The resulting construct (LK1169) was transformed into B. subtilis MO1099, yielding strain LK1173, and the integration was verified.
For overexpression of GreA, the LK371 construct that contained a GST-GreA fusion was prepared. The B. subtilis greA gene was amplified with primers GREA1 and GREA2 and inserted into the pGEX-1 expression vector.
Supercoiled plasmids for in vitro transcriptions were prepared with Wizard Midipreps purification system (Promega) and subsequently phenol-chloroform extracted, precipitated with ethyl alcohol (EtOH), and dissolved in water.
Media and growth conditions.
For plasmid and most protein purifications, appropriate strains were grown in LB medium at 37°C. When appropriate, xylose or IPTG (isopropyl-β-d-thiogalactopyranoside) was added at 0.8% or 0.8 mM, respectively. For in vivo experiments, the cells were grown in defined MOPS (morpholinepropanesulfonic acid) medium (20) supplemented with 0.4% glucose and all 20 amino acids at 25 μg/ml. Competition experiments were carried out in the LB medium.
Protein purification.
Wild-type B. subtilis RNAP (Eδ) was purified from the MH5636 strain, and RNAP without δ (E) was purified from the LK637 strain. The purifications were performed as described previously (24). σA was overexpressed from plasmid pCD2 (29) and purified as described previously (11). δ was overproduced in strain RLG7023 and purified as described previously (6).
GreA was purified by affinity chromatography as described previously (32). Fusion protein GST-GreA was cleaved by Factor Xa, and Factor Xa was removed by Xa Removal Resin (Qiagen). Proteins were dialyzed into buffer A (50 mM Tris-Cl [pH 8.0], 100 mM NaCl, 50% glycerol, 3 mM β-mercaptoethanol).
Pulldown experiments and Western blots.
B. subtilis with either His-tagged δ (LK866) or His-tagged YdeB (LK1173) or a His tag-free strain (MO1099) was used for pulldown experiments. Equal amounts of cells were sonicated and loaded onto a nickel matrix (GE Healthcare). The proteins were eluted with 400 mM imidazole according to the manufacturer's recommendations. Subsequently, aliquots from the eluates were separated on 4% to 12% SDS-polyacrylamide gel electrophoresis (Invitrogen), transferred electrophoretically to nitrocellulose membranes, and detected with primary mouse monoclonal anti-RNAP β antibody (8RB13; Santa Cruz Biotechnology) and secondary antimouse (680 nm) antibody using the Oddyssey infrared imaging system (Li-Cor Biosciences).
Transcription in vitro.
Eδ and E were reconstituted with a saturating concentration of σA. In the case of E+δ, E that was already reconstituted with σA was subsequently reconstituted with the δ protein. The E:δ ratio was 1:5. In the case of Eδ+GreA, Eδ that was already reconstituted with σA was subsequently reconstituted with the GreA protein. The Eδ:GreA ratio was 1:30 (1 μM GreA). All reconstitutions were carried out in a glycerol storage buffer (50 mM Tris-HCl [pH 8.0], 0.1 M NaCl, 3 mM 2-mercaptoethanol, 50% glycerol) for 15 min at 30°C.
Transcriptions were carried out in 10-μl reaction volumes in transcription buffer containing 40 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 1 mM dithiothreitol (DTT), and 0.1 mg/ml bovine serum albumin (BSA). In multiple-round transcriptions, the buffer was supplemented with 150 mM KCl (Fig. 1 and 2A), and in single-round transcriptions it was supplemented with 30 mM KCl (Fig. 3). As a template, 0.8 nM supercoiled plasmid was used, into which the respective promoters (core promoter versions) were cloned via EcoRI and HindIII restriction sites as described earlier (Table 1) (28). ATP, CTP, and GTP were 200 μM; UTP was 10 μM plus 2 μM radiolabeled [α-32P]UTP. For KGTP constant determinations, the GTP concentration varied from 20 to 2,000 μM. The concentration of RNAP in in vitro transcriptions was 30 nM. All transcription experiments were done at 30°C, and transcriptions were allowed to proceed for 15 min. Transcriptions were stopped with equal volumes of formamide stop solution (95% formamide, 20 mM EDTA [pH 8.0]). Samples were loaded onto 7 M urea–6.5% polyacrylamide gels and electrophoresed. The dried gels were scanned with Molecular Imager_FX (Bio-Rad). The amounts of the 145-nucleotide-long transcripts (originating from the cloned promoters) were quantified with QuantityOne software (Bio-Rad). All calculations and data fitting were done using SigmaPlot from Jandel Scientific.
Fig 1.

δ changes the requirements of RNAP for the [iNTP] at some promoters in vitro. (A) Gels showing primary data from in vitro multiple-round transcriptions from rrnBp1 (LK7) with three types of RNAP: Eδ, E, and E+δ. Eδ is RNAP purified from wt B. subtilis containing δ; E is RNAP purified from a ΔrpoE strain without δ; E+δ is E with added purified δ in a 1:5 molar ratio. The concentration of the iNTP (GTP) varied from 20 to 2,000 μM. (B) Quantitation of the data from panel A. Open circles, Eδ; filled circles, E; open rectangles, E+δ. The plateau of the maximal transcription activity with each enzyme was set as 1. (C) Comparison of KGTP values of rrnBp1 (LK7), rrnBp2 (LK140), rrnA P1 (LK14), Pveg-10BP1 (LK4), Pveg (LK1), and BP1-10Dveg (LK9). Eδ (black bars), E (bars with diagonal lines), and E+δ (dark gray bars) are the three forms of RNAP used in the assays illustrated in panels A through C. The KGTP constant specifies the concentration of GTP required for 50% maximal transcription. The bars represent mean values of at least two independent experiments.
Fig 2.

GreA has no effect on regulation of RNAP by [iNTP], and YdeB does not interact with RNAP. (A) In vitro transcription conducted in the presence (1 μM) or absence of GreA as a function of GTP concentration. rrnBp1 was used as the promoter. A representative result is shown (the experiment was performed three times). (B) YdeB does not interact with B. subtilis RNAP in vivo. The upper panel shows Western blots of nickel affinity chromatography pulldown assays from strains containing either His-tagged YdeB (LK1173) or His-tagged δ (LK866) or no His-tagged protein (w/o His-tag; MO1099). The blot was probed with anti-β antibody. The lower panel is a loading control (Coomassie blue-stained gel), showing that equal amounts of YdeB and/or δ were pulled down and loaded onto the Western blots. A representative result is shown (the experiment was performed three times with the same result).
Fig 3.

Effects of δ on the open complex of rrnBp1 and Pveg. (A) Primary data showing the effect of delta on open-complex stability at rrnBp1 and Pveg (LK7 and LK1). Open complexes were formed with E or E+δ, and at time zero heparin was added. Heparin binds to free RNAP but not to RNAP in complex with DNA. At indicated time intervals, aliquots were withdrawn and transcription was initiated with all four NTPs. The amount of the transcript reflected the amount of the open complex. (B) Representative graph for rrnBp1 open complex decay experiments. The half-life of its open complex was 33 ± 7 s with E (open circles) and 10 ± 2 s with E+δ (filled circles). These numbers are averages and standard deviations (SDs) calculated from three independent experiments. (C) Representative graph for Pveg open-complex decay experiments. The half-life of its open complex was 33 ± 2 min with E (open circles) and 8 ± 2 min with E+δ (filled circles). These numbers are averages, and SDs were calculated from three independent experiments.
For RNAP sensitivity to the [iNTP] (Fig. 1), the data were fitted to the equation f = a × [1 − exp(−b × x)] (where f is the relative transcription, x is the concentration, and a and b are constants), from which the KGTP constants were calculated.
For open complex half-life experiments, DNA was incubated with RNAP for 15 min at 30°C, and at time zero heparin was added. Heparin functions as a competitor, sequestering free RNAP and allowing only one round of transcription (33). At selected time points, aliquots were withdrawn and added to all four NTPs to initiate transcription. For each promoter, the amount of heparin had to be determined to allow transcription of a single round and to prevent transcription reinitiation. This was done by performing multiple-round transcriptions with increasing amounts of heparin. The concentration of heparin that completely abolished this multiple-round transcription (when transcription was initiated with RNAP in the presence of heparin) and that permitted single-round transcription (when transcription was initiated from preformed open complexes by the addition of all four NTPs and heparin) was used for that particular promoter. For rrnBp1 and Pveg, the heparin concentrations were 2.5 and 7 μg/ml, respectively. The data were plotted as a function of time, fitting the equation f = a × exp(−b × x) (where f is the relative transcription, x is time, and a and b are constants), from which the complex half-lives for respective promoters were calculated.
Relative GTP concentration determination.
GTP was extracted from cells grown in the MOPS medium with 0.4% glucose supplemented with [32P]H3PO4 (20 μCi/ml). Aliquots of cells were mixed with equal volumes of 11.5 M formic acid, vortexed, and stored overnight at −20°C. After centrifugation, 4 μl of supernatant was spotted on a thin-layer chromatography (TLC) plate (Polygram CEL 300 PEI; Macherey-Nagel) and run in 1.5 M KH2PO4. Commercially obtained GTP (Roche) was run alongside as a marker. Recently, an alternative approach to NTP separation was reported (34), using a different solvent, AFC (3 M ammonium formate [pH 2.4] and 0.7 M NH4Cl). We performed the NTP separations also in the AFC solvent with results identical to those obtained when 1.5 M KH2PO4 was used (data not shown). After an overnight exposure, the spots were quantified with Molecular Imager_FX (Bio-Rad) and normalized to cell density (optical density at 600 nm [OD600]).
Starvation for amino acids.
Cells were grown in MOPS, 0.4% glucose, and 25 μg/ml of all 20 amino acids at 37°C. Starvation for amino acids was induced by the addition of 3 mM (final concentration) serine hydroxamate (SHX; Sigma-Aldrich). For relative GTP concentration or promoter activity determinations, aliquots of cells were withdrawn prior to SHX addition (time zero) and at indicated time points (5, 10, and 20 min).
Promoter activity in vivo.
Core promoters were fused at +1 to a reporter gene (lacZ) from which an mRNA with a relatively short biological half-life is transcribed (4 min). The promoter fusions were integrated at the amyE locus (20). Transcripts from all three in vivo-tested promoters were identical, eliminating potential differences in mRNA stability. The wt Pveg promoter initiates with ATP (35). Here, a +1G version that does not change its properties with respect to [iNTP] sensitivity was used (36). The amount of the lacZ-mRNA was used as a measure of promoter activity, determined by quantitative primer extension. RNA was extracted, reversely transcribed, and visualized, and the reporter gene mRNA was quantified as described previously (20). At the beginning of RNA extraction, recovery marker RNA was added (RLG6943; the recovery marker was a longer version of the reporter gene allowing to distinguish these two according to molecular mass in gels), controlling for degradation problems or pipetting errors. The gels were exposed overnight, and the bands were quantified with Molecular Imager_FX (Bio-Rad). The relative promoter activity was normalized to the amount of the recovery marker and optical density (OD600) of the cells.
Competition assays.
The strains that were used to start the competition experiments were first grown separately at 37°C until they reached an OD600 of ∼0.5. Then, 50 ml of rich medium (Luria broth) was inoculated with the same CFU of each of the two exponentially growing strains that were to compete against each other. The starting OD600 was ∼0.03. Every 24 h, an aliquot was inoculated into a fresh medium to an OD600 of ∼0.03. Before each dilution, aliquots were withdrawn for CFU enumeration. For wt (RLG7554) versus ΔrpoE (LK642) strain competitions, plating the cells on agar with Cm (2.5 μg/ml; both strains grow) or Kan (5 μg/ml; only LK642 grows) was used to determine the CFU for each strain. We also used strain LK34 as the wt strain. This strain contains promoterless lacZ at the amyE site, and we competed it against LK642. We plated the cells on agar supplemented with 40 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (Sigma) and counted the CFU for each strain by the white (wt) or blue (ΔrpoE strain) color. We obtained results identical to those shown when the Cm/Kan selection was used (data not shown). For competitions of ΔrpoE strain LK642 versus the ΔrpoE strain complemented with ectopically integrated rpoE (LK831), we used the white (LK831) or blue (LK642) visualization of CFU. In single-strain growth experiments, exponentially growing wt (RLG7554) and ΔrpoE (LK642) cells (OD600, ∼0.5) were diluted to 50 ml of LB media to an OD600 of ∼0.03. The dilutions and platings were performed as in competition experiments.
RESULTS
Delta affects the sensitivity of RNAP to [iNTP] in vitro.
During transcription initiation, RNAP first forms the closed complex with the promoter DNA; next, RNAP isomerizes and melts the DNA to form the open complex in which the two DNA strands are separated and form the transcription bubble (37). As δ was previously shown to decrease the stability of open complexes of RNAP with some promoters (10, 11), we speculated that δ may affect the sensitivity of RNAP to [iNTP]. In B. subtilis, a number of promoters were shown to be sensitive to [iNTP]. Typical representatives are rRNA promoters. All rRNA promoters in B. subtilis initiate with GTP (38), and changes in [GTP] play an important role in their regulation (20).
First, as necessary tools for subsequent studies, we created B. subtilis strains with rpoE knocked out and replaced with the kanamycin resistance gene (for details see Materials and Methods and Table 1). For in vitro studies, we purified RNAP from wt and ΔrpoE strains. RNAP was reconstituted with the main vegetative sigma factor, σA, and all promoters used in this work were σA dependent. In the text, wt RNAP containing δ is designated Eδ, RNAP without δ is designated E, and RNAP purified from a ΔrpoE strain but subsequently reconstituted with δ is designated E+δ.
Second, we tested whether δ affects promoter sensitivity to [iNTP] in vitro. We conducted multiple-round transcriptions with Eδ, E, and E+δ at increasing GTP concentrations using two sets of promoters: (i) [iNTP]-sensitive and (ii) [iNTP]-insensitive promoters. The [iNTP]-sensitive promoters included rrnBp1, rrnBp2, rrnAp1, and Pveg-10BP1. rrnBp1 and rrnAp1 are rrn promoters regulated by [iNTP]. rrnBp2 is the other promoter for the rrnB operon. It is downstream of rrnBp1, and this tandem arrangement is typical for rrn operon promoters in both Gram-positive and Gram-negative bacteria (39). The rrnp2 promoters of both B. subtilis and E. coli generally display less pronounced changes in activity than p1 promoters, consistent with their role in providing the basal, housekeeping rrn promoter activity (28, 40). Pveg-10BP1 is a synthetic promoter consisting of [iNTP]-insensitive Pveg (−39 to −14) fused to the downstream portion from rrnBp1 (−13 to + 1). We recently showed that this downstream part from rrnBp1 is responsible for the [iNTP] sensitivity of this promoter (28). For the [iNTP]-insensitive promoters, we used Pveg and BP1-10Dveg. Pveg is a promoter that forms relatively stable complexes with RNAP and is not regulated by the [iNTP], as it is saturated with a relatively low level of its iNTP (20, 28). The wt Pveg promoter initiates with ATP (35). Here, a +1G version that does not change its properties with respect to [iNTP] sensitivity was used (36). BP1-10Dveg is the reciprocal version of Pveg-10BP1 (the downstream part is from Pveg, and the upstream part from rrnBp1) (28). Figure 1 shows the dependence of the activity of the three forms of RNAP on [GTP]. KGTP is used here to quantitate the promoter's sensitivity to [iGTP]. It is the concentration of the GTP required for half-maximal transcription. Promoters that are [iNTP] sensitive and regulated by [iNTP] in vivo display relatively high KGTPs in vitro. The KGTPs of [iNTP]-sensitive promoters were significantly decreased for RNAP lacking δ, and these decreases were reversed by the addition of δ. On the other hand, in the case of the [iNTP]-insensitive promoters, their requirements for the [iNTP] were not much changed regardless of the presence or absence of δ. In the case of rrnBp2, this promoter displayed an intermediate phenotype with respect to [iNTP] sensitivity, reflected by its lower KGTP with Eδ relative to that of rrnBp1 (Fig. 1C). This is in agreement with the lower ability of the p2 promoter to change its activity with changing environmental conditions.
Next, we tested the effects of two other proteins, GreA and YdeB, on the sensitivity of B. subtilis RNAP to [iNTP]. In Gram-negative Escherichia coli, the DksA protein is required for regulation of RNAP by [iNTP] (41). No obvious sequence homologs of DksA exist in B. subtilis. However, B. subtilis contains GreA, which is a structural homolog of DksA (42). Gre proteins are transcription elongation factors that help cleave the nascent transcript in the active site of RNAP, thereby rescuing stalled elongation complexes (43). Moreover, in addition to its function in transcription elongation, GreB of E. coli was shown under some conditions to also affect the KNTP at rRNA promoters (44). B. subtilis contains only one Gre factor. The other protein, YdeB, is a homolog of CarD, an essential protein from Mycobacterium tuberculosis, where it binds to RNAP and is important for regulation of rRNA promoters. Moreover, CarD was shown to be able to functionally replace DksA in an E. coli DksA-null strain (45). Hence, we decided to test the effect of GreA and YdeB on the regulation of B. subtilis RNAP by [iNTP].
We tested the effect of GreA on KGTP of rrnBp1 with Eδ in vitro (Fig. 2A). Unlike what was seen for δ, the presence or absence of GreA had no impact on KGTP. In a control experiment, we verified that the GreA protein was active on elongation complexes (see Fig. S1 in the supplemental material). As for YdeB, we first wished to determine whether it interacts with RNAP in vivo. Therefore, we performed pulldown experiments with His-tagged YdeB followed by Western blotting with antibody against the β subunit of RNAP (Fig. 2B). While His-tagged δ (positive control) interacted with and was able to pull down significant amounts of RNAP, no such interaction was detected for YdeB (the small amounts of β detected in the YdeB pulldown were comparable to the amounts pulled down from the control strain without a His-tagged protein). This lack of interaction was consistent with the result obtained in parallel experiments in which we tested the effect of purified YdeB on KGTP of rrnBp1: no effect was observed (data not shown).
We concluded that neither GreA nor YdeB had an effect on the regulation of RNAP by [iNTP]. On the other hand, δ affected the sensitivity of RNAP to the concentration of iNTP in vitro at [iNTP]-sensitive promoters but not at [iNTP]-insensitive promoters.
Delta changes the stability of the open complex at rrnBp1 and Pveg.
As δ changed the KGTP of rrnBp1 but not of Pveg, we wanted to establish whether it was due to a differential effect of δ on the open complex stability of these two promoters. However, Fig. 3 shows that δ decreased the open complex lifetimes of both rrnBp1 and Pveg. The extent of the destabilizing effect on the open complex was about the same (3- to 4-fold) for rrnBp1 and Pveg, but the open-complex lifetime of Pveg in the presence of δ (∼8 min) was still >1 order of magnitude longer than that for rrnBp1 in the absence of δ (∼33 s), consistent with the view that only promoters with relatively unstable open complexes are [iNTP] sensitive. Taken together, the in vitro experiments suggested that δ may affect the regulation of [iNTP]-sensitive promoters in vivo.
Effects of delta on [iNTP] sensitivity of RNAP in vivo.
To assess the effect of δ on promoter activity in vivo, we followed promoter activity in the wt and ΔrpoE strains during starvation for amino acids (stringent response), as under these conditions the concentration of the effector molecule, GTP, changes. Upon the induction of the stringent response, ribosomally bound RelA senses uncharged tRNA in the A site of the ribosome and synthesizes the alarmone molecule, (p)ppGpp. (p)ppGpp, in B. subtilis, directly inhibits three enzymes involved in the GTP metabolism, thereby decreasing its intracellular concentration (23, 46): (i) IMP dehydrogenase, which converts IMP to XMP; (ii) HprT, a salvage pathway enzyme, which converts hypoxanthine to IMP and guanine to GMP; and (iii) GmK, guanylate kinase, which was proposed to convert GMP to GDP. The decrease in [GTP] leads to a drop in the activity of rRNA promoters. Thus, with respect to affecting the activity of RNAP at rRNA promoters, (p)ppGpp plays an indirect role in B. subtilis, unlike in E. coli, where (p)ppGpp directly inhibits RNAP at rRNA promoters (20, 33).
First, we extracted GTP and quantified its relative concentration in wt and ΔrpoE strains before and after the induction of the stringent response. As shown in Fig. 4A, the two strains displayed comparable changes in GTP levels.
Fig 4.

δ affects the kinetics of promoter response to amino acid starvation in vivo. (A) Quantitation of the relative changes in the intracellular GTP concentration in the wt strain (RLG7554) and the ΔrpoE strain (LK642). The cells were growing in a defined MOPS-buffered medium with all 20 amino acids in the presence of 32P. At time zero, SHX was added, inducing starvation for amino acids. At time zero (before the addition of SHX) and at indicated time intervals, aliquots of the cells were withdrawn and NTPs extracted and quantified. The graph shows the averages from two independent experiments. The bars indicate the range. (B) Kinetics of the rrnBp1 promoter response to amino acid starvation in wt (filled circles; RLG7554) and ΔrpoE (open circles; LK642) strains. The amino acid starvation was induced as described for panel A. At selected time intervals, RNA was extracted, reverse transcribed, and quantified (see Materials and Methods for details). The amount of the mRNA that originated from the rrnBp1-marker gene fusion was used as a measure of the relative promoter activity. (C) Kinetics of the rrnBp2 promoter response to amino acid starvation in wt (filled circles; RLG7553) and ΔrpoE (open circles; LK293) strains. (D) Kinetics of the Pveg promoter response to amino acid starvation in wt (filled circles; RLG7555) and ΔrpoE (open circles; LK643) strains. The insets show representative reverse-transcribed 5′-end portions of the lacZ mRNAs that originated from the tested promoters.
Second, we monitored the activity of rrnBp1, rrnBp2, and Pveg in wt and ΔrpoE strains. Relative promoter activities were determined by quantitative primer extension of lacZ mRNA transcribed from promoter fusions to lacZ (for details, see Materials and Methods). As shown in Fig. 4B, in the wt the activity of rrnBp1 decreased after the onset of the stringent response. In the mutant genetic background, however, the decrease in the rrnBp1 activity was less rapid and not as pronounced within the time frame of the experiment. The activity of rrnBp2 in the wt decreased less than the activity of rrnBp1 in the wt (Fig. 4B and C), in agreement with its lower sensitivity to [iGTP] in vitro. The difference in changes in activity between rrnBp2 in wt and in ΔrpoE strains was relatively small, and at the 20-min time point there was already no difference (Fig. 4C). The activity of Pveg, in agreement with previously published results, almost did not change during the stringent response in the wt (Fig. 4D) (36), and the same was true for this promoter in the ΔrpoE mutant (Fig. 4D).
We concluded that the absence of δ affected the response of [iNTP]-sensitive rrnBp1 to the stringent response while the effect on moderately [iNTP]-sensitive rrnBp2 was less pronounced and no effect was observed on [iNTP]-insensitive Pveg. The altered ability of [iNTP]-sensitive promoters to rapidly change gene expression could be a decisive factor for the cell when it is required to rapidly adapt to changing conditions.
Delta is essential for the cell to successfully compete.
An example of the inability of the delta-null strain to rapidly adapt is its prolonged lag phase (12; A. Rabatinová, data not shown). The difference from the wt is about a half-hour, resulting in approximately one extra generation for the wt. We also evaluated the effect of the absence of δ on the ability of the cells to grow after the induction of amino acid starvation. The wt and the ΔrpoE strains grew at comparable rates during the exponential phase. However, while the wt was capable of growing at a reduced growth rate under the amino acid-limiting conditions, the growth of the ΔrpoE strain immediately ceased (see Fig. S2 in the supplemental material). While both the aforementioned phenotypes can be described as mild, we speculated that the δ-lacking mutant may be at a grave disadvantage when facing competition from the wt parent strain under changing environmental conditions that would challenge to its limits the ability of the transcription machinery to rapidly alter gene expression. To test this hypothesis, we mixed equal CFU of exponentially growing wt and ΔrpoE cells and let them grow together. Every 24 h, the cells were diluted into fresh medium. At the start of the experiment and just before every dilution, the relative numbers of wt and ΔrpoE cells were scored by plating the cells onto agar dishes with appropriate selective additives. We observed a dramatic phenotype: the ΔrpoE strain cell numbers soon declined in the medium, being outcompeted by wt cells (Fig. 5A). Next, we mixed the ΔrpoE strain and its derivative, which was complemented with an ectopically integrated rpoE. In this case, the complemented strain easily took over the culture (Fig. 5B). Finally, using the same cycle of culturing and diluting cells, we cultured wt and ΔrpoE strains separately. In this experiment, there was no significant decline in CFU for the ΔrpoE strain (Fig. 5C).
Fig 5.
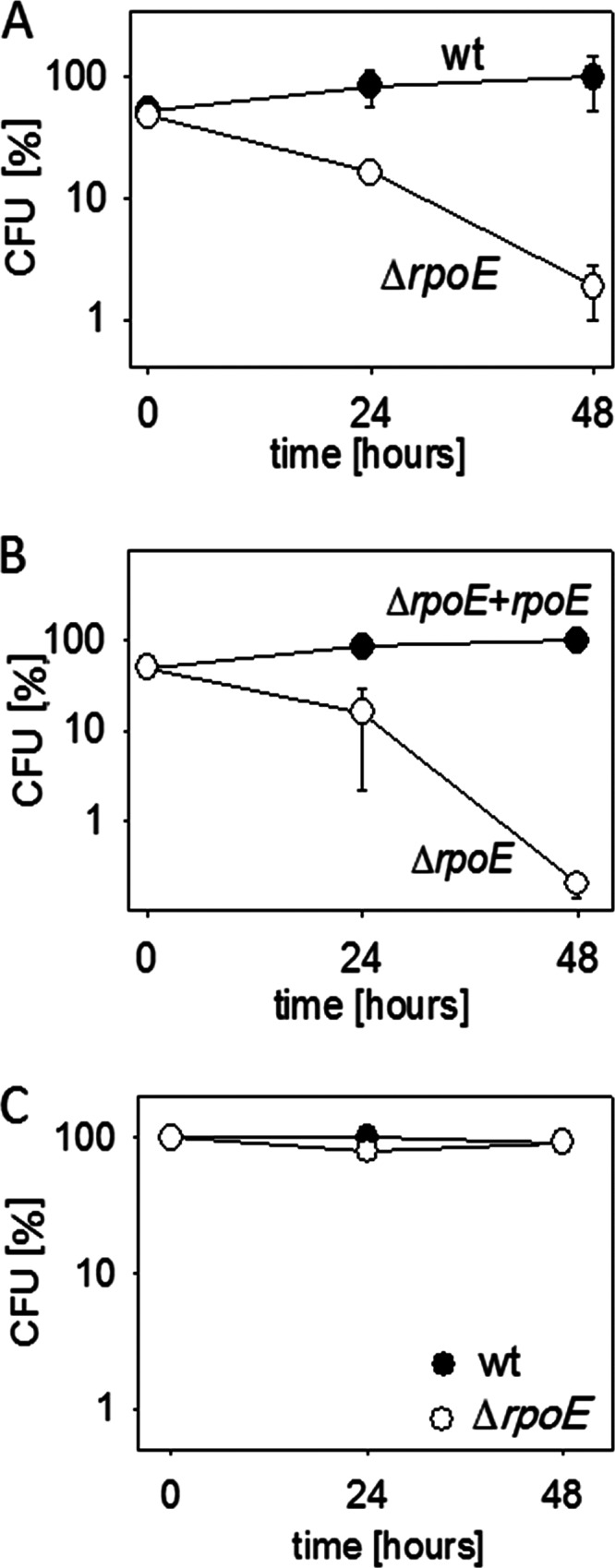
The absence of δ is detrimental to the survival of the ΔrpoE mutant in competition with the wt. The data are averages of at least two independent experiments conducted in duplicate. (A) Competition of wt B. subtilis (RLG7554) with a ΔrpoE strain (LK642). Equal numbers (CFU) of exponentially growing wt and δ-deficient (ΔrpoE) cells were mixed together in LB medium. The number of CFU for each strain at time zero was set as 50% (the sum of CFU of the two strains was 100%). Then, every 24 h, the viable cell counts for both wt and ΔrpoE strains were enumerated by plating several dilutions of the culture onto agar plates with appropriate selection markers (see Materials and Methods for details). The y axis shows the percentage of each strain in the culture. (B) Competition of the ΔrpoE strain (LK642) with its derivative containing ectopically integrated rpoE at the amyE site (LK831). The experiment was conducted as described for panel A. (C) Independent growth of wt (RLG7554) and ΔrpoE (LK642) strains. The number of wt strain CFU at each time point was set as 100%.
We concluded that although δ is not essential for the cell per se, it is indispensable for survival when facing competition.
DISCUSSION
This study describes the importance of δ for regulation of RNAP by [iNTP]. It is the first protein factor in Gram-positive bacteria identified to affect the sensitivity of RNAP to [iNTP], namely, to the regulatory metabolite GTP that plays critical roles in reprogramming of gene expression in response to stress (46). Further, the results demonstrate that δ is essential for the cell to rapidly adapt to changes in its environment. This is of utmost importance when the cell is competing with other cells, as is the case in the real world in multispecies communities, or when pathogens are battling with the host organism, as illustrated by the decrease in pathogenicity of δ-lacking Staphylococcus aureus and Streptococcus agalactiae (47, 48). Thus, although δ is not essential per se, it appears to be an indispensable part of the properly functioning transcription machinery.
What is the underlying molecular mechanism of δ?
Delta decreases the stability of the open complex. The less stable the open complex is, the higher the concentration of the iNTP required to penetrate into the transcriptionally competent complex within the available time window. Thus, δ alters the requirements of RNAP for [iNTP] at promoters when the stability of the open complex is a rate-limiting factor for transcription initiation. This appears to be relevant in vivo. The absence of δ slows down the promoter response to a change in [iNTP]. A number of promoters besides rRNA promoters are also [iNTP] sensitive in B. subtilis (36, 49, 50). Alterations to their ability to rapidly respond to changes in the [iNTP] are likely to have a cumulative and adverse effect on the cell's competitive fitness.
Previously, it was reported that δ enhances promoter selection and prevents binding of RNAP to nonspecific DNA (9). This is in agreement with the recent transcriptomic study conducted with the dental pathogen Streptococcus mutans, for which the rpoE knockout strain displayed expression of many otherwise unexpressed RNAs (51). Furthermore, as suggested by in vitro experiments (11, 52), it is possible that δ in vivo also aids the recycling of RNAP and/or affects RNAP with alternative sigma factors. Hence, the effects of δ can be multifaceted and the loss of fitness may result from a mosaic of combined effects that together prevent RNAP from functioning optimally. This defect, then, is not apparent unless the cells are challenged to their limits by the presence of a competing organism(s). Then, the absence of δ proves to be fatal.
Are there other factors that functionally overlap δ in B. subtilis?
In Gram-negative Escherichia coli, DksA potentiates the regulation of rRNA promoters by [iNTP]. DksA functions by increasing the RNAP requirements for the [iNTP] at rRNA promoters, thereby making RNAP sensitive to intracellular changes in [iNTP]. This is mediated by the DksA's destabilizing effect on the open complex (41), reminiscent of the effect of δ on RNAP. In vivo, the effect of DksA on rRNA promoter activity is more pronounced than the effect of δ, as the absence of DksA completely abolishes promoter activity change in response to amino acid starvation (41). Therefore, it is possible that other factors also modulate the promoter [iNTP] sensitivity in vivo in B. subtilis and may have functions overlapping those of δ. We have demonstrated in this study that GreA and YdeB are likely not involved in this process. Nevertheless, as gene expression regulation by [iNTP] is an important regulatory mechanism, searches for such factors are under way in our laboratory. Finally, promoter regulation by [iNTP] was reported also in eukaryotic organisms (53), and it would be interesting to determine if analogs of these prokaryotic factors exist in eukaryotes.
In conclusion, δ is an excellent example of a seemingly dispensable protein that is, in fact, essential for the cell's survival in the hostile and fiercely competitive world outside the culture flask. It shows that it does not suffice to adapt; it has to be done briskly.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from Czech Science Foundation no. 204/09/0583 (experiments with δ), P305/12/G034 (GreA, elongation complexes), and P302/11/0855 (YdeB). A.R. and J.K. were in part supported by grant numbers 648712 and 425311 (The Charles University Grant Agency). This work was also supported by the project “CEITEC— Central European Institute of Technology” (CZ.1.05/1.1.00/02.0068) from the European Regional Development Fund.
We thank T. Gaal from the University of Wisconsin and RNA Centre members for discussions.
Footnotes
Published ahead of print 29 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00188-13.
REFERENCES
- 1. Helmann JD. 2009. RNA polymerase: a nexus of gene regulation. Methods 47:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murakami KS, Darst SA. 2003. Bacterial RNA polymerases: the Wholo story. Curr. Opin. Struct. Biol. 13:31–39 [DOI] [PubMed] [Google Scholar]
- 3. Pero J, Nelson J, Fox TD. 1975. Highly asymmetric transcription by RNA-polymerase containing phage-Sp01-induced polypeptides and a new host protein. Proc. Natl. Acad. Sci. U. S. A. 72:1589–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tjian R, Losick R, Pero J, Hinnebush A. 1977. Purification and comparative properties of delta and sigma subunits of RNA-polymerase from Bacillus subtilis. Eur. J. Biochemistry 74:149–154 [DOI] [PubMed] [Google Scholar]
- 5. Lampe M, Binnie C, Schmidt R, Losick R. 1988. Cloned gene encoding the delta subunit of Bacillus subtilis RNA polymerase. Gene 67:13–19 [DOI] [PubMed] [Google Scholar]
- 6. Lopez de Saro FJ, Woody AY, Helmann JD. 1995. Structural analysis of the Bacillus subtilis delta factor: a protein polyanion which displaces RNA from RNA polymerase. J. Mol. Biol. 252:189–202 [DOI] [PubMed] [Google Scholar]
- 7. Motackova V, Sanderova H, Zidek L, Novacek J, Padrta P, Svenkova A, Korelusova J, Jonak J, Krasny L, Sklenar V. 2010. Solution structure of the N-terminal domain of Bacillus subtilis delta subunit of RNA polymerase and its classification based on structural homologs. Proteins 78:1807–1810 [DOI] [PubMed] [Google Scholar]
- 8. Doherty GP, Fogg MJ, Wilkinson AJ, Lewis PJ. 2010. Small subunits of RNA polymerase: localization, levels and implications for core enzyme composition. Microbiology 156:3532–3543 [DOI] [PubMed] [Google Scholar]
- 9. Achberger EC, Whiteley HR. 1981. The role of the delta peptide of the Bacillus subtilis RNA polymerase in promoter selection. J. Biol. Chem. 256:7424–7432 [PubMed] [Google Scholar]
- 10. Dobinson KF, Spiegelman GB. 1987. Effect of the delta-subunit of Bacillus subtilis RNA polymerase on initiation of RNA synthesis at 2 bacteriophage phi-29 promoters. Biochemistry 26:8206–8213 [DOI] [PubMed] [Google Scholar]
- 11. Juang YL, Helmann JD. 1994. The delta subunit of Bacillus subtilis RNA polymerase, an allosteric effector of the initiation and core-recycling phases of transcription. J. Mol. Biol. 239:1–14 [DOI] [PubMed] [Google Scholar]
- 12. Lopez de Saro FJ, Yoshikawa N, Helmann JD. 1999. Expression, abundance, and RNA polymerase binding properties of the delta factor of Bacillus subtilis. J. Biol. Chem. 274:15953–15958 [DOI] [PubMed] [Google Scholar]
- 13. Liu CG, Heath LS, Turnbough CL. 1994. Regulation of pyrbi operon expression in Escherichia coli by UTP-sensitive reiterative RNA synthesis during transcriptional initiation. Genes Dev. 8:2904–2912 [DOI] [PubMed] [Google Scholar]
- 14. Liu J, Turnbough CL. 1994. Effects of transcriptional start site sequence and position on nucleotide-sensitive selection of alternative start sites at the pyrC promoter in Escherichia coli. J. Bacteriol. 176:2938–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartz M, Neuhard J. 1975. Control of expression of pyr genes in Salmonella typhimurium—effects of variations in uridine and cytidine nucleotide pools. J. Bacteriol. 121:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorensen KI, Baker KE, Kelln RA, Neuhard J. 1993. Nucleotide pool-sensitive selection of the transcriptional start site in vivo at the Salmonella typhimurium pyrC and pyrD promoters. J. Bacteriol. 175:4137–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker KA, Mallik P, Pratt TS, Osuna R. 2004. The Escherichia coli Fis promoter is regulated by changes in the levels of its transcription initiation nucleotide CTP. J. Biol. Chem. 279:50818–50828 [DOI] [PubMed] [Google Scholar]
- 18. Gaal T, Bartlett MS, Ross W, Turnbough CL, Jr, Gourse RL. 1997. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science 278:2092–2097 [DOI] [PubMed] [Google Scholar]
- 19. Revyakin A, Ebright RH, Strick TR. 2004. Promoter unwinding and promoter clearance by RNA polymerase: detection by single-molecule DNA nanomanipulation. Proc. Natl. Acad. Sci. U. S. A. 101:4776–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krasny L, Gourse RL. 2004. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J. 23:4473–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lopez JM, Dromerick A, Freese E. 1981. Response of guanosine 5′-triphosphate concentration to nutritional changes and its significance for Bacillus subtilis sporulation. J. Bacteriol. 146:605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murray HD, Schneider DA, Gourse RL. 2003. Control of rRNA expression by small molecules is dynamic and nonredundant. Mol. Cell 12:125–134 [DOI] [PubMed] [Google Scholar]
- 23. Ochi K, Kandala J, Freese E. 1982. Evidence that Bacillus subtilis sporulation induced by the stringent response is caused by the decrease in GTP or GDP. J. Bacteriol. 151:1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qi Y, Hulett FM. 1998. PhoP Similar to P and RNA polymerase sigma(A) holoenzyme are sufficient for transcription of Pho regulon promoters in Bacillus subtilis: PhoP Similar to P activator sites within the coding region stimulate transcription in vitro. Mol. Microbiol. 28:1187–1197 [DOI] [PubMed] [Google Scholar]
- 25. Guerout-Fleury AM, Frandsen N, Stragier P. 1996. Plasmids for ectopic integration in Bacillus subtilis. Gene 180:57–61 [DOI] [PubMed] [Google Scholar]
- 26. Ross W, Thompson JF, Newlands JT, Gourse RL. 1990. Escherichia coli Fis protein activates ribosomal-RNA transcription in vitro and in vivo. EMBO J. 9:3733–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lewis PJ, Marston AL. 1999. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene 227:101–110 [DOI] [PubMed] [Google Scholar]
- 28. Sojka L, Kouba T, Barvik I, Sanderova H, Maderova Z, Jonak J, Krasny L. 2011. Rapid changes in gene expression: DNA determinants of promoter regulation by the concentration of the transcription initiating NTP in Bacillus subtilis. Nucleic Acids Res. 39:4598–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang BY, Doi RH. 1990. Overproduction, purification, and characterization of Bacillus subtilis RNA polymerase sigma A factor. J. Bacteriol. 172:3257–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 31. Dubnau D, Davidoff-Abelson R. 1971. Fate of transforming DNA following uptake by competent Bacillus subtilis. I. Formation and properties of the donor-recipient complex. J. Mol. Biol. 56:209–221 [DOI] [PubMed] [Google Scholar]
- 32. Sanderova H, Tiserova H, Barvik I, Sojka L, Jonak J, Krasny L. 2010. The N-terminal region is crucial for the thermostability of the G-domain of Bacillus stearothermophilus EF-Tu. Biochim. Biophys. Acta 1804:147–155 [DOI] [PubMed] [Google Scholar]
- 33. Barker MM, Gaal T, Josaitis CA, Gourse RL. 2001. Mechanism of regulation of transcription initiation by PpGpp. I. Effects of PpGpp on transcription initiation in vivo and in vitro. J. Mol. Biol. 305:673–688 [DOI] [PubMed] [Google Scholar]
- 34. Jendresen CB, Kilstrup M, Martinussen J. 2011. A simplified method for rapid quantification of intracellular nucleoside triphosphates by one-dimensional thin-layer chromatography. Anal. Biochem. 409:249–259 [DOI] [PubMed] [Google Scholar]
- 35. Whipple FW, Sonenshein AL. 1992. Mechanism of initiation of transcription by Bacillus subtilis RNA polymerase at several promoters. J. Mol. Biol. 223:399–414 [DOI] [PubMed] [Google Scholar]
- 36. Krasny L, Tiserova H, Jonak J, Rejman D, Sanderova H. 2008. The identity of the transcription +1 position is crucial for changes in gene expression in response to amino acid starvation in Bacillus subtilis. Mol. Microbiol. 69:42–54 [DOI] [PubMed] [Google Scholar]
- 37. Saecker RM, Record MT, Jr, DeHaseth PL. 2011. Mechanism of bacterial transcription initiation: RNA polymerase-promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 412:754–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Natori Y, Tagami K, Murakami K, Yoshida S, Tanigawa O, Moh Y, Masuda K, Wada T, Suzuki S, Nanamiya H, Tozawa Y, Kawamura F. 2009. Transcription activity of individual Rrn operons in Bacillus subtilis mutants deficient in(p) PpGpp synthetase genes, RelA, YjbM, and YwaC. J. Bacteriol. 191:4555–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gourse RL, Gaal T, Bartlett MS, Appleman JA, Ross W. 1996. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu. Rev. Microbiol. 50:645–677 [DOI] [PubMed] [Google Scholar]
- 40. Murray HD, Gourse RL. 2004. Unique roles of the Rrn P2 RRNA promoters in Escherichia coli. Mol. Microbiol. 52:1375–1387 [DOI] [PubMed] [Google Scholar]
- 41. Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of RRNA promoters by PpGpp and the initiating NTP. Cell 118:311–322 [DOI] [PubMed] [Google Scholar]
- 42. Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondary channel–structural framework for PpGpp-DksA synergism during transcription. Cell 118:297–309 [DOI] [PubMed] [Google Scholar]
- 43. Toulme F, Mosrin-Huaman C, Sparkowski J, Das A, Leng M, Rahmouni AR. 2000. GreA and GreB proteins revive backtracked RNA polymerase in vivo by promoting transcript trimming. EMBO J. 19:6853–6859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rutherford ST, Lemke JJ, Vrentas CE, Gaal T, Ross W, Gourse RL. 2007. Effects of DksA, GreA, and GreB on transcription initiation: insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. J. Mol. Biol. 366:1243–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. 2009. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138:146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kriel A, Bittner AN, Kim SH, Liu K, Tehranchi AK, Zou WY, Rendon S, Chen R, Tu BP, Wang JD. 2012. Direct Regulation of GTP homeostasis by (p)PpGpp: a critical component of viability and stress resistance. Mol. Cell 48:231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seepersaud R, Needham RH, Kim CS, Jones AL. 2006. Abundance of the delta subunit of RNA polymerase is linked to the virulence of Streptococcus agalactiae. J. Bacteriol. 188:2096–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watson SP, Antonio M, Foster SJ. 1998. Isolation and characterization of Staphylococcus aureus starvation-induced, stationary-phase mutants defective in survival or recovery. Microbiology 144(Part 11):3159–3169 [DOI] [PubMed] [Google Scholar]
- 49. Tojo S, Satomura T, Kumamoto K, Hirooka K, Fujita Y. 2008. Molecular mechanisms underlying the positive stringent response of the Bacillus subtilis ilv-leu operon, involved in the biosynthesis of branched-chain amino acids. J. Bacteriol. 190:6134–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tojo S, Kumamoto K, Hirooka K, Fujita Y. 2010. Heavy involvement of stringent transcription control depending on the adenine or guanine species of the transcription initiation site in glucose and pyruvate metabolism in Bacillus subtilis. J. Bacteriol. 192:1573–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xue X, Tomasch J, Sztajer H, Wagner-Dobler I. 2010. The delta subunit of RNA polymerase, RpoE, is a global modulator of Streptococcus mutans environmental adaptation. J. Bacteriol. 192:5081–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen YF, Helmann JD. 1997. DNA-melting at the Bacillus subtilis flagellin promoter nucleates near −10 and expands unidirectionally. J. Mol. Biol. 267:47–59 [DOI] [PubMed] [Google Scholar]
- 53. Kuehner JN, Brow DA. 2008. Regulation of a eukaryotic gene by GTP-dependent start site selection and transcription attenuation. Mol. Cell 31:201–211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.