

Abstract
The bitopic membrane protein VirB10 of the Agrobacterium VirB/VirD4 type IV secretion system (T4SS) undergoes a structural transition in response to sensing of ATP binding or hydrolysis by the channel ATPases VirD4 and VirB11. This transition, detectable as a change in protease susceptibility, is required for DNA substrate passage through the translocation channel. Here, we present evidence that DNA substrate engagement with VirD4 and VirB11 also is required for activation of VirB10. Several DNA substrates (oncogenic T-DNA and plasmids RSF1010 and pCloDF13) induced the VirB10 conformational change, each by mechanisms requiring relaxase processing at cognate oriT sequences. VirD2 relaxase deleted of its translocation signal or any of the characterized relaxases produced in the absence of cognate DNA substrates did not induce the structural transition. Translocated effector proteins, e.g., VirE2, VirE3, and VirF, also did not induce the transition. By mutational analyses, we supplied evidence that the N-terminal periplasmic loop of VirD4, in addition to its catalytic site, is essential for early-stage DNA substrate transfer and the VirB10 conformational change. Further studies of VirB11 mutants established that three T4SS-mediated processes, DNA transfer, protein transfer, and pilus production, can be uncoupled and that the latter two processes proceed independently of the VirB10 conformational change. Our findings support a general model whereby DNA ligand binding with VirD4 and VirB11 stimulates ATP binding/hydrolysis, which in turn activates VirB10 through a structural transition. This transition confers an open-channel configuration enabling passage of the DNA substrate to the cell surface.
INTRODUCTION
The type IV secretion systems (T4SSs) mediate the transfer of DNA and protein substrates across the envelopes of many Gram-negative and -positive bacterial species (1). The conjugation systems comprise a large subfamily of the T4SSs. These medically important systems are responsible for widespread transmission of antibiotic resistance genes and virulence genes carried by conjugative plasmids and chromosomally integrated elements. Overall, conjugation can be depicted as three distinct biochemical reactions. The DNA transfer and replication (Dtr) proteins bind a cognate origin-of-transfer (oriT) sequence and initiate processing of the DNA substrate for transfer (2–4). Next, the Dtr-oriT complex, termed the relaxosome, engages with the type IV coupling protein (T4CP) (5). Finally, the T4CP delivers the DNA substrate to a transenvelope channel comprised of the mating pair formation (Mpf) proteins for translocation across the cell envelope (6, 7). Studies in recent years have begun to define mechanistic and structural details of conjugation systems. Additionally, an accumulating body of evidence suggests that a combination of intra- and extracellular signals act to coordinate the conjugative DNA transfer reactions in space and time. The present investigations advance our understanding of a signaling mechanism required for DNA substrate transfer through the Agrobacterium tumefaciens VirB/VirD4 T4SS.
Formally, conjugation initiates when the relaxase and one or more accessory factors bind a cognate origin-of-transfer (oriT) sequence (2, 3). The relaxase nicks the DNA strand destined for transfer (T-strand) and remains covalently bound via an active-site tyrosine to the 5′ end of the T-strand. The relaxase and accessory factors carry the translocation signals for substrate docking with the T4CP, and the relaxase mediates passage of the bound T-strand through the channel. T4CPs typically possess an N-terminal transmembrane (TM) domain and a large C-terminal cytoplasmic domain with a nucleoside triphosphate binding domain (NBD) (1, 5). A combination of X-ray crystallography and electron microscopy studies of the plasmid R388-encoded TrwB T4CP established that these ATPases adopt a homohexameric, F1-F0-like structure (8, 9). T4CPs share sequence and structural similarities with the FtsK and SpoIIIE DNA translocases (10), giving rise to a proposal that, in addition to functioning as the substrate receptors, the T4CPs translocate DNA substrates through their central channels across the cytoplasmic membrane.
T4CPs interact with Mpf channels, which in Gram-negative bacteria are highly complex structures composed of one or two other ATPases, a cytoplasmic membrane translocon, and the envelope-spanning translocation channel (1, 11). Recent electron microscopy studies have advanced our understanding of Mpf channel architecture with the isolation and structural characterization of a machine subassembly termed the core complex (12, 13). This core complex, derived from the Escherichia coli pKM101-encoded T4SS, is a large, ∼1.05-MDa structure of 185 Å in width and height composed of 14 copies each of three subunits designated TraN, TraO, and TraF. The core complex is configured in two layers (I and O layers) that form a double-walled ringlike structure. The I layer, composed of the N-terminal domains of TraO and TraF, is anchored in the cytoplasmic membrane and is opened at the base by a 55-Å-diameter hole. The O layer, composed of TraN and the C-terminal domains of TraO and TraF, has a main body and a narrower cap on the outermost side of the complex that is inserted in the outer membrane. A narrow hole (10 Å) exists in the cap, resulting in a channel extending through the entire cylindrical structure. In addition to its membrane-spanning ring-shaped complexes at the cytoplasmic and outer membranes, the core has a large chamber which is thought to house the translocation channel. Another noteworthy feature is that the TraF subunit spans the entire cell envelope such that an α-helical domain termed the antennae projection (AP) extends from the O layer to form an outer membrane pore (13).
Most T4SS loci in Gram-negative bacteria code for homologs of TraN, TraO, and TraF, and biochemical evidence from studies of various systems support the notion that core complexes are common structural features of T4SSs. In the A. tumefaciens VirB/VirD4 T4SS, the TraN, TraO, and TraF homologs are VirB7, VirB9, and VirB10, respectively (1, 14). We have isolated and visualized ring-shaped complexes from A. tumefaciens that are composed minimally of these three subunits (15), and ongoing efforts are aimed at generating a high-resolution structure. This presumptive VirB core complex is envisioned to carry out two functions in relation to substrate transfer (16). First, it comprises a structural scaffold for assembly of the envelope-spanning translocation channel. By use of a chromatin immunoprecipitation (ChIP)-based, formaldehyde (FA) cross-linking assay termed transfer DNA immunoprecipitation (TrIP), we identified close contacts between the translocating T-DNA substrate and the VirD4 T4CP, VirB11 ATPase, polytopic VirB6, bitopic VirB8, core complex subunit VirB9, and VirB2 pilin (17–19). Integrating these findings with those from the pKM101 structural studies, we have proposed a general architectural model whereby VirD4 T4CP, VirB11, and VirB4 (a third ATPase also required for substrate transfer) are located at the entrance of the translocation channel and function to deliver docked substrate into the channel. The cytoplasmic membrane translocon composed minimally of VirB6 and VirB8 sits within the 55-Å ring formed by the VirB10 TM helices. Finally, VirB6 and VirB8 together with VirB9 and VirB2 pilin form the translocation channel within the chamber of the VirB7/VirB9/VirB10 core complex to mediate substrate transfer from the cytoplasmic membrane to the cell surface (20).
Previously, we reported that VirB10 undergoes a structural transition in response to an intracellular signal generated through the coordinated activities of the VirD4 and VirB11 ATPases (21). The structural transition, detectable as a change in protease susceptibility, correlates with the capacity of DNA substrates to form FA-cross-linkable contacts with VirB2 and VirB9. VirB10 thus serves not only as a structural scaffold but also functions dynamically through a conformational switch to couple cytoplasmic membrane ATP energy to gating of the translocation channel near or at the outer membrane. Consistent with this mode of action, a mutation (G272R) in a region of VirB10 located near the outer membrane was shown to lock VirB10 in the activated conformation, resulting in leakage of secretion substrates to the cell surface independently of target cell contact (22).
In the present study, we sought to further define signaling requirements for activation of VirB10. We report our discovery that, in addition to catalytically active VirD4 and VirB11, activation requires DNA substrate processing and engagement of the transfer intermediate with both ATPases. Strikingly, DNA, but not protein, substrates activate VirB10, suggesting that translocation of DNA and protein substrates through this T4SS are mechanistically distinct processes. Mutational analyses of the channel ATPases identified additional mechanistic features of DNA-ligand-induced channel activation. We discuss our findings in a broader context of known or postulated signals required for activation of conjugative DNA transfer.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in this study are listed in Table 1. Strain KA2002 (LBA4404 deleted of virD4) was constructed by deletion of the virD4 gene in A. tumefaciens strain LBA4404 by marker exchange-eviction mutagenesis using plasmid pKA126 (18) as previously described (29). A. tumefaciens strains were grown in LB supplemented with mannitol and glutamate at 28°C (42). Conditions for induction of the A. tumefaciens vir genes in AB-inducing medium (ABIM; glucose-containing minimal medium [pH 5.5], 1 mM phosphate, 200 μM acetosyringone [AS]) have been previously described (42). When necessary, medium was supplemented with antibiotics (in μg · ml−1) as follows: gentamicin (100), kanamycin (100), tetracycline (5), carbenicillin (100 or 5 for strain LBA4404), spectinomycin (500), rifampin (75), erythromycin (100), streptomycin (100).
Table 1.
Bacterial strains and plasmids
| Bacterial strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | λ−ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Gibco-BRL |
| A. tumefaciens | ||
| A348 | Strain C58 containing octopine-type Ti plasmid pTiA6NC | 23 |
| C58C1RS | Strain C58, Strr Rifr | 24 |
| KE1 | ΔvirE operon | 25 |
| At12516 | virE2− | 26 |
| Mx219 | A348 with a virF::Tn3HoHo1 mutation | 27 |
| Mx311 | A348 with a virD2::Tn3HoHo1 mutation | 27 |
| LBA4404 | T-DNA deletion strain | 28 |
| KA2000 | A348 with a ΔvirD4 mutation | 18 |
| KA2002 | LBA4404 with a ΔvirD4 mutation generated by marker exchange-eviction mutagenesis | This study |
| PC1006 | A348 with a nonpolar ΔvirB6 mutation | 29 |
| PC1010 | A348 with a nonpolar ΔvirB10 mutation | 29 |
| PC1011 | A348 with a nonpolar ΔvirB11 mutation | 29 |
| Plasmids | ||
| Vectors | ||
| pXZ153 | Kanr; IncP plasmid with Kanr gene from pUC4K | 30 |
| pBin19 | Kanr; IncP plasmid with T-DNA border repeats | 31 |
| pBSBBR | Carbr Kanr; pBSIISK+ (Crbr) ligated to pBBR1MCS2 (Kanr) | 32 |
| pBSIISK+NdeI | Carbr; pBSIISK+ containing an NdeI site at the translational start site of lacZ | 29 |
| pPC914KS | 29 | |
| pPC914KS.NcoI | 33 | |
| IncQ/CloDF plasmids | ||
| pML122ΔKanr | Genr; mobilizable RSF1010 (IncQ) derivative | 34 |
| pJB31 | Spcr; RSF1010 derivative | 35 |
| pAJ1 | pJB31 lacking mobA | 36 |
| pAJ6 | pJB31 lacking oriT | 36 |
| pClo-Leu | pBin19 derivative carrying the mob region of pCloDF13; Kanr | Gift from J. Escudero, A. Vergunst, and P. J. J. Hooykaas |
| pΔClo-LEU | pJClo deleted of CloDF13 mob region | 37 |
| pCloΔEB-LEU | pJClo deleted of nonessential mobE and mobB T4CP gene | 37 |
| pCloΔEC-LEU | pJClo deleted of nonessential mobE and mobC relaxase gene | 37 |
| virD2 plasmids | ||
| pMY1153 | pUC18CM expressing the virD operon from the Ptac promoter | 38 |
| pKA171 | Carbr; pPC914KS expressing PvirB-virD2 | This study |
| pKA172 | Carbr; pPC914KS expressing PvirB-virD2ΔC35 | This study |
| pKA250 | Kanr Carbr; pXZ153 expressing PvirB-virD4 and PvirB-virD2 | This study |
| pKA251 | Kanr Carbr; pXZ153 expressing PvirB-virD4 and PvirB-virD2ΔC35 | This study |
| virD4 plasmids | ||
| pKA9 | Carbr; pPC914KS expressing PvirB-virD4 | 18 |
| pKA21 | Kanr; pXZ153 expressing PvirB-virD4 | 18 |
| pKA42 | Kanr; pBBR1MCS2 expressing PvirB-virD4 | 18 |
| pKA84 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4Δ2–10 | This study |
| pKA85 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4Δ2–67 | This study |
| pKA43 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4Δ2-86 | 39 |
| pKA155 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4Δ42-48 | This study |
| pKA158 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4Δ54-60 | This study |
| pKA157 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4F49C | This study |
| pKA156 | Kanr Carbr; pBBR1MCS2 expressing PvirB-virD4Y51A | This study |
| virB6 plasmids | ||
| pSJB964 | Kanr Carbr; pXZ151 expressing PvirB-virB6 | 40 |
| pSJB5XXX | Kanr Carbr; pXZ151 expressing virB6.i4 alleles; XXX is the position of the residue relative to the beginning of the protein that immediately precedes the i4 mutation | 40 |
| virB11 plasmids | ||
| pXZB100 | Kanr Carbr; pXZ151 expressing PvirB-virB11 | 30 |
| pXZB102 | I265T | 30 |
| pXZB105 | L75F | 30 |
| pXZB108 | L11P/D56G/E335G | 30 |
| pXZB101 | I88T/I103T | 30 |
| pXZB103 | H269R | 30 |
| pXZB109 | I103T/M301T | 30 |
| pJCB8XXX | Kanr Carbr; pXZ151 expressing virB11.i4 alleles; XXX is the position of the residue relative to the beginning of the protein that immediately precedes the i4 mutation | 41 |
Bacterial plasmids.
Plasmids used in this study are listed in Table 1. Plasmid pKA171 expresses the full-length virD2 relaxase gene expressed from the PvirB promoter. It was constructed by PCR amplification using 5′-AAATTCCATGGCCGATCG (the NcoI site is underlined) and 5′-CCACTCGAGCGTATCGTGAAC-3′ (the XhoI site is underlined) as forward and reverse primers, respectively, and pMY1153 as a template. The PCR product was digested with NcoI and XhoI, and the resulting fragment was introduced into pPC914KS.NcoI, substituting for the virB1 and virB2′ sequences. Plasmid pKA172 expresses virD2 deleted of 35 codons from its 3′ end. It was constructed by PCR amplification using 5′-CTGCAGCCCCATATGCCTGATCGCGCTCAA-3′ (the NdeI site is underlined) and 5′-AGCTTCCGGTTGCTCGAGTCACTACTATTCTAGATGCTCGGTACCGAT-3′ (the XhoI site is underlined) as forward and reverse primers, respectively, and pMY1153 as a template. The PCR product was digested with NdeI and XhoI, and the resulting fragment was introduced into pPC914KS, resulting in expression of virD2ΔC35 from the PvirB promoter. Plasmid pKA21 expresses PvirB-virD4 from the IncP plasmid pXZ153. It was constructed by introducing PvirB-virD4 as an XbaI-KpnI fragment from pKA9 (see below) into similarly digested pXZ153. Plasmid pKA250 expresses PvirB-virD2 and PvirB-virD4 from the IncP plasmid pXZ153. It was constructed by ligating XhoI-digested pKA171 with similarly digested pKA21. Plasmid pKA251 expressing PvirB-virD2ΔC35 and PvirB-virD4 was constructed by joining XhoI-digested pKA172 and pKA21.
Plasmid pKA9 expressing PvirB-virD4 served as a template for construction of virD4 deletion and substitution mutations by QuikChange mutagenesis as described previously (18). The following plasmids were constructed with the following forward primers in parentheses and corresponding complementary primers (not shown), and the mutations of interest were confirmed by sequencing across the entire virD4 gene fragment: pKA84 deleted of N-terminal codons 2 to 10 (Δ2-10; 5′-ACTTCGCCCCAGCATATGACCCTGAGCATC-3′), pKA85 (Δ2-67; 5′-ACCGTCTTCTGGCATATGTTATCTGTTTGTC-3′), pKA155 (Δ42-48; 5′-GGTCTCATACCAAAAGATCGCTTCACCATTGAAGCCCCGGCG-3′), pKA158 (Δ52-58; 5′-GCTTTTTGGTATGAGACGTCGACCGTCTTCTGGCGTGGTTTATCT-3′), pKA157 (F49C; 5′-TTTGACGTTTTCGCATGCTGGTATGAGACCCCGCTT-3′), pKA156 (Y51A; 5′-GACGTTTTCGCCTTTTGGGCTGAGACCCCGCTTTACTTGGGT-3′).
TrIP assay.
The TrIP assay was performed essentially as previously described (15, 17). Briefly, cells from a 6-ml vir-induced culture were harvested, washed, and resuspended in 20 mM sodium phosphate buffer (pH 6.8) with formaldehyde (FA) at a final concentration of 0.1% and incubated for 20 min at 18°C with shaking. FA was then added in 0.2% increments to reach a final concentration of 1% over a 15-min period. Cells were incubated for 40 min at room temperature (RT) without shaking and then pelleted and solubilized by resuspension in 200 μl in TES buffer (50 mM Tris-Cl [pH 6.8], 2 mM EDTA, 1% β-mercaptoethanol, 1% SDS). For immunoprecipitation, protein A-Sepharose CL-4B (Pharmacia) (30-μl bed volume) was incubated with 1.1 ml of the detergent-solubilized material for 60 min at RT and centrifuged at 5,000 × g to remove protein A-Sepharose and nonspecifically bound proteins. The supernatant was incubated overnight at 4°C with anti-VirD4 or anti-VirB antibodies (Christie laboratory collection) coupled to protein A-Sepharose CL-4B. The beads were washed twice with NP1 buffer supplemented with 1% Triton X-100 and once with NP1 buffer supplemented with 0.1% Triton X-100. Immunoprecipitates were eluted by incubation for 20 min at 96°C in 20 μl of 10 mM Tris-Cl (pH 6.8). Soluble fractions (S) and immunoprecipitates (IP) were analyzed for the presence of T-DNA, pML122, or Ti plasmid by PCR amplification with primers 5′-GGGCGATTATGGCATCCAGAAAGCC and 5′-GTCGGCGGCCCACTTGGCACACAG for gene 7 present on the TLDNA, 5′-CCTGCGGATGTCAGGGCTCTCGT and 5′-TGTCCGTGCTTGCCAATCCCCG for the ophDC locus on pTiA6NC (control), 5′-CTCAGTGGTTCAAGCGGTACA and 5′-TGATAGTTCTTCGGGCTGGTT for a fragment of mobA carried on RSF1010 derivatives used in this study, and 5′-GTGAGCAAAGCCGCTGCC and 5′-AGCCAATTGATCCTGCA for a fragment of the repAB locus on pTiAch5 (control). To compare levels of T-strand by quantitative TrIP (QTrIP), immunoprecipitates were subjected to 20 cycles of PCR amplification using the primers specific for gene 7 of the T-DNA. On the 21st cycle, a single round of PCR amplification was performed with addition of 1.0 mCi of [32P]dGTP (Amersham Biosciences), as described previously (17, 18). PCR products were column purified with the QIAquick PCR purification kit (Qiagen) to remove unincorporated nucleotides. Aliquots of the eluted material were mixed with 3.5 ml of scintillation liquid (Ecolite, ICN) and counted with a Beckman Coulter. The entire TrIP protocol was repeated three times in triplicate, and the average value from a single experiment was reported.
Quantitation of Ti plasmid copy number and T-strands.
Relative numbers of T-strands generated per Ti plasmid were determined as previously described (43). Briefly, Ti plasmid copy numbers and T-strand levels were determined by subjecting equal amounts of total DNA to 20 cycles of PCR amplification using primers specific for T-DNA (gene 7 of TL-DNA) and Ti plasmid (ophDC locus) (17). As described above, on the 21st cycle, 1.0 mCi of [α-32P]dGTP was added for a single round of PCR amplification and PCR products were purified. The number of T-strands generated per molecule of Ti plasmid was obtained as a ratio of cpm of radioactivity incorporated into a T-DNA-specific amplicon (gene 7) to that obtained for the Ti plasmid-specific amplicon (ophDC), multiplied by the factor 3.76 (973/259; obtained as a ratio of number of G+C bases in PCR products of ophDC locus [973] to gene 7 [259]).
Protease susceptibility assay for VirB10 conformational change.
A. tumefaciens spheroplasts were treated with Streptomyces griseus protease (Sigma) for detection of the VirB10* proteolytic degradation product as previously described (21). Briefly, vir-induced cells were either treated with sodium arsenate (25 mM) (Allied Chemical, Morristown, NJ) for 30 min or left untreated. Cells were harvested by centrifugation, and the cell pellets were resuspended at 1:30 volume of the inducing culture in 20 mM Tris-HCl (pH 8.0), 20% (wt/vol) sucrose. Cells were converted to spheroplasts by addition of 100 μg/ml of lysozyme (Sigma), incubation for 5 min on ice, addition of 2 mM EDTA, and further incubation for 45 min on ice. Cells were then centrifuged at 5,000 × g for 5 min, and spheroplasts were gently resuspended in 20 mM Tris-HCl (pH 8.0), 10 mM MgSO4, and 20% sucrose. Spheroplasts were incubated with S. griseus protease (Sigma) at a final concentration of 100 μg/ml for 15 min on ice, harvested by mixing with an equal volume of 2 μ Laemmli's buffer, and then immediately boiled for 10 min.
Protein analyses.
Protein samples were suspended in Laemmli's buffer and subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) using glycine or tricine buffer systems as previously described (40). For detection by immunostaining, proteins were transferred onto nitrocellulose membranes, and immunoblots were developed with antibodies to the VirD2, VirD4, and VirB proteins from our laboratory collection (18, 39, 40) and goat anti-rabbit antibodies conjugated to alkaline phosphatase (Bio-Rad). Proteins were loaded on SDS-polyacrylamide gels on a per-cell (CFU) equivalent basis. Molecular mass markers were obtained from GIBCO-BRL (Grand Island, NY).
Conjugation assays.
Derivatives of RSF1010 and pCloDF13 were introduced into various A. tumefaciens donor strains by electroporation, and the resulting strains were mated with an A348Spcr on a nitrocellulose filter on an ABIM agar plate containing AS as previously described (18). Frequencies of transfer were estimated as transconjugants recovered per donor.
Virulence assays.
A. tumefaciens strains were tested for virulence by inoculating wound sites of Kalanchoe daigremontiana leaves (44). Each experiment was repeated at least three times for each strain on separate leaves.
RESULTS
Processed T-DNA but not protein substrates is required for VirB10 conformational change.
VirB10 adopts alternative protease-susceptible and -resistant conformations as a function of cellular energetic status and production of the VirD4 and VirB11 ATPases (21). As shown previously (21) and in Fig. 1, the native protein migrates as an ∼48-kDa species in SDS-polyacrylamide gels, but upon treatment of spheroplasts from the wild-type (WT) strain A348 with the S. griseus serine protease, anti-VirB10 antibodies also react with an ∼40-kDa species designated VirB10*. Neither full-length VirB10 nor the VirB10* species are detected in extracts of the similarly treated ΔvirB10 mutant, confirming the identity of the latter as a proteolytic product of VirB10. Arsenate treatment of A. tumefaciens cells, which dissipates cellular ATP, prior to conversion to spheroplasts and proteolysis abolishes formation of VirB10*. This conformational transition also requires production of catalytically active forms of the VirD4 T4CP and VirB11 ATPases, suggesting that VirB10 couples ATP energy to substrate translocation through its interactions at or near the cytoplasmic membrane with a VirD4/VirB11 complex.
Fig 1.
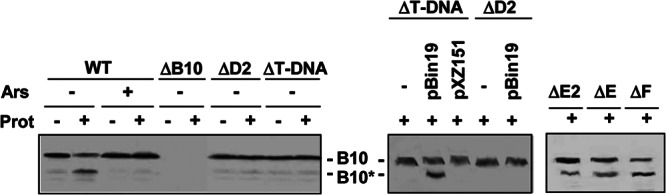
Detection of the VirB10 conformational switch by protease treatment. Strains in the left blot were pretreated with arsenate (Ars; +) or H2O (−); those in the center and right blots were pretreated with H2O only. Spheroplasts were treated with S. griseus protease (Prot; +) or left untreated (−). B10, VirB10 (∼48-kDa) B10*, VirB10* proteolytic degradation product (∼40 kDa) detected by immunostaining with anti-VirB10 antibodies. Strains, left to right: WT, A348; ΔB10, ΔvirB10 mutant PC1010; ΔD2, Mx355(pKA42) virD2::Tn3HoHo1 mutant expressing virD4 from a pBBR plasmid; ΔT-DNA, ΔT-DNA mutant LBA4404; ΔT-DNA, LBA4404 carrying IncP plasmids with (pBin19) or without (pXZ151) a T-DNA border sequence; ΔD2, Mx355(pKA42, pBin19); ΔE2, virE2 mutant At12516; ΔE, ΔvirE operon mutant KE1; ΔF, virF mutant Mx219 (virF::Tn3HoHo1).
Besides energizing VirB10, VirD4 and VirB11 bind the relaxosome or T-DNA transfer intermediate and deliver the substrate into the translocation channel (17, 18, 43). To examine whether these early DNA substrate docking reactions might be coupled to activation of VirB10, we assayed strains lacking T-DNA for generation of the VirB10* species. Strikingly, upon protease treatment of spheroplasts from the ΔT-DNA strain LBA4404, we detected only the protease-resistant, 48-kDa species (Fig. 1). Introduction of pBin19, an IncP plasmid that carries oriT-like T-DNA border sequences, restored the VirB10 protease susceptibility profile of LBA4404 to the WT pattern, whereas introduction of an IncP plasmid lacking the border sequence did not regenerate this pattern (Fig. 1). We also did not detect the VirB10* species by protease treatment of a virD2 mutant carrying T-DNA or pBin19 substrates (Fig. 1). Previously, we showed that the T-DNA substrate does not form an FA-cross-linkable contact with VirD4 in the absence of relaxase, which indicates that generation of the relaxosome (VirD2 and accessory factor VirD1 bound at the T-DNA border) or production of the VirD2–T-strand intermediate obligatorily precedes substrate docking with the T4CP (17). Our present findings further suggest that docking of the relaxosome or processed T-DNA with VirD4 is also required for activation of VirB10.
Strain LBA4404 (ΔT-DNA) produces the VirD2 relaxase, which is itself a substrate of the VirB/VirD4 channel in the absence of associated T-strand (45, 46). LBA4404 also produces several effector proteins, such as VirE2, VirE3, and VirF, that are translocated through the VirB/VirD4 T4SS independently of DNA (47, 48). The finding that protease treatment of LBA4404 spheroplasts does not generate VirB10* suggests that protein substrates do not induce the observed VirB10 conformational switch. We further assayed virE2, virE (deleted of virE1, virE2, virE3), or virF mutant strains, each of which are competent for translocation of T-DNA to plants, for generation of the VirB10* species. Treatment of spheroplasts from these strains resulted in appearance of VirB10* (Fig. 1), providing additional evidence that activation of VirB10 requires engagement of DNA, but not protein, substrates with the T4CP.
C terminus of VirD2 relaxase is required for substrate transfer and the VirB10 conformational change.
The VirD2 C terminus contains two motifs that are required for delivery of the VirD2–T-strand intermediate to plants, a bipartite nuclear localization sequence (NLS) that mediates delivery of the transfer intermediate to the plant nuclear pore and the omega (ω) sequence (DGRGG) that contributes to efficient T-DNA transfer through the VirB/VirD4 T4SS (Fig. 2A) (49). A VirD2 mutant deleted of these C-terminal motifs is completely deficient for DNA substrate transfer (Fig. 2A) (46). Conversely, fusion of a 40-residue, C-terminal fragment carrying these motifs to a reporter protein suffices for delivery of the reporter to plant cells, implying that this fragment carries the necessary information for substrate docking and translocation through the VirB/VirD4 T4SS (46). To evaluate the importance of the DNA substrate docking reaction for channel activation, we asked whether deletion of the VirD2 C-terminal translocation signal disrupted VirB10 activation. In the initial studies, we confirmed that a ΔC35 mutation did not affect relaxase-mediated processing of the T-strand. As shown previously for wild-type strain A348 (43), a ΔvirD2 mutant expressing native virD2 in trans accumulated multiple copies of processed T-strands upon induction of the vir genes (Fig. 2B). Within 8 to 12 h postinduction, cells accumulated ∼8 to 10 free T-strands per Ti plasmid, and this number increased to as many as 15 T-strands per Ti plasmid over the next 4 to 8 h. The kinetics of T-strand accumulation matched profiles observed for Vir protein accumulation as shown for VirD2 and VirB9, whereas the constitutively synthesized sugar-binding protein ChvE accumulated at comparable levels throughout the AS induction period (Fig. 2B). The virD2 mutant strain failed to generate free T-strands, and this strain expressing virD2ΔC35 generated multiple copies of free T-strands over the 16-h induction period, establishing that the ΔC35 truncation did not affect substrate processing (Fig. 2B).
Fig 2.
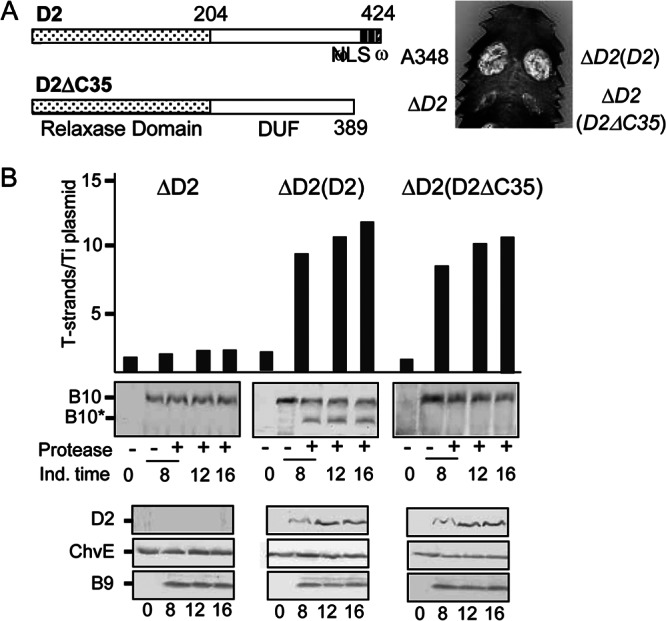
Contribution of a VirD2 C-terminal translocation signal to T-DNA transfer and VirB10 activation. (A) D2, VirD2 full length; ΔC35, C-terminal truncation mutant. Domains: relaxase, domain of unknown function (DUF), and C-terminal nuclear localization sequence (NLS), ω sequence. Numbers correspond to residues relative to the beginning of the protein. Plant leaf was inoculated with A348, wild-type strain; ΔD2 mutant, Mx355 (virD2::Tn3HoHo1 mutant) with pKA42 expressing virD4; ΔD2(D2), Mx355 with pKA250 expressing virD2 and virD4; ΔD2(D2ΔC35), Mx355 with pKA251 expressing virD2ΔC35 and virD4. (B) Numbers of T-strands per Ti plasmid in strains ΔD2, Mx355(pKA42); ΔD2(D2), Mx355(pKA250); and ΔD2(D2ΔC35), Mx355(pKA251) (see the text for details). Ind. time, T-strand levels determined at 0, 8, 12, and 16 h following vir gene induction with AS. Protease, spheroplasts were treated with S. griseus protease (+) or left untreated (−). B10, VirB10 (∼48-kDa) B10*, VirB10* proteolytic degradation product (∼40 kDa) detected by immunostaining with anti-VirB10 antibodies. Bottom, steady-state levels of D2 (VirD2), ChvE, B9 (VirB9) at the postinduction times induced as assessed by immunostaining with the respective antibodies.
Protease treatment of spheroplasts from the ΔvirD2 mutant expressing native virD2 in trans generated the VirB10* species, whereas similar treatments of spheroplasts from both the ΔvirD2 mutant and VirD2ΔC35-producing strains failed to generate this VirB10 species (Fig. 2B). Both VirD2 and VirD2ΔC35 produced from the PvirB promoter accumulated at similar levels following AS induction. We conclude from these findings that VirD2-mediated docking of the relaxosomal complex or the transfer intermediate with VirD4 is essential for activation of VirB10.
Mobilizable IncQ and pCloDF13 substrates induce VirB10 conformational change.
Next, we asked whether other DNA substrates of the VirB/VirD4 system induce the VirB10 conformational switch. This T4SS mobilizes transfer of non-self-transmissible plasmids, RSF1010 (IncQ) and pCloDF13, to recipient cells (32, 36, 37, 50). RSF1010 engages with the VirD4 T4CP and translocates along the same pathway as the T-DNA substrate through the VirB channel, although substrate specificity checkpoints have been identified by mutational analyses that allow translocation of one but not both substrates (see below) (32, 40, 41, 44). We first established that strain LBA4404 (ΔT-DNA) mobilizes transfer of the RSF1010 derivative pML122 at a frequency of ∼10−5 transconjugants (Tc)/donor, which is comparable to previously reported values (Fig. 3A) (44). LBA4404 also mobilized transfer of another RSF1010 derivative (pJB31) but not mutant forms of this plasmid lacking an intact mobA relaxase gene (pAJ1) or functional oriT sequence (pAJ6) (Fig. 3A). pCloDF13 codes for Mob functions required for nicking at oriT, but in contrast to RSF1010 and other mobilizable plasmids, pCloDF13 also codes for a VirD4-like T4CP. This T4CP interfaces with the A. tumefaciens VirB Mpf channel, as shown by the capacity of chimeric machines composed of the A. tumefaciens VirB Mpf subunits and pCloDF13-encoded MobB to mobilize plasmid transfer in the presence (32) or absence of a T-DNA substrate (Fig. 3B). As shown for RSF1010, pCloDF13 derivatives bearing mutations of the entire mob region, the mobC relaxase gene, or the mobB T4CP gene failed to translocate through the VirB T4SS (Fig. 3B).
Fig 3.

Requirement of DNA substrate processing for VirB10 activation. (A) Top, histogram shows transfer frequencies (Tc/donor, transconjugants per donor) of A348(pML122) and LBA4404 (ΔT-DNA) lacking (−) or carrying pML122 or pJB31 (IncQ plasmids) or pJB31 derivatives pAJ1 (mobA relaxase mutant) or pAJ6 (mutated oriT) to A348Spcr recipient cells. Data from a representative assay are shown along with standard deviations from replicate experiments. (A) Detection of the VirB10 conformational switch by protease treatment. Ars, arsenate (+) or H2O (−) pretreatment. Prot, S. griseus protease treatment (+) or untreated (−). B10, VirB10 (∼48-kDa) B10*, VirB10* proteolytic degradation product (∼40 kDa) detected by immunostaining with anti-VirB10 antibodies. (B) A348(pML122) and KA2002 (ΔT-DNA, ΔvirD4) carrying the plasmids indicated were assayed for plasmid mobilization and generation of the VirB10* species as described for panel A. Strains: A348(pML122); KA2002 lacking (−) or carrying pClo (pClo-Leu) or derivatives pCloΔmob (lacks entire mob region), pCloΔmobB (relaxase minus), or pCloΔmobC (T4CP minus).
Protease treatment of spheroplasts from LBA4404 carrying the IncQ plasmids pML122 or pJB31 generated the VirB10* species, establishing that these DNA substrates activate VirB10. Similar treatments of spheroplasts from LBA4404(pJB31) cells depleted of cellular ATP by treatment with arsenate or cells carrying the oriT or mobA mutant plasmids did not yield the VirB10* species. Additionally, protease treatment of spheroplasts from LBA4404 bearing a ΔvirD4 mutation (strain KA2002) did not generate the VirB10* species even when this strain carried the pML122 substrate, establishing the importance of VirD4 synthesis for the observed pML122-induced conformational switch (Fig. 3B). Finally, protease treatment of spheroplasts from strains carrying pClo (pCloDF13 derivative) generated the VirB10* species, indicating that pClo engages with its own T4CP to activate VirB10. As shown for the T-DNA and RSF1010 substrates, depletion of cellular ATP levels with arsenate abolished pClo-mediated activation of VirB10, as did mutations of the mob region or the mobC relaxase or mobB T4CP genes (Fig. 3B).
The VirD4 periplasmic domain is necessary for substrate transfer and the VirB10 conformational change.
Our findings thus far suggest that DNA substrates induce a conformational change in VirB10 through engagement with the VirD4 T4CP. To further explore the requirements for energy coupling, we determined effects of VirD4 mutations on the VirB10 conformational status. The importance of VirD4 ATP binding or hydrolysis for activation of VirB10 was shown previously through our studies of a Walker A mutant (K152Q) (21). In view of the topological models experimentally generated for VirD4 and VirB10 (Fig. 4A) (51, 52) and evidence that homologs of these proteins interact via their N-terminal regions (53, 54), we sought to test whether the N-terminal transmembrane (TM) domain of VirD4 might participate in the activation reaction. A VirD4 mutant deleted of the TM domain (Δ2-86) interacts with T-DNA but fails to deliver the substrate to the VirB11 ATPase (Fig. 4B) (18), likely because this domain fails to interact productively with VirB10 or other machine subunits. As expected, protease treatment of the VirD4Δ2-86-producing strain failed to generate the VirB10* species (Fig. 4C).
Fig 4.

Effects of VirD4 mutations on DNA transfer and VirB10 activation. (A) Schematic showing VirD4, VirB11, and VirB10 membrane topologies. The N-terminal 85 residues of VirD4 are depicted, with residues that are not conserved (black), similar (gray), or invariant (gray with black outline) among homologs associated with T4SSs in related rhizobial species. Underlined residues denote deletion mutations Δ42-48 and Δ52-58; residues F49 and Y51 were substituted with Cys, and K152 in the Walker A motif was substituted with Gln. (B) A348 (WT) or K2001 (ΔvirD4) expressing variant forms of VirD4 were assayed for T-DNA transfer through the VirB/VirD4 T4SS. Table lists: strains, VirD4 variants; D4-B9, VirD4 and VirB subunits with a detectable (+) or undetectable (−) T-DNA interaction as monitored by TrIP; Vir, T-DNA transfer to plants as monitored by virulence (+, virulent; −, avirulent). Mutant strains were grouped into 3 classes: (i) permissive, (ii) blocked T-DNA transfer from VirD4 to VirB11, (iii) blocked T-DNA transfer from VirB11 to VirB6. (C) A348 and KA2000 (ΔvirD4) strains lacking (−) or producing D4 (native VirD4) or the VirD4 mutant derivatives shown were assayed for steady-state accumulation of the VirD4 variants (top) and B10 (native VirB10) and B10* (VirB10*) upon protease treatment of spheroplasts (bottom) by immunostaining with the anti-VirD4 or -VirB10 antibodies.
We next examined the importance of subdomains within the VirD4 TM domain to substrate transfer and VirB10 activation. Accordingly, we analyzed the effects of deletion mutations comprising (i) the extreme N-terminal cytoplasmic region (Δ2-10), (ii) the cytoplasmic region plus the first transmembrane helix and the periplasmic loop (Δ2-67), and (iii) two 7-residue deletions in the first and second halves of the periplasmic loop (Δ42-48, Δ52-58) (Fig. 4). The latter deletions were constructed to test the functional importance of two regions of the periplasmic loop, one (Δ52-58) comprised mainly of highly conserved residues among close VirB10 homologs associated with other Rhizobial species T4SSs and a less-well-conserved sequence (Δ42-48) (see Fig. S1 in the supplemental material). We also constructed two substitution mutations of invariant aromatic residues (F49C, Y51C).
All of the mutant proteins accumulated at levels comparable to the native protein, suggesting that observed phenotypes are not likely due to problems of protein instability (Fig. 4C). We assayed mutant strains for the capacity to incite virulence on plants and to deliver the T-DNA through the VirB/VirD4 channel by use of the TrIP assay (17). Interestingly, the mutations grouped into three classes. One mutation (Δ2-10) designated class I was permissive for substrate transfer, as defined both by restoration of virulence of a ΔvirD4 mutant strain and formation of FA-cross-linked contacts between the T-DNA and the VirD4/VirB channel subunits. Class II mutations phenocopied the Δ2-86 mutation in rendering the mutant proteins competent for binding of the DNA substrate but not for substrate transfer to VirB11. Mutations in this class included the two large TMD deletions (Δ2-67, Δ2-86) and deletion of highly conserved residues (Δ52-58) in the periplasmic domain. The class III mutations phenocopied the K152Q Walker A mutation; the mutant proteins interacted with the T-DNA and transferred the substrate to VirB11 but not to the cytoplasmic membrane proteins VirB6 and VirB8. Mutations in this class included three periplasmic domain mutations, Δ42-48, F49C, and Y51C. Strikingly, therefore, the VirD4 periplasmic domain contributes to substrate transfer reactions between the T4CP, VirB11, and the VirB6 and VirB8 channel subunits. Additionally, distinct regions of the periplasmic domain differentially modulate these early transfer reactions.
We assayed for effects of the NTD mutations on the VirB10 conformational status and found that all mutations that disrupted T-DNA transfer abolished generation of the VirB10* species (Fig. 4C). These findings indicate that the VirD4 periplasmic domain contributes not only to early-stage transfer reactions in the cytoplasm but also to activation of VirB10 and, therefore, a later-stage reaction required for substrate transfer through the distal portion of the channel.
VirB11 mutations exert correlative effects on substrate transfer and VirB10 conformation.
Next, we sought to further define the contribution of VirB11 to VirB10 activation by characterizing two sets of previously isolated mutations. The first set was isolated originally as dominant negative suppressors of WT virB11 gene function (30, 41). These mutations fall into two classes, those that block WT gene function in merodiploid strains and are nonfunctional when expressed in the absence of the WT gene (dominant, nonfunctional) and those that retain functionality when expressed in the absence of the WT gene (dominant, functional).
As expected, ΔvirB11 strains producing the dominant, functional mutant proteins displayed a WT DNA transfer profile as monitored with the TrIP assay (Fig. 5A). We also showed with a semiquantitative variation of the TrIP assay termed QTrIP (17) that complementation of a ΔvirB11 mutant with wild-type virB11 fully restored T-DNA transfer to levels detected in the WT strain A348, and strains producing the dominant, functional mutant proteins similarly efficiently transferred the substrate through the channel (Fig. 5A). Treatment of spheroplasts from strains producing the dominant, functional mutants also generated the VirB10* species, indicating that the mutant proteins coordinate with VirD4 both for DNA transfer and VirB10 activation (Fig. 5B).
Fig 5.

Effects of VirB11 substitution mutations on T-DNA transfer and VirB10 activation. (A) Top, transfer of T-DNA to native and mutant forms of VirB11, as assessed with the TrIP assay. Strains: PC1011 (ΔvirB11) lacking (−) or producing B11 (native VirB11) or the mutant derivatives shown. S, supernatant from the IP reaction; IP, immunoprecipitated material recovered with anti-VirB11 antibodies (αB11); Ct, Ti plasmid control amplicon; T-DNA, T-DNA amplicon. Bottom, results of QTrIP assays showing levels of processed T-DNA (T-strand) recovered by precipitation with anti-VirD4, -VirB11, -VirB6, and -VirB9 antibodies relative to strain A348 (WT strain), as described previously (17). Results are presented as a percentage of the T-DNA detected in the immunoprecipitates from WT A348. Schematics summarize effects of dominant, nonfunctional mutations in blocking DNA transfer at specific stages of the translocation pathway. WT, no stage-specific block (conferred by native VirB11 and the dominant, functional mutant proteins shown). A ΔvirB10 mutation (ΔB10) blocks later-stage transfer from VirB6/VirB8 to VirB2/VirB9 (17). (B) PC1011 (ΔvirB11) lacking (−) or producing B11 (native VirB11) or the mutant derivatives shown were assayed for accumulation of B10 (native VirB10) and B10* (VirB10*) upon protease treatment of spheroplasts by immunostaining with the anti-VirB10 antibodies. Tra, T-DNA transfer to plants as monitored by virulence (+, virulent; −, avirulent,); Pil, production of detectable (+) or undetectable (−) levels of pili.
In contrast, the ΔvirB11 strains producing two dominant, nonfunctional mutant proteins (I88T/I103T, H269R) were blocked in the earliest step of T-DNA transfer from VirD4 to VirB11, as shown with the TrIP and QTrIP assays (Fig. 5A). Protease treatment of these strains also failed to generate the VirB10* species (Fig. 5B). These mutant proteins thus fail to interact productively with VirD4 both for DNA transfer and activation of VirB10. Interestingly, however, one dominant, nonfunctional mutant protein (L11P/D56G/E335G) did support DNA transfer from VirD4 to the membrane proteins VirB6 and VirB8 but not to VirB2 and VirB9 (Fig. 5A). Yet, the corresponding mutant strain did not generate the VirB10* species, indicative of a failure to induce the conformational switch (Fig. 5B). This mutant protein therefore appears to engage productively with VirD4 to support the early substrate transfer reactions but nonproductively with the T4CP for activation of VirB10, which is required for later-stage transfer from VirB6 and VirB8 to VirB2 and VirB9 (17, 21).
Finally, we determined that the I265T mutation as well as a second mutation (L18.i4, see below) conferred the interesting phenotype of inducing the VirB10 structural transition and supporting T-DNA transfer but not pilus biogenesis (Fig. 5B) (41). Conversely, as shown previously, a virD4 mutant fails to activate VirB10 or transfer DNA but elaborates pili (18, 55). These findings establish that the observed DNA ligand- and ATP-driven conformational change of VirB10 is not required for pilus production.
Differential effects of VirB11 insertion mutations on substrate transfer and VirB10 activation.
The second set of mutations was generated by insertion of a 4-residue epitope (HMVD; i4) along the length of VirB11. In contrast to that described above, these mutations were generally recessive and classified as permissive or loss-of-function according to their effects on T-DNA transfer to plants (41). As expected, strains producing the permissive mutations displayed the WT pattern of T-DNA channel subunit interactions as monitored with the TrIP assay (Fig. 6). Correspondingly, protease treatment of these strains generated the VirB10* species indicative of VirB10 activation. In contrast, strains producing the loss-of-function mutations showed a block in T-DNA transfer from VirD4 to VirB11, and these strains failed to generate the VirB10* species (Fig. 6).
Fig 6.
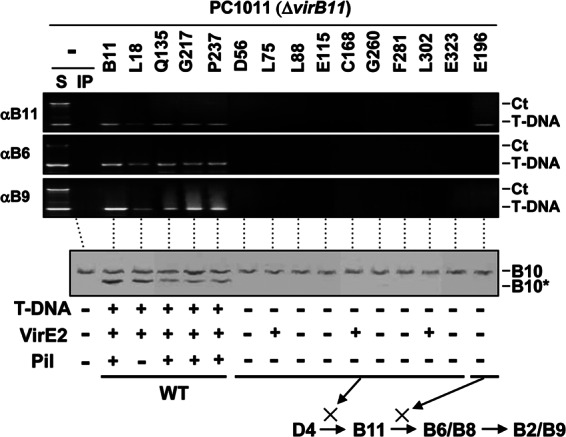
Effects of VirB11.i4 mutations on T-DNA transfer and VirB10 activation. Top, transfer of T-DNA to native and mutant forms of VirB11 and to VirB6 and VirB9, as assessed with the TrIP assay. Strains: PC1011 (ΔvirB11) lacking (−) or producing B11 (native VirB11) or the i4 mutant proteins shown. S, supernatant from the IP reaction; IP, immunoprecipitated material recovered with anti-VirB11, -VirB6, or -VirB9 antibodies (αB11, αB6, αB9). For PC1011 strains producing native or mutant forms of VirB11 (the i4 mutation is inserted after the residue listed), amplicons from the immunoprecipitate (IP) fractions are shown. Ct, control, chromosomal DNA amplicon; T-DNA, T-DNA amplicon. Bottom, PC1011 (ΔvirB11) lacking (−) or producing B11 (native VirB11) or the mutant derivatives were assayed for accumulation of B10 (native VirB10) and B10* (VirB10*) upon protease treatment of spheroplasts by immunostaining with the anti-VirB10 antibodies. T-DNA, T-DNA transfer to plants as monitored by virulence (+, virulent; −, avirulent); VirE2, transfer of VirE2 to plant cells as monitored with a mixed infection assay (+, transfer; −, no transfer). Pil, production of detectable (+) or undetectable (−) levels of pili. Schematics summarize effects of mutations in blocking DNA transfer at specific stages of the translocation pathway. WT, no stage-specific block.
Interestingly, strains producing a subset of the loss-of-function mutations retained the ability to deliver VirE2 to plant cells despite the block in T-DNA transfer (Fig. 6) (41). The mutant proteins (L75.i4, C168.i4, L302.i4) conferring this substrate discrimination phenotype failed to interact with the T-DNA but nevertheless supported transfer of VirE2 to plant cells as determined with a mixed-infection assay (41). Thus, protein substrates such as VirE2 neither activate VirB10 (see Fig. 1) nor require VirB10 activation for translocation through the VirB/VirD4 T4SS (Fig. 6).
The T-DNA Tra− mutations could exert their effects by disrupting VirB11 interactions with VirD4 or with the T-DNA substrate. To distinguish between these possibilities, we tested whether these mutations also disrupted translocation of another substrate, the RSF1010 derivative pML122. Interestingly, the three mutations that were permissive for VirE2 translocation (L75.i4, C168.i4, L302.i4) also were permissive for transfer of the IncQ plasmid (Fig. 6 and 7). In strains carrying both DNA substrates, these discrimination mutations still selectively arrested T-DNA transfer at the VirD4 to VirB11 step without disrupting IncQ plasmid translocation (Fig. 7). In these strains, engagement of the IncQ plasmid substrate with VirD4 and VirB11 also resulted in activation of VirB10, as evidenced by generation of the VirB10* species upon protease treatment (Fig. 7). In contrast, strains carrying both DNA substrates and virB11 alleles with mutations that completely blocked transfer of all tested DNA substrates (E115.i4, F281.i4) at the VirD4 to VirB11 step also failed to activate VirB10 (Fig. 7).
Fig 7.
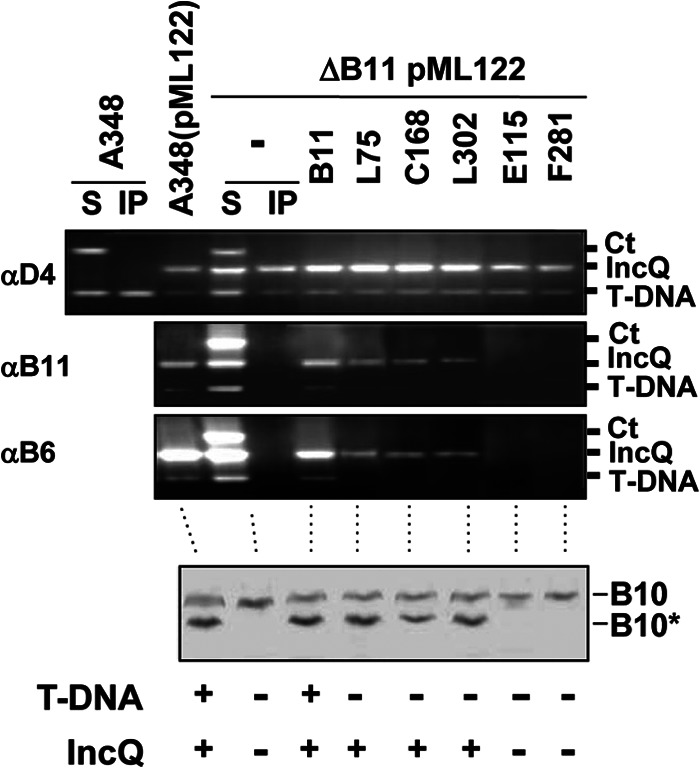
Effects of VirB11 substrate discrimination mutations on DNA transfer and VirB10 activation. Upper, transfer of T-DNA and IncQ plasmid pML122 to VirD4, native and mutant forms of VirB11, and VirB6 as assessed with the TrIP assay. Strains: A348 (WT strain) without and with pML122 (IncQ plasmid); PC1011(pML122) (ΔvirB11 mutant carrying pML122) without (−) or with plasmids expressing B11 (native VirB11) or i4 mutant proteins (the i4 mutation is inserted after the residue listed). S, supernatant from the IP reaction; IP, immunoprecipitated material recovered with anti-VirD4, -VirB11, or -VirB6 antibodies (αD4, αB11, αB6). For PC1011 strains producing native or mutant forms of VirB11, amplicons from the immunoprecipitate (IP) fractions are shown. Ct, control, Ti plasmid amplicon; IncQ, pML122 plasmid amplicon; T-DNA, T-DNA amplicon. Lower, A348(pML122) and PC1011(pML122) strains were assayed for accumulation of B10 (native VirB10) and B10* (VirB10*) upon protease treatment of spheroplasts by immunostaining with the anti-VirB10 antibodies. T-DNA, T-DNA transfer to plants as monitored by virulence (+, virulent; −, avirulent); IncQ, transfer of IncQ plasmid pML122 to agrobacterial recipients.
Together, these findings establish that the VirB11 mutant proteins conferring the T-DNA Tra−, IncQ Tra+ phenotype are in fact capable of engaging productively with VirD4 to mediate IncQ plasmid translocation and VirB10 activation. These mutant proteins thus appear to block T-DNA transfer through a lack of a productive interaction with the T-DNA transfer intermediate. The data further suggest that activation of VirB10 requires not only coordination of the VirD4 and VirB11 ATPases but also productive engagement of a DNA substrate with both VirD4 and VirB11. The VirB11 mutant proteins conferring the substrate discrimination phenotype are capable of activating VirB10, but only if they establish a productive interaction with the IncQ plasmid substrate.
VirB6 channel blocking mutations do not affect VirB10 conformational change.
Finally, we asked whether channel subunits acting downstream of the ATPases in the translocation pathway might also regulate the activation reaction. VirB6 mutations also have been isolated that either block substrate transfer altogether or selectively inhibit T-DNA transfer at specific stages in the translocation pathway while permitting translocation of other substrates (19, 40). We tested for effects of these mutations on activation of VirB10 and found that none blocked generation of the VirB10* species (Fig. 8B). These findings strongly indicate that the early-stage reactions involving DNA substrate engagement with VirD4 and VirB11 and productive contacts between the two ATPases are necessary and sufficient for inducing the VirB10 conformational switch.
Fig 8.
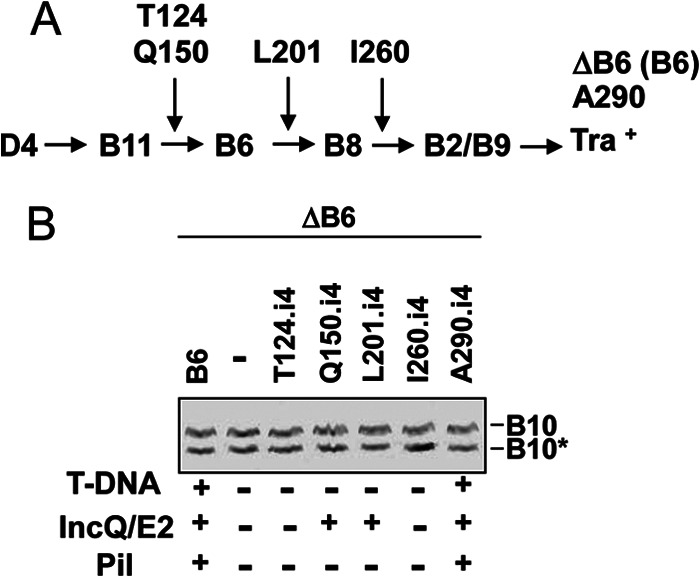
Effects of i4 mutations in VirB6 on T-DNA transfer and VirB10 activation. (A) Schematic showing steps in the T-DNA transfer pathway blocked by VirB6.i4 mutant proteins (the i4 mutation is inserted at the residue listed) when produced in the nonpolar ΔvirB6 mutant PC1006. ΔB6(B6) (PC1006 producing native VirB6) and A290 (A290.i4 mutant) display a WT T-DNA translocation phenotype (see reference 19). (B) PC1006 (ΔvirB6 mutant) lacking (−) or producing native VirB6 (B6) or the i4 mutant proteins were assayed for accumulation of B10 (native VirB10) and B10* (VirB10*) upon protease treatment of spheroplasts by immunostaining with the anti-VirB10 antibodies. T-DNA, T-DNA transfer to plants as monitored by virulence (+, virulent; −, avirulent); IncQ/E2, transfer of IncQ plasmid pML122 to agrobacterial recipients or VirE2 to plant cells as monitored by a mixed infection assay; Pil, production of detectable (+) or undetectable (−) levels of pili (see reference 40).
DISCUSSION
The A. tumefaciens VirB7/VirB9/VirB10 core complex serves as a structural scaffold for biogenesis of the VirB/VirD4 translocation channel and pilus (11, 12, 15). In the assembled core complex, VirB10 adopts a topology that is unique among bacterial proteins by spanning the entire cell envelope. This makes VirB10 ideally suited for sensing of both intra- and extracellular signals required for activation of the translocation channel (13, 52). Previously, we gained evidence that VirB10 undergoes a structural transition in response to sensing of ATP utilization by the VirD4 and VirB11 ATPases (21), and in the present study we now have shown that a second type of intracellular signal, docking of a DNA substrate with the channel ATPases, also is required for activation of VirB10. As a framework for this discussion, Fig. 9 presents a model depicting the requirements for activation of VirB10 and DNA translocation. The DNA transfer pathway through the VirB/VirD4 channel, as defined by TrIP, involves substrate docking with the VirD4 T4CP and delivery to VirB11 and then passage across the cytoplasmic membrane via a presumptive VirB6/VirB8 translocon and the periplasm and outer membrane via a channel formed at least in part by VirB2 and VirB9 (17). We propose here that two intracellular signals, DNA ligand binding and ATP energy utilization, induce the VirB10 structural transition, which opens a channel gate at or near the outer membrane enabling substrate passage to the cell surface. Although ligand- and energy-induced channel gating has been described for small-molecule and macromolecular transport systems (56–59), this is the first report indicating that the similar signals activate gating of the T4SS channel.
Fig 9.

Model depicting the DNA substrate translocation pathway and activating intracellular signals. Transfer stages: (1) DNA transfer initiates by engagement of the relaxosome or the relaxase–T-strand transfer intermediate with the VirD4 T4CP, (2) the VirD4 T4CP delivers the substrate to VirB11, (3) VirB11 delivers the substrate to the cytoplasmic membrane translocon comprised of VirB6 and VirB8, (4) the translocon delivers the substrate across the cytoplasmic membrane, and (5) substrate passes through the distal portion of the transfer channel comprised of VirB2 and VirB9 (17). Intracellular activating signals: (A) DNA substrate docking with VirD4 and delivery to VirB11 and (B) VirD4 and VirB11 ATP hydrolysis activities. These signals stimulate a conformational switch in VirB10 that is required for gating or assembly of the distal portion of the translocation channel.
Our initial studies established that engagement of DNA, but not protein, substrates with the VirD4 T4CP is a prerequisite for VirB10 activation. We supplied evidence that processing and docking of three different DNA substrates, T-DNA, RSF1010, and pCloDF13, with VirD4 (or MobB in the case of pCloDF13) were required for inducing VirB10's structural transition (Fig. 1 and 3). Furthermore, both components of the DNA substrate, the relaxase and the DNA, were essential for inducing the transition. Although we did not definitively establish that a direct relaxase-T4CP interaction is required for channel activation, several studies have supplied strong evidence that relaxases interact with cognate T4CPs and that such an interaction is required for DNA translocation. With our TrIP assay, for example, we previously determined that VirD2 synthesis is essential for establishment of a detectable VirD4–T-DNA cross-link (17). Here, we further showed that a catalytically active form of VirD2 did not suffice for establishment of the VirD4–T-DNA interaction, but that an intact C terminus was also required (Fig. 2). The C-terminal region of VirD2 carries an NLS and an ω sequence, the latter of which resembles charged signals carried by T4SS effector proteins (45–48). This C-terminal region has been shown to mediate the delivery of a fused reporter protein to plant cells, confirming that it carries sufficient information for docking with and translocation through the VirB/VirD4 T4SS (45, 46). Finally, studies of several conjugation systems have provided compelling evidence that relaxases interact directly with their cognate T4CPs (60–62). Taken together, therefore, our data strongly indicate that VirD2 mediates DNA substrate docking with VirD4 via its C terminus and that this interaction is essential for activation of VirB10.
In general, relaxosome docking with the T4CP is considered important for coupling of the oriT nicking, T-strand-unwinding, and translocation reactions in space and time (3). In A. tumefaciens, it is conceivable that relaxosomes of the RSF1010 and CloDF13 plasmid substrates engage with VirD4 for coupled processing and translocation. However, the findings that A. tumefaciens cells accumulate multiple copies of the VirD2–T-strand transfer intermediate per Ti plasmid (Fig. 2) (43) and, furthermore, that the ParA-like accessory factor VirC1 contributes to the recruitment of free VirD2–T-strand complexes to VirD4 (43) suggest that the initiating signal for T-DNA transfer is probably not the relaxosome docking reaction per se but rather engagement of the transfer intermediate with VirD4. Whether other accessory factors involved in the substrate recruitment reaction, e.g., ParA-like VirC1 or factors designated Vbp's (43, 63), might also contribute to substrate activation of the VirB/VirD4 channel remains an intriguing question for further investigation.
We were initially surprised that ligand-induced activation of VirB10 is a property of DNA and not protein transfer through the T4SS. In fact, it is possible that both substrates induce structural changes in the channel but that our protease susceptibility assay detects only a DNA ligand-induced change. However, monomeric proteins differ appreciably as substrates compared with relaxase–single-stranded DNA (ssDNA) particles, and it is reasonable to expect that different mechanisms exist for their translocation. With regard to DNA translocation, recently it was reported that the R388-encoded TrwB is a DNA-dependent ATPase whose catalytic activity and oligomeric state are also modulated by the Dtr accessory factor TrwA (64, 65). These findings raise the intriguing possibility of an ordered reaction whereby docking of the relaxosome or transfer intermediate stimulates ATP hydrolysis activity, which induces a change in conformation or oligomeric state in the T4CP and, in turn, a conformational change in VirB10. The conformational switch in VirB10 might confer an open-channel configuration required for passage of long ssDNA polymers. It should also be noted that, whereas the DNA transfer pathway was defined by TrIP (17), the route of protein translocation through the T4SS is completely unknown, and different models have been proposed (18, 19, 66). Of particular interest is the shoot-and-pump model, which posits that DNA and protein substrates are delivered across the membrane via two different translocases, DNA through the lumen of the T4CP hexamer and protein monomers through an Mpf-encoded translocase, e.g., VirB6/VirB8 (66).
Our mutational analyses established the importance of VirD4's periplasmic domain for both early-stage DNA transfer reactions and VirB10 activation (Fig. 4). VirD4 interacts with VirB10 (18, 52), and homologs of these subunits encoded by the R388 and R27 (IncH) plasmid transfer systems also have been shown to interact via their N-terminal regions (53, 54, 67). These findings support a proposal that periplasmic domains of VirD4 and VirB10 interact directly for coupling of ATP energy with the conformational switch; if so, the periplasmic domain mutations might exert their effects on energy activation by disrupting this interaction. Why disruption of a VirD4-VirB10 interaction would impact early-stage DNA transfer from VirD4 to VirB11 or the VirB6/VirB8 channel subunits is not immediately obvious. However, previously we showed that synthesis of VirB10 and other core complex subunits is necessary even for the VirD4 to VirB11 transfer step (18), implying that core complex interactions with VirD4 and/or VirB11 are essential for establishment of productive contacts between the ATPases themselves. The VirD4 periplasmic domain mutations might disrupt a T4CP-VirB10 interaction necessary for formation or stabilization of the presumptive core/VirD4/VirB11 ternary complex. Finally, although we favor the notion that the periplasmic domain mutations disrupt a VirD4-VirB10 binding partner interface, we cannot exclude the possibility that the mutations exert their effects indirectly through global disruption of VirD4's conformational or oligomeric state. Extensive biochemical studies by the de la Cruz and Alkorta groups showing the importance of TrwB's N-terminal TMD to its activity, structure, and stability are consistent with such a possibility (68–72).
The VirB11 family members are structurally dynamic hexamers that undergo major conformational changes upon ATP binding (73–75). Such ATP-induced conformational changes do not seem to play a role in DNA substrate transfer from VirD4 to VirB11, since a VirB11 Walker A mutant does not disrupt this transfer step (18). However, phenotypes of the ATP binding site mutations clearly establish that ATP binding/hydrolysis is required for the VirB10 structural transition (Fig. 5) (21). Additionally, we found that the capacity of VirB11 to interact with substrate DNA was strictly correlated with activation of VirB10 (Fig. 5 to 7). This correlation held true in our characterization of a large collection of substitution and i4 mutations but was best illustrated by analyses of the substrate discrimination mutations (Fig. 7). In the absence of the IncQ plasmid substrate, the discrimination mutations blocked both T-DNA transfer from VirD4 to VirB11 and the VirB10 structural transition. In the presence of the IncQ plasmid substrate, the mutations still blocked T-DNA transfer but permitted translocation of the IncQ plasmid, and in this genetic context the VirB11 mutant proteins supported activation of VirB10. These phenotypes strongly indicate that engagement of the DNA substrate with VirB11 is essential for activation of VirB10 and that the substrate discrimination mutations selectively block VirB11 from interacting productively with the T-DNA substrate. How VirB11 discriminates between substrates is unknown, but the mechanism could involve reported differences in the translocation signals carried by the VirD2 and MobA relaxase components of the T-DNA and RSF1010 transfer intermediates (46, 76) or accessory factors shown to be important for substrate processing or recruitment to the T4SS, e.g., VirD1 or ParA-like VirC1 for T-DNA or MobB for RSF1010 (43, 76).
We located the VirB11 mutations on a structural model derived from the Brucella suis VirB11 crystal structure (74). B. suis VirB11 is considered a reliable structural prototype for the VirB11 family of ATPases (see Fig. S2 in the supplemental material) (74). The B. suis VirB11 monomer consists of N-terminal and C-terminal domains (designated NTD and CTD) joined by a linker region. The protein assembles as a hexameric double-ring structure, with each ring formed by extensive interactions among adjacent NTDs and CTDs, respectively. The hexamer is further stabilized by a domain swap in which the linker region of one monomer fits into a groove of the CTD in an adjacent monomer. Not surprisingly, the permissive mutations generally mapped within nonstructured loops, whereas the nonpermissive mutations mapped in the ATP binding pocket or in structural folds in the NTD or CTD or linker region (see Fig. S3A and B in the supplemental material). Most of the nonpermissive mutations can therefore be expected to disrupt assembly or conformational flexibility of the hexamer, whereas mutations on the surface or in the lumen of the hexamer could directly interfere with VirB11 binding to VirD4, another partner protein, or substrate. The substrate discrimination mutations are located near the Walker A motif (C168.i4), within the lumen of the hexamer (L302.i4), or within the NTD (see Fig. S3B). In view of these structural assignments, it is intriguing to speculate that a combination of ATP-mediated conformational changes and the central lumen of the VirB11 hexamer contribute directly to the recognition and processing of DNA substrates for delivery through the inner membrane translocase.
Finally, it is interesting to note that the DNA ligand and ATP energy signals characterized in this study, while necessary, are not sufficient for channel activation and substrate transfer. In the 1970s, studies of the F transfer system supplied evidence that an extracellular signal, generated upon formation of pilus-mediated contact with a recipient cell, stimulated early DNA processing reactions in the donor cell (77). The nature of this contact-mediated signal remains unidentified, but in view of our present findings and evidence that VirB10-like proteins span the entire cell envelope, it is enticing to suggest that such a signal is transmitted across the donor cell envelope via the VirB10 sensor subunit. Zechner and her colleagues also recently determined that bacteriophage R17, whose receptor is the F-like R1-encoded pilus, enters the cell only if the relaxosome is docked with a catalytically active form of the TraD T4CP (78). Phage R17 binding appears to supply a requisite extracellular signal, possibly by mimicking recipient cell contact, which together with the DNA substrate-T4CP docking signal activates the channel to allow phage uptake. Thus, a combination of intracellular signals and a contact-mediated extracellular signal might generally activate T4SS channels via conformational effects on VirB10-like subunits for transmission of nucleic acid substrates in either direction across the cell envelope.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Christie laboratory for helpful discussions.
These studies were supported by NIH GM48476.
Footnotes
Published ahead of print 5 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00114-13.
REFERENCES
- 1. Alvarez-Martinez CE, Christie PJ. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 73:775–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de la Cruz F, Frost LS, Meyer RJ, Zechner EL. 2010. Conjugative DNA metabolism in Gram-negative bacteria. FEMS Microbiol. Rev. 34:18–40 [DOI] [PubMed] [Google Scholar]
- 3. Zechner EL, Lang S, Schildbach JF. 2012. Assembly and mechanisms of bacterial type IV secretion machines. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:1073–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong JJ, Lu J, Glover JN. 2012. Relaxosome function and conjugation regulation in F-like plasmids—a structural biology perspective. Mol. Microbiol. 85:602–617 [DOI] [PubMed] [Google Scholar]
- 5. Gomis-Ruth FX, de la Cruz F, Coll M. 2002. Structure and role of coupling proteins in conjugal DNA transfer. Res. Microbiol. 153:199–204 [DOI] [PubMed] [Google Scholar]
- 6. Schroder G, Lanka E. 2005. The mating pair formation system of conjugative plasmids—a versatile secretion machinery for transfer of proteins and DNA. Plasmid 54:1–25 [DOI] [PubMed] [Google Scholar]
- 7. Wallden K, Rivera-Calzada A, Waksman G. 2010. Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 12:1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomis-Ruth FX, Coll M. 2001. Structure of TrwB, a gatekeeper in bacterial conjugation. Int. J. Biochem. Cell Biol. 33:839–843 [DOI] [PubMed] [Google Scholar]
- 9. Hormaeche I, Alkorta I, Moro F, Valpuesta JM, Goni FM, De La Cruz F. 2002. Purification and properties of TrwB, a hexameric, ATP-binding integral membrane protein essential for R388 plasmid conjugation. J. Biol. Chem. 277:46456–46462 [DOI] [PubMed] [Google Scholar]
- 10. Gomis-Ruth FX, Sola M, de la Cruz F, Coll M. 2004. Coupling factors in macromolecular type-IV secretion machineries. Curr. Pharm. Des. 10:1551–1565 [DOI] [PubMed] [Google Scholar]
- 11. Fronzes R, Christie PJ, Waksman G. 2009. The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7:703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fronzes R, Schafer E, Wang L, Saibil HR, Orlova EV, Waksman G. 2009. Structure of a type IV secretion system core complex. Science 323:266–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G. 2009. Structure of the outer membrane complex of a type IV secretion system. Nature 462:1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sarkar MM, Husnain SI, Jakubowski SJ, Christie PJ. 2013. Isolation of bacterial type IV machine subassemblies, p 187–204 In Delcour A. (ed), The bacterial cell surface: methods and protocols, methods in molecular biology. Humana Press, Inc., New York, NY: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Christie PJ. 2009. Structural biology: translocation chamber's secrets. Nature 462:992–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cascales E, Christie PJ. 2004. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science 304:1170–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atmakuri K, Cascales E, Christie PJ. 2004. Energetic components VirD4, VirB11 and VirB4 mediate early DNA transfer reactions required for bacterial type IV secretion. Mol. Microbiol. 54:1199–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jakubowski SJ, Krishnamoorthy V, Cascales E, Christie PJ. 2004. Agrobacterium tumefaciens VirB6 domains direct the ordered export of a DNA substrate through a type IV secretion system. J. Mol. Biol. 341:961–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thanassi DG, Bliska JB, Christie PJ. 2012. Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol. Rev. 36:1046–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cascales E, Christie PJ. 2004. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc. Natl. Acad. Sci. U. S. A. 101:17228–17233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Banta LM, Kerr JE, Cascales E, Giuliano ME, Bailey ME, McKay C, Chandran V, Waksman G, Christie PJ. 2011. An Agrobacterium VirB10 mutation conferring a type IV secretion system gating defect. J. Bacteriol. 193:2566–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu J, Oger PM, Schrammeijer B, Hooykaas PJ, Farrand SK, Winans SC. 2000. The bases of crown gall tumorigenesis. J. Bacteriol. 182:3885–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Larebeke N, Genetello C, Schell J, Schilperoort RA, Hermans AK, Van Montagu M, Hernalsteens JP. 1975. Acquisition of tumour-inducing ability by non-oncogenic agrobacteria as a result of plasmid transfer. Nature 255:742–743 [DOI] [PubMed] [Google Scholar]
- 25. McBride KE, Knauf VC. 1988. Genetic analysis of the virE operon of the Agrobacterium Ti plasmid pTiA6. J. Bacteriol. 170:1430–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fullner KJ, Lara JC, Nester EW. 1996. Pilus assembly by Agrobacterium T-DNA transfer genes. Science 273:1107–1109 [DOI] [PubMed] [Google Scholar]
- 27. Stachel SE, An G, Flores C, Nester EW. 1985. A Tn3 lacZ transposon for the random generation of beta-galactosidase gene fusions: application to the analysis of gene expression in Agrobacterium. EMBO J. 4:891–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ooms G, Hooykaas PJJ, Van Veen RJM, Van Beelen P, Regensburg-Tunik R, Schilperoort RA. 1982. Octopine Ti-plasmid deletion mutants of Agrobacterium tumefaciens with emphasis on the right side of the T-region. Plasmid 7:15–19 [DOI] [PubMed] [Google Scholar]
- 29. Berger BR, Christie PJ. 1994. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J. Bacteriol. 176:3646–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou XR, Christie PJ. 1997. Suppression of mutant phenotypes of the Agrobacterium tumefaciens VirB11 ATPase by overproduction of VirB proteins. J. Bacteriol. 179:5835–5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee LY, Gelvin SB. 2008. T-DNA binary vectors and systems. Plant Physiol. 146:325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cascales E, Atmakuri K, Liu Z, Binns AN, Christie PJ. 2005. Agrobacterium tumefaciens oncogenic suppressors inhibit T-DNA and VirE2 protein substrate binding to the VirD4 coupling protein. Mol. Microbiol. 58:565–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rashkova S, Spudich GM, Christie PJ. 1997. Characterization of membrane and protein interaction determinants of the Agrobacterium tumefaciens VirB11 ATPase. J. Bacteriol. 179:583–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fullner KJ, Nester EW. 1996. Temperature affects the T-DNA transfer machinery of Agrobacterium tumefaciens. J. Bacteriol. 178:1498–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beaupre CE, Bohne J, Dale EM, Binns AN. 1997. Interactions between VirB9 and VirB10 membrane proteins involved in movement of DNA from Agrobacterium tumefaciens into plant cells. J. Bacteriol. 179:78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stahl LE, Jacobs A, Binns AN. 1998. The conjugal intermediate of plasmid RSF1010 inhibits Agrobacterium tumefaciens virulence and VirB-dependent export of VirE2. J. Bacteriol. 180:3933–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Escudero J, Den Dulk-Ras A, Regensburg-Tuink TJ, Hooykaas PJ. 2003. VirD4-independent transformation by CloDF13 evidences an unknown factor required for the genetic colonization of plants via Agrobacterium. Mol. Microbiol. 47:891–901 [DOI] [PubMed] [Google Scholar]
- 38. Yanofsky MF, Porter SG, Young C, Albright LM, Gordon MP, Nester EW. 1986. The virD operon of Agrobacterium tumefaciens encodes a site-specific endonuclease. Cell 47:471–477 [DOI] [PubMed] [Google Scholar]
- 39. Atmakuri K, Ding Z, Christie PJ. 2003. VirE2, a type IV secretion substrate, interacts with the VirD4 transfer protein at cell poles of Agrobacterium tumefaciens. Mol. Microbiol. 49:1699–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jakubowski SJ, Krishnamoorthy V, Christie PJ. 2003. Agrobacterium tumefaciens VirB6 protein participates in formation of VirB7 and VirB9 complexes required for type IV secretion. J. Bacteriol. 185:2867–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sagulenko E, Sagulenko V, Chen J, Christie PJ. 2001. Role of Agrobacterium VirB11 ATPase in T-pilus assembly and substrate selection. J. Bacteriol. 183:5813–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou X-R, Christie PJ. 1999. Mutagenesis of Agrobacterium VirE2 single-stranded DNA-binding protein identifies regions required for self-association and interaction with VirE1 and a permissive site for hybrid protein construction. J. Bacteriol. 181:4342–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Atmakuri K, Cascales E, Burton OT, Banta LM, Christie PJ. 2007. Agrobacterium ParA/MinD-like VirC1 spatially coordinates early conjugative DNA transfer reactions. EMBO J. 26:2540–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jakubowski SJ, Cascales E, Krishnamoorthy V, Christie PJ. 2005. Agrobacterium tumefaciens VirB9, an outer-membrane-associated component of a type IV secretion system, regulates substrate selection and T-pilus biogenesis. J. Bacteriol. 187:3486–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vergunst AC, van Lier MC, den Dulk-Ras A, Grosse Stuve TA, Ouwehand A, Hooykaas PJ. 2005. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc. Natl. Acad. Sci. U. S. A. 102:832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Kregten M, Lindhout BI, Hooykaas PJ, van der Zaal BJ. 2009. Agrobacterium-mediated T-DNA transfer and integration by minimal VirD2 consisting of the relaxase domain and a type IV secretion system translocation signal. Mol. Plant Microbe Interact. 22:1356–1365 [DOI] [PubMed] [Google Scholar]
- 47. Vergunst AC, Schrammeijer B, den Dulk-Ras A, de Vlaam CM, Regensburg-Tuink TJ, Hooykaas PJ. 2000. VirB/D4-dependent protein translocation from Agrobacterium into plant cells. Science 290:979–982 [DOI] [PubMed] [Google Scholar]
- 48. Schrammeijer B, Dulk-Ras Ad A, Vergunst AC, Jurado Jacome E, Hooykaas PJ. 2003. Analysis of Vir protein translocation from Agrobacterium tumefaciens using Saccharomyces cerevisiae as a model: evidence for transport of a novel effector protein VirE3. Nucleic Acids Res. 31:860–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shurvinton CE, Hodges L, Ream W. 1992. A nuclear localization signal and the C-terminal omega sequence in the Agrobacterium tumefaciens VirD2 endonuclease are important for tumor formation. Proc. Natl. Acad. Sci. U. S. A. 89:11837–11841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fullner KJ. 1998. Role of Agrobacterium virB genes in transfer of T complexes and RSF1010. J. Bacteriol. 180:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Das A, Xie YH. 1998. Construction of transposon Tn3phoA: its application in defining the membrane topology of the Agrobacterium tumefaciens DNA transfer proteins. Mol. Microbiol. 27:405–414 [DOI] [PubMed] [Google Scholar]
- 52. Jakubowski SJ, Kerr JE, Garza I, Krishnamoorthy V, Bayliss R, Waksman G, Christie PJ. 2009. Agrobacterium VirB10 domain requirements for type IV secretion and T pilus biogenesis. Mol. Microbiol. 71:779–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gilmour MW, Gunton JE, Lawley TD, Taylor DE. 2003. Interaction between the IncHI1 plasmid R27 coupling protein and type IV secretion system: TraG associates with the coiled-coil mating pair formation protein TrhB. Mol. Microbiol. 49:105–116 [DOI] [PubMed] [Google Scholar]
- 54. Llosa M, Zunzunegui S, de la Cruz F. 2003. Conjugative coupling proteins interact with cognate and heterologous VirB10-like proteins while exhibiting specificity for cognate relaxosomes. Proc. Natl. Acad. Sci. U. S. A. 100:10465–10470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lai EM, Chesnokova O, Banta LM, Kado CI. 2000. Genetic and environmental factors affecting T-pilin export and T-pilus biogenesis in relation to flagellation of Agrobacterium tumefaciens. J. Bacteriol. 182:3705–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Merianos HJ, Cadieux N, Lin CH, Kadner RJ, Cafiso DS. 2000. Substrate-induced exposure of an energy-coupling motif of a membrane transporter. Nat. Struct. Biol. 7:205–209 [DOI] [PubMed] [Google Scholar]
- 57. Fanucci GE, Coggshall KA, Cadieux N, Kim M, Kadner RJ, Cafiso DS. 2003. Substrate-induced conformational changes of the periplasmic N terminus of an outer-membrane transporter by site-directed spin labeling. Biochemistry 42:1391–1400 [DOI] [PubMed] [Google Scholar]
- 58. Letoffe S, Delepelaire P, Wandersman C. 1996. Protein secretion in Gram-negative bacteria: assembly of the three components of ABC protein-mediated exporters is ordered and promoted by substrate binding. EMBO J. 15:5804–5811 [PMC free article] [PubMed] [Google Scholar]
- 59. Thanabalu T, Koronakis E, Hughes C, Koronakis V. 1998. Substrate-induced assembly of a contiguous channel for protein export from E. coli: reversible bridging of an inner-membrane translocase to an outer membrane exit pore. EMBO J. 17:6487–6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szpirer CY, Faelen M, Couturier M. 2000. Interaction between the RP4 coupling protein TraG and the pBHR1 mobilization protein Mob. Mol. Microbiol. 37:1283–1292 [DOI] [PubMed] [Google Scholar]
- 61. Thomas J, Hecht DW. 2007. Interaction of Bacteroides fragilis pLV22a relaxase and transfer DNA with Escherichia coli RP4-TraG coupling protein. Mol. Microbiol. 66:948–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen Y, Zhang X, Manias D, Yeo HJ, Dunny GM, Christie PJ. 2008. Enterococcus faecalis PcfC, a spatially localized substrate receptor for type IV secretion of the pCF10 transfer intermediate. J. Bacteriol. 190:3632–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guo M, Jin S, Sun D, Hew CL, Pan SQ. 2007. Recruitment of conjugative DNA transfer substrate to Agrobacterium type IV secretion apparatus. Proc. Natl. Acad. Sci. U. S. A. 104:20019–20024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tato I, Zunzunegui S, de la Cruz F, Cabezon E. 2005. TrwB, the coupling protein involved in DNA transport during bacterial conjugation, is a DNA-dependent ATPase. Proc. Natl. Acad. Sci. U. S. A. 102:8156–8161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tato I, Matilla I, Arechaga I, Zunzunegui S, de la Cruz F, Cabezon E. 2007. The ATPase activity of the DNA transporter TrwB is modulated by protein TrwA: implications for a common assembly mechanism of DNA translocating motors. J. Biol. Chem. 282:25569–22576 [DOI] [PubMed] [Google Scholar]
- 66. Llosa M, Gomis-Ruth FX, Coll M, de la Cruz F. 2002. Bacterial conjugation: a two-step mechanism for DNA transport. Mol. Microbiol. 45:1–8 [DOI] [PubMed] [Google Scholar]
- 67. de Paz HD, Sangari FJ, Bolland S, Garcia-Lobo JM, Dehio C, de la Cruz F, Llosa M. 2005. Functional interactions between type IV secretion systems involved in DNA transfer and virulence. Microbiology 151:3505–3516 [DOI] [PubMed] [Google Scholar]
- 68. Hormaeche I, Iloro I, Arrondo JL, Goni FM, de la Cruz F, Alkorta I. 2004. Role of the transmembrane domain in the stability of TrwB, an integral protein involved in bacterial conjugation. J. Biol. Chem. 279:10955–10961 [DOI] [PubMed] [Google Scholar]
- 69. Vecino AJ, Segura RL, Ugarte-Uribe B, Aguila S, Hormaeche I, de la Cruz F, Goni FM, Alkorta I. 2010. Reconstitution in liposome bilayers enhances nucleotide binding affinity and ATP-specificity of TrwB conjugative coupling protein. Biochim. Biophys. Acta 1798:2160–2169 [DOI] [PubMed] [Google Scholar]
- 70. Vecino AJ, de la Arada I, Segura RL, Goni FM, de la Cruz F, Arrondo JL, Alkorta I. 2011. Membrane insertion stabilizes the structure of TrwB, the R388 conjugative plasmid coupling protein. Biochim. Biophys. Acta 1808:1032–1039 [DOI] [PubMed] [Google Scholar]
- 71. Vecino AJ, Segura Rde L, de la Arada I, de la Cruz F, Goni FM, Arrondo JL, Alkorta I. 2012. Deletion of a single helix from the transmembrane domain causes large changes in membrane insertion properties and secondary structure of the bacterial conjugation protein TrwB. Biochim. Biophys. Acta 1818:3158–3166 [DOI] [PubMed] [Google Scholar]
- 72. Gomis-Ruth FX, Moncalian G, Perez-Luque R, Gonzalez A, Cabezon E, de la Cruz F, Coll M. 2001. The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature 409:637–641 [DOI] [PubMed] [Google Scholar]
- 73. Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E, Buhrdorf R, Fischer W, Haas R, Waksman G. 2003. VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO J. 22:1969–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hare S, Bayliss R, Baron C, Waksman G. 2006. A large domain swap in the VirB11 ATPase of Brucella suis leaves the hexameric assembly intact. J. Mol. Biol. 360:56–66 [DOI] [PubMed] [Google Scholar]
- 75. Savvides SN. 2007. Secretion superfamily ATPases swing big. Structure 15:255–257 [DOI] [PubMed] [Google Scholar]
- 76. Parker C, Meyer RJ. 2007. The R1162 relaxase/primase contains two, type IV transport signals that require the small plasmid protein MobB. Mol. Microbiol. 66:252–261 [DOI] [PubMed] [Google Scholar]
- 77. Kingsman A, Willetts N. 1978. The requirements for conjugal DNA synthesis in the donor strain during flac transfer. J. Mol. Biol. 122:287–300 [DOI] [PubMed] [Google Scholar]
- 78. Lang S, Kirchberger PC, Gruber CJ, Redzej A, Raffl S, Zellnig G, Zangger K, Zechner EL. 2011. An activation domain of plasmid R1 TraI protein delineates stages of gene transfer initiation. Mol. Microbiol. 82:1071–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.