

Abstract
Avian pathogenic Escherichia coli (APEC) infection causes avian colibacillosis, which refers to any localized or systemic infection, such as acute fatal septicemia or subacute pericarditis and airsacculitis. The RfaH transcriptional regulator in E. coli is known to regulate a number of phenotypic traits. The direct effect of RfaH on the virulence of APEC has not been investigated yet. Our results showed that the inactivation of rfaH significantly decreased the virulence of APEC E058. The attenuation was assessed by in vivo and in vitro assays, including chicken infection assays, an ingestion and intracellular survival assay, and a bactericidal assay with serum complement. The virulence phenotype was restored to resemble that of the wild type by complementation of the rfaH gene in trans. The results of the quantitative real-time reverse transcription-PCR (qRT-PCR) analysis and animal system infection experiments indicated that the deletion of rfaH correlated with decreased virulence of the APEC E058 strain.
INTRODUCTION
Isolates of extraintestinal pathogenic Escherichia coli (ExPEC) cause infection in nearly every organ and anatomical site in humans and animals. Among ExPEC strains, avian pathogenic E. coli (APEC) strains are responsible for serious extraintestinal diseases of poultry, causing high morbidity and mortality in chickens and turkeys, leading to great economic losses (1, 2). APEC infection causes a variety of severe infections, including acute fatal septicemia, subacute pericarditis, and airsacculitis. Most often, APEC strains infect chickens, turkeys, ducks, and other avian species through fecal dust via the respiratory tract. APEC strains possess genes coding for various virulence factors for colonizing and invading the host, including adhesins, toxins, polysaccharide coatings, protectins, invasins, and iron acquisition systems (3, 4). Epidemiological studies have shown that APEC isolates predominantly belong to the O1, O2, and O78 serogroups (5, 6).
From attachment and colonization to the host cells to systemic invasion, bacteria sense the environment and regulate the expression of virulence genes that are required for effective pathogenesis. A complex regulatory network exists in E. coli that mediates this response to environmental signals (7, 8). In E. coli and many other bacterial species, a regulatory protein, RfaH, acts as a transcriptional antiterminator that reduces the polarity of long operons encoding cell components (9, 10). RfaH was first discovered as a regulator of lipopolysaccharide (LPS) synthesis in Salmonella enterica (11) and E. coli (12). Later, RfaH was shown to be essential for the expression of other cell components encoded on long operons in E. coli, including the expression of F plasmid (13), different capsules (14–16), and hemin uptake receptor (17), as well as the toxins alpha-hemolysin (18, 19) and cytotoxic necrotizing factor 1 (20). RfaH-dependent operons share a short cis-acting element termed ops (for operon polarity suppressor) that is essential to allow RfaH to function (21). How all these virulence factors evolved to utilize the same core regulatory mechanism still awaits discovery.
Previous research showed that disruption of the rfaH gene in uropathogenic E. coli strain 536 results in a significant decrease in virulence (22). As rfaH seems to be conserved among various bacteria, its role in the regulation of virulence of other ExPEC pathogens has been suggested. The purpose of this investigation was to assess the hypothesis that rfaH is critical to the virulence of APEC E058. To that end, an isogenic rfaH mutant of APEC O2 strain E058 was constructed using lambda Red recombination as described previously (23). The mutant was tested for its contribution to APEC E058 pathogenicity, including in vivo and in vitro assays to reveal the pathogenic traits.
MATERIALS AND METHODS
Bacterial strains, primers, and growth conditions.
The strains and plasmids used in this study are listed in Table 1, and the primers are listed in Table 2. Bacteria were routinely cultured in Luria Bertani (LB) broth at 37°C with aeration. Antibiotics were added at the following concentrations: chloramphenicol (Cam), 30 μg/ml, and ampicillin (Amp), 60 μg/ml.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E058 | Wild-type avian E. coli serotype O2 | 41 |
| E058ΔrfaH | E058ΔrfaH::cat | This study |
| ReE058ΔrfaH | complementation of E058ΔrfaH | This study |
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Invitrogen |
| Plasmids | ||
| pMD18-T Vector | TA cloning vector; Amp | TaKaRa |
| pGEM-T Easy Vector | TA cloning vector; Amp | Promega |
| pMD-rfaH | rfaH cloned into pMD 18-T vector | This study |
| pMD-rfaH-cat | Cat-resistant gene inserted into pMD-rfaH | This study |
| pT-PnativerfaH | pGEM-T Easy carrying rfaH ORF and its putative native promoter | This study |
| pKD46 | Amp; expresses λ Red recombinase | 23 |
| pKD3 | cat gene, template plasmid | 23 |
Table 2.
Primers designed and used in this study
| Primer | Primer sequence (5′–3′) | Target gene, plasmid, or region |
|---|---|---|
| Targets of primers for gene mutagenesis | ||
| HF | CTCGGATCCTTAGAGTTTGCGGAACTC (BamHI site underlined) | rfaH |
| HR | CTCAAGCTTTTACTGTACTGCAAGCGC (HindIII site underlined) | rfaH |
| SHF | CTCGATATCGCCGTATCCTGGTGATAA (EcoRV site underlined) | pMD-rfaH |
| SHR | CTCGATATCGTGGTATGAATCACTTCC (EcoRV site underlined) | pMD-rfaH |
| CF | CTCGATATCTTGTGTAGGCTGGAGCTGCT (EcoRV site underlined) | pKD3 |
| CR | CTCGATATCATGGGAATTAGCCATGGTCC (EcoRV site underlined) | pKD3 |
| RHF | CTGCGGCAGTTTATCAAACAG | Upstream region of rfaH |
| RHR | GAAGCGATTATTGAGCTGGCC | Downstream region of rfaH |
| TF | ATGGGCTACTCATCACCG | tatD |
| TR | CACCAGGCAGTTTATCCAG | tatD |
| UF | CGAAAACCCTAAAGGCTACT | ubiD |
| UR | CCACTCCTGATAATCCAACG | ubiD |
| Targets of primers for real-time PCR | ||
| rfaH RT-F | TCGGGCTCGGTGAAA ATG | rfaH |
| rfaH RT-R | ATCGACGCCGTATCCTGGTG | rfaH |
| chuA RT-F | TACCGACCCAACCAACAG | chuA |
| chuA RT-R | GTCCCGAACGCCAAAAT | chuA |
| kpsM RT-F | TGCCAGACATCTCGTTCC | kpsM |
| kpsM RT-R | TCGCTTCAATAGCACCAAT | kpsM |
| traT RT-F | CAGCAATCAAGAAGCGTAAC | traT |
| traT RT-R | TTCGCCTGAATCCAGTAGTA | traT |
| iutA RT-F | CGATGCCACCTTGCTTGA | iutA |
| iutA RT-R | CAGCCCGTTACTGACGAATG | iutA |
| iucD RT-F | AACAACCTTATTTACCACCCTG | iucD |
| iucD RT-R | TCTGTCCTCCACCAACCAC | iucD |
| iroN RT-F | GGCGATACGCAAAACAGT | iroN |
| iroN RT-R | CCCAGTCCCAGATACCATT | iroN |
| iss RT-F | AACACCAAAGGAAACCATCA | iss |
| iss RT-R | CCGAGCAATCCATTTACGA | iss |
| tsh RT-F | GGCGCTTACTTTGATGTGAT | tsh |
| tsh RT-R | GCTGTTACGACGCATTGAGA | tsh |
| vat RT-F | GCTTTGGTTCGCTCGTGTT | vat |
| vat RT-R | ATCTGCCTGGATGGTTGTGTT | vat |
| cvaC RT-F | AATGTCTCCATCCGGTTTA | cvaC |
| cvaC RT-R | TCTTCCCGCAGCATAGTT | cvaC |
| astA RT-F | AAA AGTCGGCTGGTGGAA | astA |
| astA RT-R | CTGCGTGGCATTTGAGGA | astA |
Construction of rfaH deletion mutant.
Deletion of rfaH from the chromosome of APEC E058 was performed using gene replacement methods based on the lambda Red recombinase system (23). E058 was initially electroporated with pKD46 to express Red recombinase. The E058ΔrfaH strain was constructed as follows: the rfaH gene (GenBank accession number M94889.1) was amplified by PCR using the primers HF and HR (Table 2). The products were cloned into the pMD18-T simple vector to form pMD-rfaH. To insert the chloramphenicol acetyltransferase (cat) cassette into rfaH, reverse PCR was adopted. The reverse PCR product was amplified from pMD-rfaH using the primers SHF and SHR (Table 2). The cat cassette was obtained from pkD3 using primers CF and CR (Table 2). The cat cassette was then inserted into the rfaH genes at the EcoRV site.
Reverse transcription-PCR analysis.
To determine whether the insertion had a polar effect on the upstream or downstream genes, total RNA was extracted from log-phase bacteria of strains E058 and E058ΔrfaH using the RNAiso Plus kit (TaKaRa, Dalian, China) according to the manufacturer's instructions. Contaminating DNA was removed from the samples, and cDNA synthesis was performed using the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa) according to the manufacturer's instructions. Primer sets for PCR amplification of the target genes tatD (TF/TR), rfaH (HF/HR), and ubiD (UF/UR) in cDNA samples are listed in Table 2. In parallel, PCRs were performed with genomic DNA as a positive control and RNA samples without activation of reverse transcription (RT) as a negative control. The PCR products were resolved on 0.8% agarose gels and visualized by ethidium bromide staining.
Complementation of the rfaH mutant.
To complement the deleted rfaH, the entire rfaH gene, including its natural putative promoter, was amplified and subcloned into the pGEM-T Easy vector using primer pairs RHF and RHR (Table 2), forming pT-PnativerfaH, and the resulting plasmid was transformed into E. coli DH5α. Positive colonies were selected and identified by PCR and sequencing, and the purified recombinant plasmid pT-PnativerfaH was transformed to the mutant strain E058ΔrfaH.
Bactericidal assay with SPF chicken serum.
Complement-sufficient specific-pathogen-free (SPF) chicken serum was prepared and pooled from 10 SPF chickens. A bactericidal assay was performed in a 96-well plate as described previously but with the following modifications (24). SPF chicken serum was diluted to 5, 12.5, 25, 50, and 100% in pH 7.2 phosphate-buffered saline (PBS). Bacteria (10 μl containing 106 CFU) were inoculated into reaction wells containing 190 μl of the diluted SPF chicken serum, 100% heat-inactivated SPF chicken serum, or PBS alone and then incubated at 37°C for 30 min. Serial dilutions (1:10) of each well were plated onto LB agar plates. The resulting colonies were counted after 24 h of incubation.
Virulence assay in vivo.
To assess attenuation of the rfaH mutant, groups of 10 1-day-old chickens were infected with the wild-type strain and its isogenic rfaH mutant, as well as the complementation strain. The birds were treated in the experiments in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals of the People's Republic of China (Approved by the State Council on 31 October 1988). Strains were grown to exponential phase, collected, washed twice in PBS, and then adjusted to the appropriate doses. Birds were inoculated via the air sac with 0.1 ml of each bacterial suspension containing 107 CFU bacteria. The number of bacteria contained in the inoculum was confirmed by plating on LB agar. Negative controls were injected with PBS. Mortality was monitored until 7 days postinfection (p.i.).
Colonization and persistence of the mutant.
Animals were infected with the wild-type strain, the mutant, and the complementation strain to determine their colonization and persistence abilities during systemic infection. Briefly, 5-week-old SPF chickens (white leghorn; Jinan SPAFAS poultry Co. Ltd., Jinan, China) were inoculated via the left thoracic air sac with 108 CFU of each bacterial suspension. After 24 h, 15 chickens from each group were euthanized and examined for macroscopic and histological lesions. The hearts, livers, spleens, lungs, and kidneys of the birds were collected, weighed, and triturated. The numbers of bacteria were determined by plating serial dilutions of the homogenates onto LB agar plates.
Coinfection assays.
For in vivo competitive-coinfection assays, 5-week-old white leghorn SPF chickens were inoculated with cultures of the wild-type strain, E058, and its mutant, E058ΔrfaH, mixed at a ratio of 1:1 (1 × 108 CFU for each strain) via the left air sac. The chickens were provided with food and water ad libitum; 24 h after infection, the hearts, livers, spleens, lungs, and kidneys of the inoculated chickens were collected, weighed, and homogenized, and serial dilutions were plated on LB medium with or without chloramphenicol for selection of mutant or total bacteria, respectively.
Ingestion and intracellular-survival assay.
Ingestion and intracellular-survival tests were performed principally as described previously (25). The avian macrophage cell line HD-11 was cultured in Dulbecco's modified Eagle medium (DMEM) (Gibco, New York, NY) with 10% fetal bovine serum (FBS) (PAA, Pasching, Australia) in 24-well cell culture plates. The cells were maintained at 37°C in a 5% CO2 environment, and the plates contained ∼2 × 105 cells per well. The plates were incubated for 24 h prior to the ingestion assay. Bacteria were added to the cells at a multiplicity of infection (MOI) of 100 for 1 h to allow ingestion. The wells were washed with PBS, and the appropriate cell culture medium containing 100 μg/ml gentamicin was added for 1.5 h to kill extracellular bacteria. At this time (T0) and at additional, different incubation periods (6, 12, and 24 h), cells were washed using PBS and lysed with 1 ml 0.1% Triton X-100 for 5 min at room temperature. Released bacteria were diluted and plated for viable counts. The ingestion ratio was determined by dividing the number of ingested bacterial cells at T0 by the number of bacteria in the initial inoculation. Intracellular growth was expressed as the change (n-fold) in the bacterial number at an additional incubation time point relative to the initial number of ingested bacteria (T0).
RNA isolation and qRT-PCR.
To determine whether the cell culture medium or interaction with the host can induce expression of the transcriptional regulator RfaH, quantitative RT (qRT)-PCR was applied to analyze the rfaH expression of E058 under both in vitro and in vivo conditions. For in vitro measurements, E058 was cultured with aeration to the logarithmic phase in DMEM with 10% fetal bovine serum or in LB medium. Cultures were pelleted (10 min; 2,500 × g; 4°C) and stored at −70°C for RNA isolation. For in vivo samples, 1-day-old chickens (n = 10) were infected with 108 CFU of E058 by the air sac route. Five hours following challenge with E058, 9 ml blood was collected with 1 ml anticoagulant (0.5% sodium citrate). The anticoagulant and blood were centrifuged (5 min; 500 × g; 4°C), and the supernatants containing bacteria were collected. The supernatants were centrifuged (10 min; 2,500 × g; 4°C), and the bacterial pellet was frozen at −70°C until RNA extraction. RNA isolation and cDNA synthesis were performed using the RNAiso Plus kit and the PrimeScript RT reagent kit with gDNA Eraser according to the manufacturer's instructions (TaKaRa). qRT-PCR was performed to determine the rfaH transcription level of E058 under both in vitro and in vivo conditions using SYBR premix Ex Taq (TaKaRa) and rfaH RT-F/R primers (Table 2). Data were normalized to the housekeeping gene gapA transcript.
To analyze how gene expression was affected by the loss of RfaH, we performed qRT-PCR analyses for various virulence genes in APEC. Overnight cultures of strains were diluted 1:100 in fresh LB until the logarithmic phase of growth. Following RNA isolation and cDNA synthesis, qRT-PCR was performed to determine the transcription levels of the virulence genes using SYBR premix Ex Taq and gene-specific primers (Table 2), and the data were normalized to the gapA transcript.
Statistical analysis.
Differences between groups were analyzed using the Statistical Package for the Social Sciences (SPSS version 15.0; SPSS, Chicago, IL). P values of less than 0.05 were considered to be significant.
RESULTS
Deletion of rfaH does not affect the growth kinetics of APEC E058.
An rfaH deletion of E058 was created using gene replacement methods based on the lambda Red recombinase system. Part (126 bp) of the rfaH gene (489 bp) was deleted, and the cat cassette was inserted. To determine whether the insertion had a polar effect on the upstream or downstream genes, total RNA samples extracted from the parental E058 and E058ΔrfaH were analyzed by RT-PCR using primer sets designed for tatD, rfaH, and ubiD (Table 2). Compared to the parental E058, the insertion of the cat gene in E058ΔrfaH only disrupted the transcription of the rfaH gene but had no influence on the expression of the genes upstream and downstream of rfaH (Fig. 1).
Fig 1.
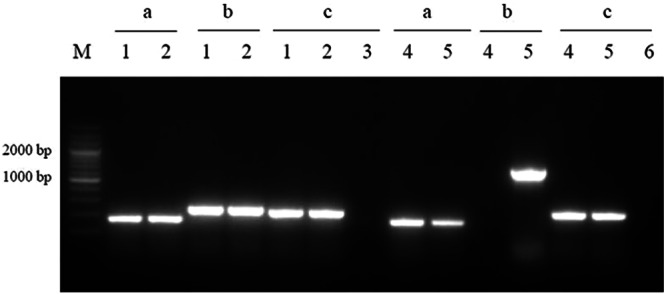
Detection of tatD, rfaH, and ubiD gene expression in E058 and E058ΔrfaH by RT-PCR. The RT-PCRs were performed using the following templates: cDNA derived from total RNA of E058 (lanes 1) and E058ΔrfaH (lanes 4) and genomic DNA from E058 (lanes 2) and E058ΔrfaH (lanes 5). Reaction sets contained the following primers: for lanes labeled “a,” TF/TR; for lanes labeled “b,” HF/HR; and for lanes labeled “c,” UF/UR. The negative controls (lanes 3 and 6) used total RNA as the template without activation of RT. A 200-bp marker (TaKaRa) was used as the molecular size standard (M).
For complementation, the open reading frame (ORF) and natural putative promoter of rfaH were amplified from E058 and cloned into the pGEM-T Easy vector. The resulting recombinant plasmid, pT-PnativerfaH, was transformed into the mutant strain E058ΔrfaH, yielding the complementation strain, ReE058ΔrfaH.
Compared with the parental strain, the colonies of the isogenic E058ΔrfaH mutant on the LB agar plates were similar to those of the parental strain, and the growth rate in LB broth in logarithmic phase was also similar to that of the parental strain (data not shown).
The rfaH deletion attenuates APEC E058 in birds.
APEC E058 is a virulent avian pathogenic strain that could cause typical avian colibacillosis, with bacteria invading air sacs, blood, pericardial fluid, and the typical fibrinous lesions. To investigate whether RfaH plays a role in the virulence of the strain, groups of 10 1-day-old birds were infected with 1 × 107 CFU of the wild-type strain and its isogenic rfaH mutant, as well as with the complementation strain, and the mortality of the birds was observed for 7 days after the challenge; the mortality rates were 100%, 0%, and 100%, respectively (Table 3). Loss of rfaH resulted in abolishment of virulence of E058 in birds, while trans-complementation of the mutant strain with rfaH completely restored its virulence. The results provide evidence that RfaH is an important virulence factor in the pathogenesis of APEC infection.
Table 3.
Death rates elicited by APEC E058 and its isogenic rfaH mutant after air sac infection
| Strain | No. dead/total no. (%) after air sac infection of birds (1 × 107 CFU) |
|---|---|
| E058 | 10/10 (100) |
| E058ΔrfaH | 0/10 (0) |
| ReE058ΔrfaH | 10/10 (100) |
RfaH protein is required for serum resistance of APEC E058.
Resistance to serum has been associated with E. coli causing infections in poultry and extraintestinal infections in other species. Our results showed that loss of the regulatory protein RfaH results in high susceptibility to chicken serum in strain E058ΔrfaH (Fig. 2).
Fig 2.
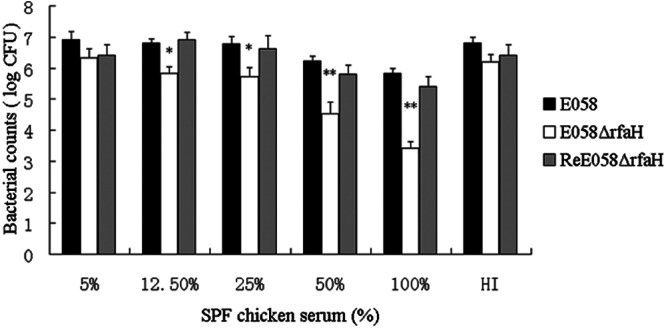
Bactericidal activities of SPF chicken serum against wild-type strain E058 and an isogenic mutant. HI represents 100% of heat-inactivated SPF chicken serum. The data represent an average of three trials. The error bars indicate standard deviations. The asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01).
Mutation in rfaH reduces the survival ability of APEC E058 within chicken macrophages.
Analysis of the capability of the rfaH mutant to be ingested by avian macrophages was carried out using an ingestion assay in the avian macrophage HD-11 cell line. The rfaH mutant showed a highly increased ingestion ratio of macrophages compared to the wild-type strain or the complementation strain (Fig. 3).
Fig 3.
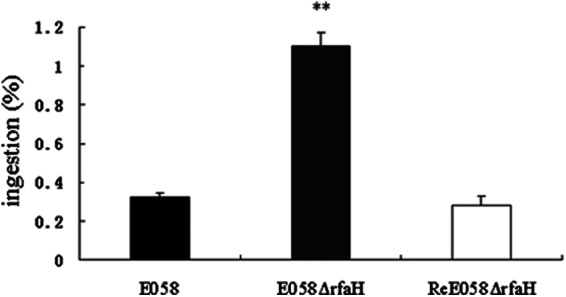
Ingestion of wild-type strain E058, isogenic mutant E058ΔrfaH, and complementation strain ReE058ΔrfaH by HD-11 cells. The values represent the average data from three independent experiments. The error bars indicate standard deviations. **, statistically significant differences between the wild-type and mutant strains (P < 0.01).
To determine whether the loss of RfaH has an influence on intracellular survival, the yield of wild-type strain E058 was compared to that of its rfaH mutant recovered following an additional 6, 12, and 24 h of incubation. At 6, 12, and 24 h p.i. (beginning at T0), the numbers of wild-type bacteria recovered from macrophages were 1.47%, 1.75%, and 2.45% of the inoculum, which were 4-, 5-, and 7-fold higher than those of the primary ingested bacteria (T0) (Fig. 4), respectively. In contrast, the rfaH mutant showed impaired growth in macrophages, with numbers representing 2.15%, 0.36%, and 0.11% of the inoculum at 6, 12, and 24 h p.i., which were 2-fold higher at 6 h p.i. and 3- and 10-fold lower at 12 and 24 h p.i. than the primary ingested bacteria (T0), respectively (Fig. 4). trans-complementation of the rfaH mutation restored the ability of the rfaH mutant to grow in macrophages at wild-type-strain levels, with numbers representing 1.05%, 1.44%, and 1.95% of the inoculum at 6, 12, and 24 h p.i., respectively. The rfaH mutant exhibited significantly reduced intracellular yields relative to its parental wild-type or complementation strain at 12 and 24 h p.i. (P < 0.01).
Fig 4.
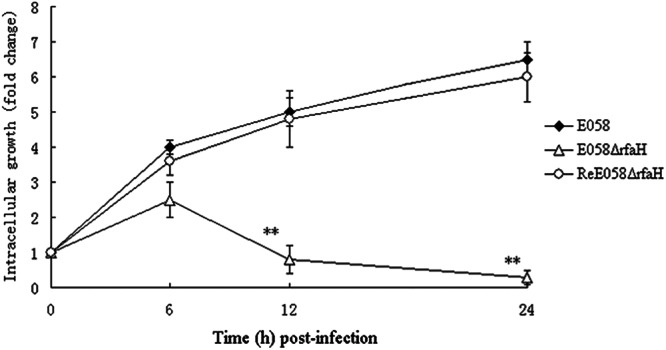
Intracellular growth of bacteria in HD-11 chicken macrophages. The intracellular growth rates of wild-type strain E058, the isogenic mutant E058ΔrfaH, and the complementation strain ReE058ΔrfaH were compared over a 24-h period. Intracellular bacterial growth is shown as the change in the number of intracellular bacteria following an additional 6, 12, and 24 h of incubation compared to the primary ingested bacteria (T0). Standard errors of the mean (SEM) for three independent experiments are shown. The asterisks indicate statistically significant differences (**, P < 0.01).
Effect of rfaH during systemic infection in vivo.
To investigate the effect of the rfaH mutation on bacterial pathogenesis, we carried out a colonization and persistence assay or coinfection challenge in a chicken model. To evaluate virulence, the bacterial loads in various tissues were determined. In the colonization and persistence assay, 5-week-old chickens were inoculated via the air sac with the wild-type strain, its isogenic mutant, and the complementation strain. From the inoculation site, the virulent strain is typically able to invade and infect deeper tissues, generates gross lesions, and causes a systemic infection. However, in this model, the attenuated strain was impaired in its capacity to colonize deeper tissues. Mutants in rfaH exhibited significantly reduced colonization compared to the wild type in the organs tested (P < 0.01) (Fig. 5). Conversely, the recovered complementation strain in all organs tested was restored to a level similar to that of the wild-type (P > 0.05) (Fig. 5).
Fig 5.

Colonization and persistence of the wild-type strain E058 (●), E058ΔrfaH (■), and ReE058ΔrfaH (▲) during systemic infection. The data are presented as log10 CFU · g of tissues−1. The horizontal bars indicate the mean log10 CFU · g−1. Each data point represents a sample from an individual chicken. Statistically significant differences as determined by the Mann-Whitney test are indicated by asterisks (**, P < 0.01). The error bars indicate standard deviations.
The competitive-coinfection model demonstrated that E058ΔrfaH was outcompeted by the wild-type strain. As shown in Fig. 6, the rfaH mutant strain was recovered at significantly lower levels than wild-type E058 in all tested organs (P < 0.01). These results indicate that the role of RfaH is critical to allow APEC strain E058 to compete for colonization in vivo.
Fig 6.
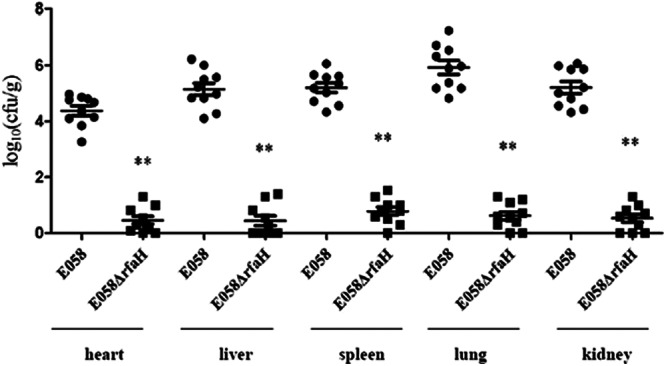
In vivo competition assays. E058 (●) and mutant E058ΔrfaH (■) were inoculated simultaneously. The data are presented as log10 CFU · g of tissues−1. The horizontal bars indicate the mean log10 CFU · g−1. Each data point represents a sample from an individual chicken. Statistically significant differences in values between E058 and its mutant are indicated by asterisks (**, P < 0. 01). The error bars indicate standard deviations.
Expression profile of related virulence genes.
The results of the cell assays showed a difference in survival ability between the wild-type and mutant strain, which suggests that the cell culture medium or interaction with host cells can induce expression of RfaH. However, our results showed that the expression of rfaH in E058 in vivo or following culture in the cell culture medium in vitro was unaltered compared to that in E058 cultured in LB medium (P > 0.05) (data not shown).
Since RfaH has been described as a factor that positively affects gene expression, the transcription profile of virulence genes associated with capsule, iron uptake, serum resistance, autotransporter, colicin V, and the F pilus were analyzed upon rfaH inactivation by qRT-PCR in vitro. The transcription levels of the kpsM, chuA, iucD, iutA, iroN, iss, vat, tsh, cvaC, astA, and traT genes were quantified by qRT-PCR. As shown in Fig. 7, the transcription levels of the virulence genes chuA, traT, kpsM, iss, and cvaC were significantly decreased in the rfaH mutant by 0.06, 0.02, 0.05, 0.49, and 0.56 times, respectively (P < 0.01), while the other associated virulence genes were not influenced compared to the wild-type strain (P > 0.05). The expression levels of these virulence genes were restored in the complementation strain (Fig. 7).
Fig 7.
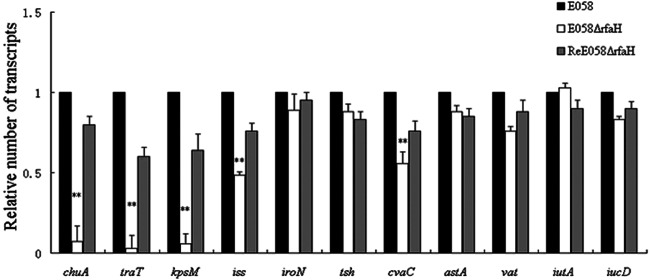
Quantitative RT-PCR analysis of associated virulence gene transcription levels in strains E058, E058ΔrfaH, and ReE058ΔrfaH. RNA was isolated from strains cultured in LB. Transcript levels were measured in cDNA preparations from each strain and normalized to the gapA level, and the results are shown as fold changes relative to the wild-type level. The asterisks indicate statistically significant differences (**, P < 0.01). The error bars indicate standard deviations.
DISCUSSION
APEC is a subset of extraintestinal pathogenic E. coli and shares virulence traits with strains isolated from human cases of neonatal meningitis, urinary tract infections, and septicemia. Thus, APEC strains represent a high risk of zoonotic infection (26), and their virulence gene pool may contribute to the emergence of other ExPEC strains (27). APEC O2 strain E058 is a highly virulent isolate involved in the development of colisepticemia, cellulitis, and respiratory disease, and it belongs to one of the most prevalent serogroups that cause avian colibacillosis.
Bacterial pathogens use specific or global regulators to mediate adaptive responses to the different environments and stresses encountered within the host (28). The infection process requires rapid adaptation to the host environment by alteration of gene expression. RfaH is a transcriptional antiterminator that reduces the polarity of long operons encoding secreted and surface-associated cell components (LPS, capsules, exotoxins, hemin uptake receptor ChuA, and F pilus) involved in the virulence of E. coli pathogens.
Since all the structures influenced by RfaH are potential virulence factors, we proposed that mutation of the rfaH gene results in a decrease in APEC virulence. Indeed, in a chicken infection model, the virulence of wild-type strain E058 was almost completely abolished through the loss of the rfaH gene. The rfaH mutant did not exhibit a growth defect, as determined by the generation time in LB at 37°C, indicating that the attenuation observed was not due to a general growth defect. Our results indicated that RfaH regulon activity is largely responsible for APEC attenuation. This statement is based on the fact that the rfaH mutant was affected in both in vivo and in vitro assays.
Our results demonstrated that the resistance to serum is impaired through the inactivation of rfaH. This is significant, since there is a correlation between resistance to the bactericidal effects of serum and the capacity of APEC strains to cause septicemia and mortality (29, 30). Serum resistance has been a virulence parameter for APEC. Many phenotype traits contribute to E. coli's serum resistance, including lipopolysaccharide, type 1 fimbriae, capsule, O antigen, and outer membrane proteins (29, 31–33), including OmpA, TraT, and Iss (34–36). APEC strains more often contain ColV plasmids that encode serum resistance (37). We have shown that loss of the regulatory protein RfaH results in high susceptibility to chicken serum in strain E058ΔrfaH, which might be explained by decreased expression of traT and iss (Fig. 7) due to inactivation of the rfaH gene.
The most common form of APEC infection in poultry is characterized by initial respiratory tract colonization, followed by a systemic spread to other parts of the body. Avian air sacs do not have cellular defense mechanisms and depend initially on the influx of heterophils, followed by macrophages as a cellular defense (38, 39). The ability to persist within macrophages provides a survival advantage to APEC strains by abrogating elimination by the host immune responses. Moreover, the pathogenic APEC strains are more resistant to killing by chicken macrophages in vitro than the less pathogenic strains are (40). Therefore, using the HD-11 chicken macrophage line, we examined the effects of mutation of rfaH on bacterial survival within cultured macrophages. The rfaH mutant bacteria appeared to be more susceptible to engulfment by the macrophages, while the wild-type or complemented bacteria appeared to resist engulfment, as shown in the ingestion assay (Fig. 3). We have shown that the absence of RfaH results in decreased intracellular growth of APEC in macrophage cells (Fig. 4). As intracellular growth provides a survival advantage to APEC, the phenotype may contribute to virulence attenuation of rfaH mutants.
By using an avian experimental model, we demonstrated that inactivation of the rfaH gene of E058 leads to attenuation of virulence, since the rfaH mutant showed significantly decreased colonization compared with the wild-type strain in all organs tested in the single-strain challenge model (Fig. 5), implying that RfaH plays a critical role in the virulence of APEC E058 in chickens. The E058ΔrfaH complementation strain restored virulence and colonized the internal organs of inoculated birds to the same extent as the wild-type strain (Fig. 5). Chickens that were challenged with wild-type strain E058 developed severe bilateral airsacculitis, severe hypertrophy and congestion of the spleen, and moderate perihepatitis. In contrast, the mutant E058ΔrfaH caused no conspicuous changes in the tissues mentioned above. The coinfection model provides more sensitivity to differences in the colonization or virulence of the rfaH mutant and those of the wild-type strain without involving individual differences of the host. Our results provide evidence that, in contrast to the wild-type strain, the rfaH mutant was significantly outcompeted in the chickens (Fig. 6). These results indicate that the ability to produce RfaH is critical to allow APEC E058 to compete for colonization in vivo. Meanwhile, the results of the qRT-PCR analysis demonstrated that the inactivation of rfaH decreased the transcription level of virulence genes involved in the capsule, hemin uptake, serum resistance, and colicin V synthesis (Fig. 7), which may contribute to the reduced colonization and proliferation capacities of APEC E058.
Taken together, our results confirm that the RfaH regulon plays a major role in APEC E058 virulence and associated traits.
ACKNOWLEDGMENTS
We gratefully acknowledge Xin'an Jiao in the Jiangsu Key Laboratory of Zoonosis at our university for kindly providing the cell line HD-11.
This work was supported by National Natural Science Foundation of China Grants 31272559, 30972196, 30771604, and 30471281; National Program for High Technology Research and Development in China Grant 2003 AA 222141; and Special Fund for Agroscientific Research in the Public Interest 201303044 to Song Gao. This work is also supported by a program for Changjiang Scholars and Innovative Research Teams in Universities (PCSIRT0978) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Footnotes
Published ahead of print 15 March 2013
REFERENCES
- 1. Russo TA, Johnson JR. 2000. Proposal for a new inclusive designation for extraintestinal pathogenic isolates of Escherichia coli: ExPEC. J. Infect. Dis. 181:1753–1754 [DOI] [PubMed] [Google Scholar]
- 2. Mokady D, Gophna U, Ron EZ. 2005. Virulence factors of septicemic Escherichia coli strains. Int. J. Med. Microbiol. 295:455–462 [DOI] [PubMed] [Google Scholar]
- 3. Johnson JR. 1991. Virulence factors in Escherichia coli urinary tract infection. Clin. Microbiol. Rev. 4:80–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao L, Gao S, Huan H, Xu X, Zhu X, Yang W, Gao Q, Liu X. 2009. Comparison of virulence factors and expression of specific genes between uropathogenic Escherichia coli and avian pathogenic E. coli in a murine urinary tract infection model and a chicken challenge model. Microbiology 155:1634–1644 [DOI] [PubMed] [Google Scholar]
- 5. Cheville NF, Arp LH. 1978. Comparative pathologic findings of Escherichia coli infection in birds. J. Am. Vet. Med. Assoc. 173:584–587 [PubMed] [Google Scholar]
- 6. Harry EG, Hemsley LA. 1965. The association between the presence of septicaemia strains of Escherichia coli in the respiratory and intestinal tracts of chickens and the occurrence of coli septicaemia. Vet. Rec. 77:35–40 [PubMed] [Google Scholar]
- 7. Harel J, Martin C. 1999. Virulence gene regulation in pathogenic Escherichia coli. Vet. Res. 30:131–155 [PubMed] [Google Scholar]
- 8. Grebe TW, Stock JB. 1999. The histidine protein kinase superfamily. Adv. Microb. Physiol. 41:139–227 [DOI] [PubMed] [Google Scholar]
- 9. Artsimovitch I, Landick R. 2002. The transcriptional regulator RfaH stimulates RNA chain synthesis after recruitment to elongation complexes by the exposed nontemplate DNA strand. Cell 109:193–203 [DOI] [PubMed] [Google Scholar]
- 10. Bailey MJ, Hughes C, Koronakis V. 1997. RfaH and the ops element, components of a novel system controlling bacterial transcription elongation. Mol. Microbiol. 26:845–851 [DOI] [PubMed] [Google Scholar]
- 11. Lindberg AA, Hellerqvist CG. 1980. Rough mutants of Salmonella typhimurium: immunochemical and structural analysis of lipopolysaccharides from rfaH mutants. J. Gen. Microbiol. 116:25–32 [DOI] [PubMed] [Google Scholar]
- 12. Creeger ES, Schulte T, Rothfield LI. 1984. Regulation of membrane glycosyltransferases by the sfrB and rfaH genes of Escherichia coli and Salmonella typhimurium. J. Biol. Chem. 259:3064–3069 [PubMed] [Google Scholar]
- 13. Sanderson KE, Stocker BA. 1981. Gene rfaH, which affects lipopolysaccharide core structure in Salmonella typhimurium, is required also for expression of F-factor functions. J. Bacteriol. 146:535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clarke BR, Pearce R, Roberts IS. 1999. Genetic organization of the Escherichia coli K10 capsule gene cluster: identification and characterization of two conserved regions in group III capsule gene clusters encoding polysaccharide transport functions. J. Bacteriol. 181:2279–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rahn A, Whitfield C. 2003. Transcriptional organization and regulation of the Escherichia coli K30 group 1 capsule biosynthesis (cps) gene cluster. Mol. Microbiol. 47:1045–1060 [DOI] [PubMed] [Google Scholar]
- 16. Stevens MP, Hanfling P, Jann B, Jann K, Roberts IS. 1994. Regulation of Escherichia coli K5 capsular polysaccharide expression: evidence for involvement of RfaH in the expression of group II capsules. FEMS Microbiol. Lett. 124:93–98 [DOI] [PubMed] [Google Scholar]
- 17. Nagy G, Dobrindt U, Kupfer M, Emody L, Karch H, Hacker J. 2001. Expression of hemin receptor molecule ChuA is influenced by RfaH in uropathogenic Escherichia coli strain 536. Infect. Immun. 69:1924–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bailey MJ, Koronakis V, Schmoll T, Hughes C. 1992. Escherichia coli HlyT protein, a transcriptional activator of haemolysin synthesis and secretion, is encoded by the rfaH (sfrB) locus required for expression of sex factor and lipopolysaccharide genes. Mol. Microbiol. 6:1003–1012 [DOI] [PubMed] [Google Scholar]
- 19. Leeds JA, Welch RA. 1996. RfaH enhances elongation of Escherichia coli hlyCABD mRNA. J. Bacteriol. 178:1850–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landraud L, Gibert M, Popoff MR, Boquet P, Gauthier M. 2003. Expression of cnf1 by Escherichia coli J96 involves a large upstream DNA region including the hlyCABD operon, and is regulated by the RfaH protein. Mol. Microbiol. 47:1653–1667 [DOI] [PubMed] [Google Scholar]
- 21. Bailey MJ, Hughes C, Koronakis V. 2000. In vitro recruitment of the RfaH regulatory protein into a specialised transcription complex, directed by the nucleic acid ops element. Mol. Gen. Genet. 262:1052–1059 [DOI] [PubMed] [Google Scholar]
- 22. Nagy G, Dobrindt U, Schneider G, Khan AS, Hacker J, Emody L. 2002. Loss of regulatory protein RfaH attenuates virulence of uropathogenic Escherichia coli. Infect. Immun. 70:4406–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zaleski A, Scheffler NK, Densen P, Lee FK, Campagnari AA, Gibson BW, Apicella MA. 2000. Lipooligosaccharide P(k) (Galalpha1-4Galbeta1-4Glc) epitope of Moraxella catarrhalis is a factor in resistance to bactericidal activity mediated by normal human serum. Infect. Immun. 68:5261–5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gahring LC, Heffron F, Finlay BB, Falkow S. 1990. Invasion and replication of Salmonella typhimurium in animal cells. Infect. Immun. 58:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson TJ, Wannemuehler Y, Johnson SJ, Stell AL, Doetkott C, Johnson JR, Kim KS, Spanjaard L, Nolan LK. 2008. Comparison of extraintestinal pathogenic Escherichia coli strains from human and avian sources reveals a mixed subset representing potential zoonotic pathogens. Appl. Environ. Microbiol. 74:7043–7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ewers C, Antao EM, Diehl I, Philipp HC, Wieler LH. 2009. Intestine and environment of the chicken as reservoirs for extraintestinal pathogenic Escherichia coli strains with zoonotic potential. Appl. Environ. Microbiol. 75:184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mekalanos JJ. 1992. Environmental signals controlling expression of virulence determinants in bacteria. J. Bacteriol. 174:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. La Ragione RM, Woodward MJ. 2002. Virulence factors of Escherichia coli serotypes associated with avian colisepticaemia. Res. Vet. Sci. 73:27–35 [DOI] [PubMed] [Google Scholar]
- 30. Mellata M, Dho-Moulin M, Dozois CM, Curtiss R, III, Brown PK, Arne P, Bree A, Desautels C, Fairbrother JM. 2003. Role of virulence factors in resistance of avian pathogenic Escherichia coli to serum and in pathogenicity. Infect. Immun. 71:536–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goldman RC, Joiner K, Leive L. 1984. Serum-resistant mutants of Escherichia coli O111 contain increased lipopolysaccharide, lack an O antigen-containing capsule, and cover more of their lipid A core with O antigen. J. Bacteriol. 159:877–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ngeleka M, Harel J, Jacques M, Fairbrother JM. 1992. Characterization of a polysaccharide capsular antigen of septicemic Escherichia coli O115:K “V165”:F165 and evaluation of its role in pathogenicity. Infect. Immun. 60:5048–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whitfield C, Roberts IS. 1999. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 31:1307–1319 [DOI] [PubMed] [Google Scholar]
- 34. Weiser JN, Gotschlich EC. 1991. Outer membrane protein A (OmpA) contributes to serum resistance and pathogenicity of Escherichia coli K-1. Infect. Immun. 59:2252–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nemeth J, Muckle CA, Lo RY. 1991. Serum resistance and the traT gene in bovine mastitis-causing Escherichia coli. Vet. Microbiol. 28:343–351 [DOI] [PubMed] [Google Scholar]
- 36. Nolan LK, Giddings CW, Horne SM, Doetkott C, Gibbs PS, Wooley RE, Foley SL. 2002. Complement resistance, as determined by viable count and flow cytometric methods, and its association with the presence of iss and the virulence of avian Escherichia coli. Avian Dis. 46:386–392 [DOI] [PubMed] [Google Scholar]
- 37. Nolan LK, Horne SM, Giddings CW, Foley SL, Johnson TJ, Lynne AM, Skyberg J. 2003. Resistance to serum complement, iss, and virulence of avian Escherichia coli. Vet. Res. Commun. 27:101–110 [DOI] [PubMed] [Google Scholar]
- 38. Mellata M, Dho-Moulin M, Dozois CM, Curtiss R, III, Lehoux B, Fairbrother JM. 2003. Role of avian pathogenic Escherichia coli virulence factors in bacterial interaction with chicken heterophils and macrophages. Infect. Immun. 71:494–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Toth TE, Siegel P, Veit H. 1987. Cellular defense of the avian respiratory system. Influx of phagocytes: elicitation versus activation. Avian Dis. 31:861–867 [PubMed] [Google Scholar]
- 40. Pourbakhsh SA, Boulianne M, Martineau-Doize B, Fairbrother JM. 1997. Virulence mechanisms of avian fimbriated Escherichia coli in experimentally inoculated chickens. Vet. Microbiol. 58:195–213 [DOI] [PubMed] [Google Scholar]
- 41. Gao S, Liu XF, Zhang RK, Jiao XA, Wen QY, Wu CX, Tang YM, Zhu XB, Li C, Chen J, Cui HP. 1999. The isolation and identification of pathogenic Escherichia coli isolates of chicken origin from some regions in China. Acta Vet. Et. Zootech. Sin. 30:164–171 [Google Scholar]