

Abstract
The adaptation of microorganisms to their environment is controlled by complex transcriptional regulatory networks (TRNs), which are still only partially understood even for model species. Genome scale annotation of regulatory features of genes and TRN reconstruction are challenging tasks of microbial genomics. We used the knowledge-driven comparative-genomics approach implemented in the RegPredict Web server to infer TRN in the model Gram-positive bacterium Bacillus subtilis and 10 related Bacillales species. For transcription factor (TF) regulons, we combined the available information from the DBTBS database and the literature with bioinformatics tools, allowing inference of TF binding sites (TFBSs), comparative analysis of the genomic context of predicted TFBSs, functional assignment of target genes, and effector prediction. For RNA regulons, we used known RNA regulatory motifs collected in the Rfam database to scan genomes and analyze the genomic context of new RNA sites. The inferred TRN in B. subtilis comprises regulons for 129 TFs and 24 regulatory RNA families. First, we analyzed 66 TF regulons with previously known TFBSs in B. subtilis and projected them to other Bacillales genomes, resulting in refinement of TFBS motifs and identification of novel regulon members. Second, we inferred motifs and described regulons for 28 experimentally studied TFs with previously unknown TFBSs. Third, we discovered novel motifs and reconstructed regulons for 36 previously uncharacterized TFs. The inferred collection of regulons is available in the RegPrecise database (http://regprecise.lbl.gov/) and can be used in genetic experiments, metabolic modeling, and evolutionary analysis.
INTRODUCTION
Transcription regulation is one of the main mechanisms in prokaryotes for quickly switching their metabolism in changing environments. Bacteria use two major mechanisms to control target gene expression. First, the most common mechanism is switching transcription levels via proteins called transcription factors (TFs) that can specifically recognize TF binding sites (TFBSs) in response to different intracellular or environmental conditions (1). Second, sequence-specific RNA regulatory elements located in noncoding upstream gene regions are able to respond to intracellular metabolites and control the expression of downstream genes (2). Both mechanisms result in either repression or activation of target genes. A set of genes directly controlled by the same TF (or by RNA elements from the same structural family) are considered to belong to a regulon. All regulons together in the same organism form the transcription regulatory network (TRN).
A TRN is usually represented as a graph in which nodes represent genes and edges represent regulatory interactions. A general topology of microbial TRNs can be presented as a network in which a few global TFs regulate a large portion of the genes and the majority of local TFs regulate a small number of operons. However, despite the accumulated knowledge about microbial TRNs, it is still a major challenge to identify the complete TRN in an individual organism. Traditional experimental techniques for studying transcriptional regulation, such as DNase I footprinting, electromobility shift assays, and beta-galactosidase fusion assays, have limitations in productivity and are restricted to a few model organisms (3). High-throughput experimental techniques, such as the chromatic immunoprecipitation approach, the genomic SELEX, and microarray technology, have been successfully used to explore transcriptional responses of thousands of genes in several bacteria. However, for these techniques, it is necessary to determine the conditions under which the studied TFs are active. Also, regulatory cascades, coregulation, and other indirect effects on regulation create noise that makes directly observed regulatory responses too complex for analysis (4, 5).
The recent availability of a large number of complete genomes promoted the development of new computational approaches for TRN reconstruction from genomic data (6). The template-based methods rely on the assumption that orthologous TFs maintain regulation of orthologous target genes. Thus, a TRN in a new organism is obtained by simple propagation of TF target gene pairs from known TRNs. However, this approach cannot predict new TFBSs or check the conservation of binding sites for orthologous genes (7–9). The expression data-driven approaches are used to infer TRNs from sets of RNA expression measurements in cells grown under different conditions (10). The computation-driven approach allows identification and clustering of conserved cis-regulatory DNA motifs; however, the assignment of such motifs to particular TFs remains a challenge (11–13). Finally, the knowledge-driven comparative-genomics approach (14), which uses a combination of the motif identification computational algorithms, the genome context techniques, and available experimental data from the literature, was successfully applied to the analysis of various regulatory systems in several groups of microorganisms (15, 16). The combined reconstruction of certain metabolic pathways and transcriptional regulons was previously used to infer regulons involved in the central metabolism of sugars, amino acids, nucleotides, metals, and cofactors and resulted in the prediction of numerous novel functions for previously uncharacterized genes (17–19).
The Gram-positive bacterium Bacillus subtilis is an important model for studying the sporulation, cell differentiation, stress response, and social behavior of bacteria. B. subtilis is most commonly found in soil environments, where it is associated with decaying organic material or plant roots (20). Also, B. subtilis can live in the gastrointestinal tract of animals (21). As a model organism, B. subtilis has been intensively studied, resulting in the characterization of numerous transcriptional factors and regulons for central metabolic pathways and cellular processes (22, 23). The DBTBS database (23) accumulates the experimental knowledge on transcriptional regulation in B. subtilis, including information on 120 TFs, their cognate binding sites, and target genes. In addition to TFs, at least 4% of B. subtilis genes are controlled via cis-regulatory RNA elements, such as T boxes and other metabolite-sensing riboswitches (2). Despite such a large amount of data on regulatory mechanisms involving genes of the central metabolism and stress response pathways, many TF regulons in B. subtilis have been insufficiently studied, providing an incomplete knowledge of the range of target genes and associated TFBSs. For instance, nearly 20% of the experimentally studied TFs lack information on their cognate binding sites. Moreover, previously studied regulators constitute nearly half of the estimated total number of TFs in B. subtilis (24).
A few attempts have been made to infer B. subtilis regulons by bioinformatics approaches. In one study, phylogenetically conserved DNA motifs were identified in the genomes of B. subtilis, B. halodurans, and B. stearothermophilus and further clustered to predict putative regulons; however, the identities of TFs that potentially bind to these motifs remained unknown (25). In another study, the integration of expression data analysis with identification of common DNA motifs was used to construct a regulatory network for 27 TFs and 8 sigma factors (26). In the present work, we applied the knowledge-driven comparative-genomics approach and available genomes from the order Bacillales to infer and capture the largest transcriptional network in B. subtilis, which includes 129 TF regulons and 24 regulons controlled by regulatory RNAs. These include 28 TF regulons with previously unknown DNA binding motifs and 36 novel regulons for previously uncharacterized TFs. For each reconstructed regulon, we tentatively predicted both target genes/operons and cognate TF binding sites. The updated TRN of B. subtilis can be used for experimental research on transcriptional regulation and metabolic modeling.
MATERIALS AND METHODS
Eleven complete genomes of bacteria of the order Bacillales were downloaded from MicrobesOnline (27) (Fig. 1). The phylogenetic tree for the studied Bacillales species was built using the 16S-23S RNA concatenate sequence by the maximum-likelihood algorithm implemented in the PHYLYP package (28).
Fig 1.

Phylogenetic tree and distribution of TFs in the studied bacteria from the order Bacillales. The hatched bars show total numbers of TFs per genome. The crosshatched bars show numbers of orthologs of B. subtilis TFs. The tree was built using phylogenetic analysis of the concatenated 16S-23S rRNA sequences from the studied genomes.
TF repertoire.
A set of B. subtilis TFs was created by analysis of corresponding collections in the DBD (29), MiST2 (30), and DBTBS (23) databases, as well as using the previously determined B. subtilis TF repertoire (24). Sigma factors and RNA binding proteins were excluded from this collection. Orthologous TFs in Bacillales spp. were identified by the bidirectional best-hit criterion with a 30% identity threshold using the Smith-Waterman algorithm implemented in the GenomeExplorer software (31) and confirmed via phylogenetic genomic context analysis in the MicrobesOnline tree browser (27). TF families were assigned using known TF domain architectures from the CDD (32), Pfam (33), and P2TF (34) databases.
TF regulon reconstruction.
For identification of TFBSs and regulon reconstruction, we used the comparative-genomics approach (14) implemented in the RegPredict Web server (35). RegPredict allows the simultaneous analysis of multiple microbial genomes and integrates information on gene orthologs, operon predictions, and functional gene annotations. We used three major workflows for TF regulon inference: (i) propagation of previously known regulons with known DNA binding motifs to all studied species, (ii) propagation of previously known regulons with unknown DNA binding motifs to all studied species, and (iii) ab initio prediction of novel regulons. The three workflows differ in the step of training set collection. For previously known regulons, we collected known TFBSs from the DBTBS database (23) and from the literature. To identify novel TFBS motifs, we collected sets of upstream regions for potential target genes and their orthologs in organisms possessing the studied TF. Potential target genes for novel regulons were identified via genomic context analysis of a TF gene and its orthologs using MicrobesOnline (27). This method is based on the observations that functionally related genes tend to cluster on chromosomes (36) and that TFs often control genes located nearby on the chromosome (e.g., TFs are often autoregulated). To infer TFBS motifs, we used the Discover Profile tool in RegPredict (35) and confirmed the predicted motifs by multiple alignments of orthologous upstream DNA regions by the MUSCLE algorithm (37).
Regulons for the inferred TF DNA motifs were reconstructed using the RegPredict Web server (35) as previously described (16). Briefly, each genome was scanned with the constructed DNA motif, and genes with candidate regulatory sites in upstream regions were selected. Each predicted regulatory interaction was analyzed for conservation across the Bacillales genomes using the Clusters of Coregulated Orthologous Operons (CRONs) in RegPredict (35). This approach helps to significantly diminish overprediction rates. In many cases, a gene encoding a TF was located adjacent to the TF-controlled operon in a divergent orientation, thus forming a divergon. Both operons in such divergons were included in the reconstructed TF regulon, since they share the same candidate TFBS. Thus, in the reconstructed regulatory network, a large set of TFs is autoregulated due to this common type of genetic organization of a TF and its target operon.
Biological functions of regulated genes were tentatively predicted in each regulon by a combination of a sequence similarity search against the Swiss-Prot section of the UniProtKB database (27), domain architecture analysis (38), and using functional gene annotations from the SEED (39) and KEGG (40) databases.
RNA element regulon reconstruction.
RNA regulatory elements (riboswitches) were identified using the probabilistic covariance models, which include both RNA secondary structure and sequence consensus (38). The covariance models of riboswitches were taken from the Rfam database (41) and used to scan genomes with the Infernal program (42). The identified candidate RNA regulatory sites were uploaded into the RegPredict Web server (35), and the respective RNA regulons were reconstructed using the same approach as for TF regulons.
RESULTS AND DISCUSSION
Repertoire of transcription factors in Bacillales.
In order to reconstruct the B. subtilis regulons operated by TFs, we collected the entire repertoire of known and putative TFs encoded in its genome (see Table S1 in the supplemental material). Among 258 TFs identified in B. subtilis, 114 regulators were studied experimentally according to the DBTBS database and the literature. Within the latter group of experimentally studied TFs, 82 regulators have information on their cognate TFBSs, whereas DNA binding sites for the remaining 32 regulators have not been characterized. Analysis of the distribution of B. subtilis TFs by protein families showed that 60% of regulators belong to 10 major families (MarR, TetR, LysR, Xre, GntR, OmpR, AraC, LacI, MerR, and LuxR), whereas the remaining 40% of the proteins were assigned to one of 40 other families with a characteristic DNA binding domain.
To analyze conservation of B. subtilis TFs, we searched for their orthologs in 10 complete genomes of Bacillales species selected for comparative analysis (see Table S1 in the supplemental material). As expected, the number of identified TF orthologs decreases with an increase of the phylogenetic distance between B. subtilis and related species (Fig. 1). The largest numbers of orthologous TFs were detected in three closely related species—B. licheniformis (191 TFs), B. amyloliquefaciens (182 TFs), and B. pumilis (156 TFs). We found 19 regulators in B. subtilis that lack orthologs among the studied Bacillales species. Among these species-specific TFs, two stress response regulators, AzbB and Mta, have been characterized while others have no assigned function. The core set of TFs conserved in all studied Bacillales genomes includes 26 regulators that control the metabolism of amino acids and nitrogen (ArgR, CodY, and CymR), carbohydrates (CcpA, CggR, and RbsR), biotin cofactor (BirA), fatty acids (FapR), nucleotides (NrdR and PurR), metal homeostasis (Fur, MntR, and Zur), respiration (ResD and Rex), sporulation (AbrB, Spo0A, SpoIIID, and SpoVT), and stress responses (CsoR, CtsR, HrcA, LexA, LiaR, and PerR), as well as the chromosomal replication initiation regulator DnaA.
Comparative analysis of transcriptional regulation in Bacillales.
For regulon inference in the Bacillales group of genomes, we utilized a comparative-genomic approach implemented in the RegPredict Web server (35). For reconstruction of TF-operated regulons, we used a position weight matrix (PWM)-based approach for inferring potential TFBSs in each analyzed genome and implemented a consistency check approach to verify the predicted regulatory interactions via multispecies comparisons. Depending on the availability of experimental data, we applied one of the following strategies for initial PWM construction: (i) align all known TF binding sites or, if TFBSs are unknown, extract intergenic regions of all known TF-regulated genes (for previously characterized TFs) and (ii) extract intergenic regions of potentially coregulated genes determined through the genome context analysis of a TF gene (for uncharacterized TFs). TFBS motifs were identified, and the respective PWMs were constructed by a pattern recognition program in the RegPredict server. Identification of RNA regulatory elements in the target genomes was performed by using their probabilistic models uploaded from the Rfam database.
As a result, the reconstructed regulatory network in B. subtilis includes 129 regulons operated by TFs and 24 regulons controlled by RNA elements. The reconstructed regulons are contained in the Bacillales collection of regulons in the RegPrecise database (43). A detailed description of the reconstructed transcriptional regulons is provided below and summarized in Tables S2 to S4 in the supplemental material.
(i) Reconstruction of experimentally studied TF regulons.
First, we focused on reconstruction of regulons operated by TFs that were previously experimentally investigated in B. subtilis (see Table S2 in the supplemental material). For this group of TFs, we propagated the previously known regulatory interactions to other Bacillales species and expanded the existing regulons by prediction of new regulon members by the comparative-genomics approach. For 66 regulators in this category, we used available experimental information about their TFBSs in B. subtilis to build initial PWMs and further searched the genomes to identify (i) similar binding sites upstream of orthologous genes in other species and (ii) additional sites in B. subtilis. For the remaining 28 TFs with previously unknown binding sites, we report for the first time the identities of their cognate TFBSs. Most of the discovered TFBS motifs are characterized by a dyad symmetry structure (Table 1), suggesting the bound form of the respective TFs is a dimer.
Table 1.
Novel TF binding DNA motifs discovered in Bacillales genomesa

Novel previously uncharacterized TFs are marked with an asterisk.
As a result, we investigated 93 known TF regulons that control a large variety of cell functions, including stress responses, sporulation, respiration, metal homeostasis, metabolism of fatty acids, and nucleic bases (see Table S3 in the supplemental material). Almost half of the reconstructed regulons (43 TFs) belong to carbohydrate and nitrogen metabolism. The latter functional group of regulators includes three global regulators that sense the nutritional and metabolic status of the cell, namely, CodY, CcpA, and TnrA (Table 2).
Table 2.
New predicted targets for three global regulons of carbohydrate and nitrogen metabolism in B. subtilis
| TF regulon | Predicted target operons |
|---|---|
| TnrA | pucABCDE, pucH, argCJBD-carAB-argF, ureABCEFD, pucI-ywoF, gltC, yuiABC, ydaB, yumC, rapJ, braB, yqzL-recO, ysnE, yclG, yclF, pgsBCAE, yrbD, yvgT |
| CcpA | ylbBC, mrp, yqgQ-glcK, yesOPQRSTUVWXYZ, yhaR, cspD, ywcJ, citH, ganB, cstA, nupC-pdp, ywfI, glcDF, sucCD, ykoM, ylbP, araR, abnB, gatCAB, yqgX, glpTQ, drm-punA, apbA-yllA, pmi, cycB-ganPQ-lacA, ytcPQ, kduID, ysbAB, glsA1-glnT, sdhCAB-ysmA, fruRKA, yqgY, yqgW, msmREFG-melA, ndk, lutRABC, odhAB, sacPA, mtlARFD, xsa, yngIHHBGFE |
| CodY | acuABC, serA, opuE, yuiA, yocS, yuxJ-pbpD-yuxK, ycjHGF, sspOP, oppABCDF, gatCAB, amhX, hom-thrCB, spoIIQ, hpr, spoVS, yoyD-yodF, kapD, spoVG, opuBA-BB-BC-BB, opuCA-CB-CC-CD, kinE-ogt, cueR, yhdT, yocR, murQR-ybbF-amiE-nagZ-ybbC, ytkC, rocR, ybxG, iscS-thiI, msmRE-amyDC-melA, braB, metE2, citR, metIC, citZ-icd-mdh, glnQHMP, yjcL, yjnA, yhaA, phoB, nasDEF, frlBO-yurNM-frlD, gamR, mcpC, yuaE, yuaFG, rocD, cotR, ispA, yusZ, yhjCB, rok, adeC, slp |
TnrA is a global regulator that controls transcription during growth under nitrogen-limited conditions. The predicted candidate TnrA binding sites allowed us to expand the regulon by 18 new genes involved in nitrogen metabolism (Table 2). In particular, we found candidate TnrA sites upstream of B. subtilis genes involved in purine degradation (pucABCDE, pucH, and pucI) and putative amino acid transporters (braB, yclF, and yrbD). For another global nutritional regulator, CodY, that monitors branched-chain amino acids and GTP pools in the cell, we predicted 54 new regulon members in B. subtilis (Table 2). The predicted CodY-regulated genes mostly belong to amino acid metabolism (serA, gom-thrCB, metE2, metIC, frlBO-yurNM, and rocD), amino acid transport (opuE, oppABCDF, ybxG, braB, and glnQHMP), and sporulation (sspOP, spoIIQ, hpr, spoVS, and kinE-ogt). Finally, we found 41 new target operons under the control of the global regulator of carbon metabolism CcpA (Table 2). A large number of newly predicted CcpA-regulated genes are involved in catabolic pathways for various carbon sources, including rhamnogalacturonan (yesOPQRSTUVWXYZ), multiple sugars (msmEFG), nucleosides (drm-punA), galacturonate (kduID), fructose (fruRKA), lactate (lutABC), glycolate (glcCD), and mannitol (mtlARFD). Conserved CcpA sites were found upstream of three operons encoding enzymes of the citric acid cycle—succinate dehydrogenase (sdhCAB), succinyl-coenzyme A (CoA) ligase (sucCD), and 2-oxoglutarate dehydrogenase (odhAB).
Novel potential target operons were also predicted for other previously studied TFs in B. subtilis. The SOS response regulon LexA was expanded by the DNA binding protein HBsu, the sporulation membrane proteins YtrH and YtrI, and the ATP-dependent helicase YqhH. The arginine regulon ArgR (AhrC) was predicted to control the arginine biosynthesis genes argGH and the arginine transport operon artPQM. Among novel members of the ferric uptake regulon Fur, we found two proteins potentially involved in iron utilization—a ferredoxin, Fer, and a ferredoxin reductase, YumC. Similarly, the zinc uptake regulon Zur was expanded by a putative zinc-binding protein ZinT (YrpE). The anaerobic metabolism regulon Fnr was enriched with the lactate utilization operon, ldh-lctP, and a putative formate-nitrite transporter, ywcJ. The biotin biosynthesis regulon BirA includes two additional operons encoding two paralogs of the biotin transporter BioY. The purine regulon PurR was expanded by the serine/threonine permease SteT, which could provide serine as a source of one-carbon fragments for the serine hydroxymethyltransferase GlyA. The lactate utilization regulon LutR includes not only the lactate dehydrogenase operon lutABC, but also the lactate permease gene lutP.
(ii) Inference of novel TF regulons.
Ab initio inference and comparative-genomics analysis of potential regulons for previously uncharacterized TFs allowed us to expand the B. subtilis regulatory network with 36 novel regulons (Table 3). All of these regulons except HisR and NrdR were assigned to a TF by a combination of two types of genomic-context evidence: (i) positional clustering of target genes and TFs on the chromosome and (ii) autoregulation of a TF by a cognate TFBS. Based on the functional roles of regulated genes, the inferred TF regulons can be classified into two major groups that control carbohydrate metabolism (12 regulons) and stress response/drug resistance (15 regulons). In addition, the HisR and NrdR regulons control the metabolism of amino acids and nucleotides, respectively. The functional roles of the remaining 7 hypothetical regulons remain to be determined. Below, we briefly describe the functional content for the selected subset of novel TF regulons.
Table 3.
Contents of novel reconstructed TF regulons in B. subtilis
| TF regulona | Target operon(s) | Functional roleb |
|---|---|---|
| BglR (YydK) | yyzE-bglA, bglR | C |
| BglZ (YkvZ) | bglC, bglZ | C |
| FruR | fruRKA | C |
| GamR (YbgA) | gamAP, gamR | C |
| GlcR | glcR-ywpJ | U |
| HisR (YerC) | hisZGDBHAFI, yuiF | A |
| IolR1 (DegA) | iolX, yrbE | C |
| LytT | lrgAB | R |
| MdxR (YvdE) | mdxD, mdxEFG-malAKL-pgcM | C |
| MsmR | msmREFG-melA | C |
| MurR (YbbH) | murQR-ybbF-amiE-nagZ-ybbC | C |
| NrdR | nrdIEF-ymaB | N |
| RbsR | rbsRKDACB | C |
| RhaR (YulB) | rhaEWRBMA | C |
| RhgR (YesS) | yesOPQ-urhG-rhgR-yesTUVWXYZ | C |
| RmgR (YtdP) | ytePRSTU | C |
| YbzH | ybcL, ybzH | R |
| YcxD | ycxC, ycxD | R |
| YczG | ycnE-nfrA2, yczG | R |
| YdfD-YisV | yisV, ydfD, yisU, ydfC | R |
| YdfF | ydfE, ydfF | U |
| YdfL | ydfK | R |
| YhcF | yhcEFGHI | R |
| YhdI-YdeL | yhdJ, yhdI, ydeK, ydeL | U |
| YhgD | yhgDE | U |
| YisR | yisQ | R |
| YizB | yizB-yitQR | U |
| YrkD | yrkD, yrkEFHIJ | R |
| YtcD | ytbDE, ytcD | U |
| YuaC | gbsAB | R |
| YvbF-YvaV | opuB(ABCD), yvbF, opuC(ABCD), yvaV | R |
| YvfU | yvfRSTU | U |
| YwbI | cidA-ywbG, ywbI | R |
| YwrC | ywrCBA | R |
| YybA | yybA, paiAB, yyaTS | R |
| YybR-YdeP | ydeP, yybR, ydeQ, ppaC, yfkO | R |
For novel regulators that were tentatively named in this work for the first time, the original TF name is given in parentheses.
Functional roles of target genes: A, amino acid metabolism; C, carbon metabolism; T, metabolite transport; N, nucleotide metabolism; O, cofactor metabolism; R, stress responses and drug resistance; U, unknown.
(a) Carbohydrate metabolism.
Carbohydrates are the most extensively used carbon and energy sources for heterotrophic bacteria. In B. subtilis, about 2 dozen regulons are known to be involved in carbohydrate metabolism (44).
Three predicted TF regulons control genes for utilization of rhamnose and rhamnose-containing polymers (Fig. 2). A DeoR-like regulator, YulB (herein named RhaR), is encoded within the putative rhamnose catabolic operon in B. subtilis. A conserved 19-bp tandem-repeat DNA motif identified upstream of the rhamnose operons in the Bacillales was tentatively assigned to the putative rhamnose-responsive regulator RhaR. Previous transcriptional analysis of B. subtilis revealed three gene clusters induced by growth on type I rhamnogalacturonan (45). For two of these gene clusters, yesOPQRSTUVWXYZ and ytePQRST, we inferred unique regulatory motifs of tandem-repeat symmetry and assigned these motifs to the hypothetical AraC family regulators YesS and YtdP encoded within these clusters. Analysis of functional content suggests that both predicted TF regulons play a role in the breakdown and utilization of rhamnogalacturonan, and thus, we renamed these TFs RhgR and RmgR.
Fig 2.

Novel reconstructed regulons for rhamnose metabolism in Bacillales. The genes in each regulated operon are shown by arrows; TF genes are in black, ABC transporters are in dark gray, catabolic enzymes and other genes present in B. subtilis are in light gray, and genes lacking orthologs in B. subtilis are in white. TF binding sites are shown by circles (RhaR) (A), triangles (RhgR) (B), and squares (RmgR) (C). Regulons upregulated by growth on type I rhamnogalacturonan are marked with asterisks.
Phylogenetic distribution of the three reconstructed rhamnose-related regulons differs between Bacillales (Fig. 2). The RhaR and RmgR regulons coexist in five analyzed species, whereas RmgR was also found in B. clausii and B. pumilus, which lack RhaR. The RhgR regulon was found in only three Bacillales genomes, where it coexists with the RhaR and RmgR regulons. The gene content of the reconstructed regulons is only partially conserved between B. subtilis and other analyzed genomes. For instance, the rhamnose utilization regulon RhaR in Oceanobacillus iheyensis includes a new gene encoding a predicted rhamnose permease (herein named RhaY), whereas the bifunctional rhamnose catabolic enzyme RhaEW is replaced by the RhaD aldolase. The only conserved members of the rhamnogalacturonan regulons RhgR and RmgR are two predicted rhamnose oligosaccharide ABC transporters (Fig. 2).
The LacI family regulator DegA (renamed IolR1) was predicted to control various inositol catabolic genes in the Bacillales (Fig. 3). In B. subtilis, the inferred 18-bp palindromic DNA motif coregulates the predicted inositol derivative dehydrogenase gene yrbE and the scyllo-inositol dehydrogenase gene iolX, which is located next to the degA gene. The major myo-inositol catabolic operon is regulated by the DeoR family regulator IolR in most of the studied Bacillus species, including B. subtilis, where it was studied experimentally (46). However, an IolR ortholog is absent from Geobacillus kaustophilus, where the IolR1 regulon is expanded to include the myo-inositol catabolic operon. In B. halodurans, which lacks IolR but has the IolR1 regulon, we predicted a nonorthologous LacI family regulon (herein named IolR2) characterized by a distinct 14-bp palindromic DNA motif that coregulates the inositol catabolic genes. This example illustrates a level of complexity and interchangeability in the regulation of sugar catabolic pathways, even within the same taxonomic group.
Fig 3.

Interconnections between inferred TF regulons for inositol metabolism in the Bacillales. Genes from each regulon are shown by arrows; TF genes are in blue and black, predicted inositol transporters are in pink, and inositol dehydrogenases are in green; orthologous genes are marked by the same letter inside arrows. TF binding sites of IolR, IolR1, and IolR2 are shown by triangles, circles, and squares, respectively. The functional roles of the regulated genes in B. subtilis are as follows: iolS, oxidoreductase; iolA, methylmalonate-semialdehyde dehydrogenase; iolB, 5-deoxy-glucuronate isomerase; iolC, 5-keto-2-deoxygluconokinase; iolD, 3D-(3,5/4)-trihydroxycyclohexane-1,2-dione hydrolase; iolE, inosose dehydratase; iolF, myo-inositol transporter; iolG, myo-inositol 2-dehydrogenase; iolH, putative sugar-phosphate epimerase; iolI, inosose isomerase; iolJ, 5-keto-2-deoxy-d-gluconate-6 phosphate aldolase; iolX, scyllo-inositol 2-dehydrogenase; yrbE, predicted inositol derivative dehydrogenase.
The B. subtilis genome encodes several glycosyl hydrolases that are able to hydrolyze plant-derived aryl β-glucosides (47). We inferred two novel regulons, YydK from the GntR family (herein named BglR) and YkvZ from the LacI family (herein named BglZ), that potentially control expression of two aryl-phospho-β-d-glucosidases, BglA and BglC, respectively. Expression of the bglA gene is induced by the aryl-β-d-glucoside salicin (48), suggesting that BglR could potentially sense salicin 6-phosphate as an effector.
Two novel predicted regulons control amino sugar catabolic pathways. The RpiR family regulator YbbH (herein named MurR) was predicted to bind a 17-bp palindromic DNA motif located in the upstream region of the potential N-acetylmuramic acid utilization operon in B. subtilis and two other bacilli (Tables 2 and 3). The first gene of this operon encodes the MurQ etherase that catalyzes the cleavage of the d-lactyl ether bond of N-acetylmuramic acid 6-phosphate, producing N-acetylglucosamine-6-phosphate and d-lactate. The ybbF-encoded phosphotransferase system (PTS) can potentially feed the pathway by transportation and phosphorylation of exogenous N-acetylmuramic acid. Based on this tentative metabolic reconstruction, we predict that N-acetylmuramate-6-phosphate is the most probable effector molecule for MurR. The predicted glucosamine regulator GamR belongs to the GntR family and is encoded by the ybgA gene, which is located in a divergon with the glucosamine utilization operon gamAP in B. subtilis. Interestingly, orthologous GamR regulons in other Bacillales genomes include genes encoding a chitinase, a putative 6-phospho-β-glucosidase, and a β-glucoside-like PTS system, suggesting their potential involvement in chitin/chitosan utilization (see Table S3 in the supplemental material).
The LacI family TF YvdE (herein named MdxR) was predicted to control the intracellular maltogenic amylase mdxD and the maltodextrin utilization operon mdxEFG-malAKL-pgcM (49). Another LacI family transcriptional regulator, MsmR, controls an operon encoding a potential multiple-sugar ABC transporter (MsmEFG) and cytoplasmic α-galactosidase (MelA). The ribose and fructose utilization operons are controlled by RbsR and FruR regulators from the LacI and DeoR TF families, respectively. Both regulons are extremely conserved in the analyzed genomes, suggesting their functional indispensability for the whole Bacillaceae group.
(b) Amino acid metabolism.
In B. subtilis and other Bacillales, most amino acid biosynthesis and transport pathways (with the exception of the arginine regulon controlled by ArgR and the cysteine regulon controlled by CymR) are controlled at the level of RNA by aminoacyl tRNA-specific T-box regulatory elements (50). However, the histidine biosynthesis operon is not regulated by T boxes, and the respective regulatory mechanism is unknown. Histidine is one of the most expensive amino acids in the cell (51), and thus, it should be expected that its biosynthesis is tightly regulated. We identified a new conserved 20-bp palindromic DNA motif upstream of the histidine biosynthesis his operons in all studied Bacillales genomes except B. cereus (Table 1). A search for additional targets revealed a similar conserved DNA site upstream of the yuiF gene encoding a putative amino acid transporter from the NhaC family. Based on reconstruction of similar histidine regulons in Staphylococcus spp., it was previously proposed that the identified histidine regulatory motif is a binding site of the hypothetical regulator YerC (herein named HisR) (15). The predicted histidine-responsive repressor HisR is homologous to the TrpR family of tryptophan-sensing repressors. The inferred HisR regulon coregulates the histidine biosynthesis his operon and the putative histidine uptake permease yuiF.
(c) Stress responses.
In its natural environment, B. subtilis faces various stresses, most of which are caused by diverse toxic compounds and antibiotics excreted by plants and by other microorganisms. The inferred regulatory network includes 8 novel TF regulons that control putative drug efflux transporters and antibiotic resistance genes (YbzH, YdfD/YisV, YybR/YdeP, YhcF, YdfL, YisR, YcxD, and YwrC). Two reconstructed regulons, YczG and YrkD, include reductases and oxygenases involved in degradation of aromatic and other toxic compounds. The predicted LytTR and YwbI regulons include the LrgAB and CidA proteins, which are known to affect the activities of extracellular murein hydrolases and potentially trigger programmed cell death in response to antibiotics and environmental stresses.
The hypothetical MarR family TF YybA in B. subtilis was predicted to control the paiAB and yyaTS genes that are potentially involved in the polyamine degradation and export pathway. Polyamines, such as spermine and spermidine, are small cations that have an influence on a wide range of biological processes, and thus, the intracellular levels of these cations are tightly regulated (52). PaiA was characterized as a spermine/spermidine acetyltransferase that catalyzes the first reaction in the polyamine degradation pathway (53). Although in B. subtilis and B. amyloliquefaciens, the regulatory gene yybA clusters with the yyaT acetyltransferase gene, the yybA orthologs belong to the same operon as the paiA acetyltransferase gene in most other Firmicutes. This suggests that the two acetyltransferases have related functions in the polyamine degradation pathway.
Two novel TF regulons were predicted to be involved in the osmotic-stress response. Choline is a metabolic precursor for the compatible solute glycine betaine. First, we analyzed the potential regulation of two paralogous operons, opuB and opuC, encoding choline uptake ABC transporters (54). Both operons are preceded by the divergently arranged yvbF and yvaV genes encoding paralogous TFs from the MarR family. Analysis of intergenic regions of these two divergons revealed a common 25-bp palindromic DNA motif that is conserved in multiple Bacillales spp. The novel DNA binding motif was tentatively assigned to the paralogous YvbF and YvaV regulators, which share 83% sequence identity. The second novel regulon involved in osmoprotection is controlled by the MarR family regulator YuaC. The yuaC gene is located in a conserved gene cluster with the gbsBA operon, which encodes a two-step pathway of glycine betaine synthesis from choline (55). An 18-bp palindromic DNA motif identified upstream of the gbsBA genes in five Bacillales spp. was tentatively assigned to the YuaC regulator. Since the gbsBA operon in B. subtilis is known to be induced by the presence of choline (55), we propose that YuaC functions as a choline-responsive repressor.
(iii) Identification of regulons for RNA regulatory elements.
For reconstruction of RNA regulons, we used known RNA regulatory motifs from the Rfam database (41) to scan intergenic regions and analyzed the genomic context of predicted regulatory RNAs using the RegPredict Web server (see Table S3 in the supplemental material). As a result, we identified 668 RNA regulatory elements from 22 Rfam families distributed in 11 studied genomes of Bacillales (see Table S4 in the supplemental material). The B. subtilis genome contains 65 RNA elements that represent all the Rfam families studied in this work with the single exception of the GEMM RNA motif (Table 4).
Table 4.
Content of B. subtilis regulons controlled by RNA elements
| RNA regulona | Target operon(s)b | Functional rolec |
|---|---|---|
| Cobalamin (B12 element) | btuFC-cbiZ-pduO | O |
| FMN (RFN element) | ribDEBAH, ribU | O |
| TPP (THI element) | thiC, thiT, ylmB, tenAI-thiOSGFD, ykoFEDC | O |
| Glycine | gcvTP1P2 | A |
| Lysine (L box) | yvsH, lysC | A |
| SAM (S box) | cysHP-sat-cysCG-sirBC, metK, metE3, metE2, metNPQ, metIC, metF, metE, mtnWXBD, mtnKA, yoaDCB | A |
| T box (various amino acid specificities) | alaS, hisS-aspS, cysES, glyQS, ileS, leuS, ilvBHC-leuABCD, yvbW, pheST, proI, proAB, serS, thrZS, trpS, rtpA-ycbK, tyrSZ, valS | A |
| PreQ1 | queCDEF | N |
| Purine (G box) | pbuG, purEKBCSQLFMNHD, pbuE, nupG, xpt-pbuX | N |
| PyrR | pyrBC-carAB-pyrKDFE, pyrP, pyrR | N |
| Ribosomal leaders | rplJL, rplM-rpsI, rplS, infC-rpmI-rplT, rplU-ysxB-rpmA | D |
| glmS | glmS | C |
| ykoK (M box) | mgtE | M |
| ykkC-yxkD | ykkCD, yxkD | R |
| ydaO | ktrAB, ydaO | U |
| ylbH | ylbH-coaD | U |
| yybP-ykoY | ykoY, yybP | U |
Regulons that operate by known metabolite-sensing riboswitches and aminoacyl-tRNA-sensing T boxes are underlined.
Novel predicted targets of known riboswitch regulons are underlined.
Functional roles of target genes: A, amino acid metabolism; R, stress response; C, amino sugar metabolism; M, metal homeostasis; N, nucleic base metabolism; O, cofactor metabolism; D, ribosomal proteins; U, unknown.
The inferred RNA regulons that operate by known metabolite-sensing riboswitches control central biosynthetic pathways for cofactors (cobalamin, riboflavin, and thiamine), nucleobases (nucleoside queuosine and purines), amino acids (lysine, methionine/cysteine, and glycine), glucosamine, and metal homeostasis (magnesium). The expanded known riboswitch regulons in B. subtilis include the thiamine biosynthesis and transport genes (TPP regulon), the riboflavin transporter gene ribU (FMN regulon), the predicted lysine permease gene yvsH (L-box regulon), the methionine metabolism and sulfate reduction genes (SAM regulon), and the nucleoside permease gene nupG (G-box regulon).
The T-box RNA elements were initially identified in the regulatory leader regions of aminoacyl-tRNA synthetase genes, and some amino acid biosynthetic genes in B. subtilis and related bacteria control gene expression via a unique transcription antitermination mechanism (56). The T box serves as a riboswitch that binds directly to a specific uncharged tRNA and thus measures the amino acid availability in the cell. In B. subtilis, we found 19 T-box elements characterized by 13 different amino acid specificities that control 15 different aminoacyl-tRNA synthetases; the proline, cysteine, and branched-chain amino acid biosynthesis genes; the amino acid permease gene yvbW; and the rtpA-ycbK operon involved in tryptophan-dependent regulation (see Table S4 in the supplemental material).
Finally, we reconstructed 8 novel RNA-controlled regulons that were not previously studied in B. subtilis. Ribosomal operon leaders from five Rfam families (L10, L13, L19, L20, and L21) regulate the respective ribosomal protein operons (Table 4). The ykkC-yxkD element controls the multidrug efflux transporter genes yxkD and ykkCD. The ylbH RNA motif controls rRNA small-subunit methyltransferase D (ylbH) and phosphopantetheine adenylyltransferase (coaD) genes. The yybP-ykoY motif regulates the uncharacterized genes ykoY and yybP.
Interconnections in the regulatory network of B. subtilis.
The reconstructed TRN of B. subtilis provides novel insights into the interplay between several different regulons operated by both TFs and RNA motifs. To estimate the regulatory network connectivity, we first analyzed regulatory cascades between various TFs and then identified a subset of target operons under the control of at least two regulators. Finally, we estimated the number of TFs that are subject to autoregulation.
In our B. subtilis TRN model, 55 TFs are involved in 50 regulatory cascades (Fig. 4). Most of these cascades (70%) involve three global TFs—CcpA, CodY, and TnrA—that regulate other local TFs. The catabolite control protein CcpA forms the largest number of regulatory cascades with local regulators of carbohydrate catabolism. In addition, CcpA coregulates TFs controlling metabolic pathways of lactate utilization (LutR), acetoin utilization (AcoR), citrate transport (CitT), the citric acid cycle (CcpC), and phosphate starvation (PhoP). CodY monitors the general nutrition state of the cell and regulates TFs of competence (ComK), sporulation (SpoVG, SpoVS, and Hpr), amino acid and nitrogen metabolism (MurR, RocR, PutR, and GamR), and the citric acid cycle (CitR). The nitrogen assimilation regulator TnrA coregulates three TFs of nitrogen metabolism (GltC, PucR, and GlnR), as well as the sporulation regulator KipR and the pleiotropic regulator of degradative enzymes DegU.
Fig 4.
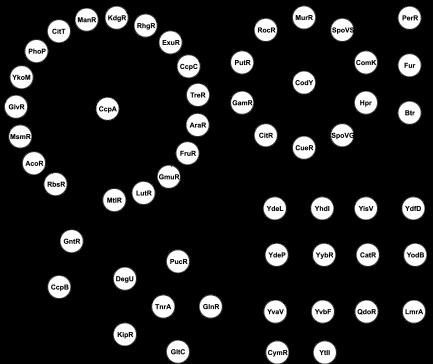
Regulatory cascades in B. subtilis TRN. TFs are shown as circles. The directions of regulatory interactions are shown by arrows. TF autoregulation is shown by curved arrows.
Twelve regulators form pairs of TFs with mutual regulation. In each pair, both TFs belong to the same protein family and have highly similar DNA binding motifs. Three of these TF pairs—TnrA/GlnR (57), YodB/CatR (58), and LmrA/QdoR (59)—were studied in B. subtilis, and their ability to bind to each other's sites was demonstrated. Moreover, these TF paralogs are able to respond to the same class of stimuli, as TnrA and GlnR sense the nitrogen status, whereas LmrA and QdoR respond to flavonoids. These observations suggest that the remaining three pairs of hypothetical TFs (YdeL/YhdI, YisV/YdfD, and YdeP/YybR) also respond to similar effectors.
The collection of inferred B. subtilis regulons contains at least 54 regulons (for 51 TFs and 3 regulator RNAs) that have at least one operon under the simultaneous control of at least two regulators (see Fig. S1 in the supplemental material). Ninety percent of all operons that were found under the control of multiple TFs belong to the global regulons CcpA, CodY, and TnrA. Most of these operons are regulated by a local TF and one of the global regulators. However, we also found overlaps between two global TFs, such as CodY/TnrA (6 target operons) and CodY/CcpA (7 target operons). The branched-chain amino acid biosynthesis operon ilvBHC-leuABCD, controlled by three global regulators (CcpA, CodY, and TnrA) and by the leucine-specific T-box regulatory RNA, is the most regulated operon in the current TRN model. Among the predicted regulons, 7 operons are under the overlapping control of three TFs, whereas ∼70 operons are coregulated by two regulons.
The autoregulation of a TF is a common regulatory-network motif in B. subtilis: 92 out of 129 studied TFs were predicted to control their own expression. Among the autoregulated TFs, 37 regulators belong to the feed-forward loop network motifs, where a target operon is regulated by two TFs and, in addition, one of these TFs is also regulated by another TF(s). For instance, the global regulator CcpA is involved in the feed-forward loop motif with numerous local regulators of sugar utilization pathways (KdgR, RhgR, MtlR, LutR, FruR, AraR, ExuR, RbsR, GlvR, GntR, ManR, TreR, GmuR, and CcpC).
Conclusions.
In the present work, we implemented the knowledge-driven comparative-genomics approach to capture the existing knowledge of transcriptional regulation in B. subtilis and to expand its regulatory network by inferring novel TF regulons. The obtained TRN model includes 129 TF regulons and 24 RNA element regulons that control more than 1,000 genes in B. subtilis. Each reconstructed regulon in B. subtilis contains a list of functionally annotated target genes organized in putative operons, a set of individual TFBSs, and the corresponding consensus DNA motif and the assigned TF. Moreover, for 84 TF regulons and 24 RNA regulons, we describe known or putative effectors (or environmental signals) that trigger the respective regulator. The resulting TRNs in B. subtilis and 10 other Bacillales genomes contain nearly 1,000 target genes per genome in the reconstructed TF and RNA regulons that include genes involved in the metabolism of carbohydrates, amino acids, nucleotides, and fatty acids; metal homeostasis; stress responses; sporulation; and competence. In the future, we plan to propagate this reference collection by automatic annotation of regulatory networks in all sequenced genomes in the Bacillales taxonomic group.
The B. subtilis TRN was expanded by ∼140 new target genes for 93 previously studied TF regulons and ∼150 target genes for 36 novel TF regulons that were reported in this work for the first time. Most of the novel regulons belong to the stress response and carbohydrate metabolism functional categories, although we also discovered regulons for the pathways of histidine biosynthesis, polyamine homeostasis, osmotic-stress protection, and deoxyribonucleotide metabolism. In addition to identification of novel target genes, we report the identification of novel DNA binding motifs for 64 TF regulons. This large set of novel regulatory interactions and TF binding DNA motifs awaits future validation by targeted and high-throughput experimental techniques. For example, the recently published transcriptomes of B. subtilis cultures with three different CcpA expression levels (60) were used for validation of new CcpA targets predicted here (Table 2). Among 41 new target operons of CcpA in B. subtilis, 28 operons are significantly differentially regulated in these transcriptomics experiments.
In conclusion, the collection of regulatory interactions and regulons captured for the Bacillales taxonomic group obtained in this work complements three previously obtained collections of regulons inferred by the comparative-genomics approach for other taxonomic groups of Firmicutes—Staphylococcaceae (15), Lactobacillaceae, and Streptococcaceae (61). All four regulon collections are available for download from the RegPrecise database (http://regprecise.lbl.gov/). The reference regulon collections are useful for building predictive metabolic models with regulatory constraints and for studying the evolution of regulatory networks in groups of phylogenetically related species.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Office of Science and Office of Biological and Environmental Research of the U.S. Department of Energy under contract DE-SC0004999 with SBMRI and LBNL. Additional funding was provided by the Russian Foundation for Basic Research (12-04-33003 to D.A.R., 12-04-32098 to S.A.L., and 12-04-91332 to E.O.E.), state contract 8135 (application 2012-1.2.2-12-000-1013-079), the Ministry of Education and Science of the Russian Federation project 8049, and the Program “Molecular and Cellular Biology” of the Russian Academy of Sciences.
Footnotes
Published ahead of print 15 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00140-13.
REFERENCES
- 1. Browning DF, Busby SJ. 2004. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2:57–65 [DOI] [PubMed] [Google Scholar]
- 2. Winkler WC, Breaker RR. 2005. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 59:487–517 [DOI] [PubMed] [Google Scholar]
- 3. Minchin SD, Busby SJ. 2009. Analysis of mechanisms of activation and repression at bacterial promoters. Methods 47:6–12 [DOI] [PubMed] [Google Scholar]
- 4. Grainger DC, Lee DJ, Busby SJ. 2009. Direct methods for studying transcription regulatory proteins and RNA polymerase in bacteria. Curr. Opin. Microbiol. 12:531–535 [DOI] [PubMed] [Google Scholar]
- 5. Ishihama A. 2010. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol. Rev. 34:628–645 [DOI] [PubMed] [Google Scholar]
- 6. Babu MM, Lang B, Aravind L. 2009. Methods to reconstruct and compare transcriptional regulatory networks. Methods Mol. Biol. 541:163–180 [DOI] [PubMed] [Google Scholar]
- 7. Lozada-Chavez I, Janga SC, Collado-Vides J. 2006. Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res. 34:3434–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Janga SC, Perez-Rueda E. 2009. Plasticity of transcriptional machinery in bacteria is increased by the repertoire of regulatory families. Comput. Biol. Chem. 33:261–268 [DOI] [PubMed] [Google Scholar]
- 9. Perez JC, Groisman EA. 2009. Evolution of transcriptional regulatory circuits in bacteria. Cell 138:233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gardner TS, Faith JJ. 2005. Reverse-engineering transcription control networks. Phys. Life Rev. 2:65–88 [DOI] [PubMed] [Google Scholar]
- 11. McCue LA, Thompson W, Carmack CS, Lawrence CE. 2002. Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res. 12:1523–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qin ZS, McCue LA, Thompson W, Mayerhofer L, Lawrence CE, Liu JS. 2003. Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat. Biotechnol. 21:435–439 [DOI] [PubMed] [Google Scholar]
- 13. Liu J, Xu X, Stormo GD. 2008. The cis-regulatory map of Shewanella genomes. Nucleic Acids Res. 36:5376–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodionov DA. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107:3467–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ravcheev DA, Best AA, Tintle N, Dejongh M, Osterman AL, Novichkov PS, Rodionov DA. 2011. Inference of the transcriptional regulatory network in Staphylococcus aureus by integration of experimental and genomics-based evidence. J. Bacteriol. 193:3228–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodionov DA, Novichkov PS, Stavrovskaya ED, Rodionova IA, Li X, Kazanov MD, Ravcheev DA, Gerasimova AV, Kazakov AE, Kovaleva GY, Permina EA, Laikova ON, Overbeek R, Romine MF, Fredrickson JK, Arkin AP, Dubchak I, Osterman AL, Gelfand MS. 2011. Comparative genomic reconstruction of transcriptional networks controlling central metabolism in the Shewanella genus. BMC Genomics 12(Suppl. 1):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu Y, Ding Y, Ren C, Sun Z, Rodionov DA, Zhang W, Yang S, Yang C, Jiang W. 2010. Reconstruction of xylose utilization pathway and regulons in Firmicutes. BMC Genomics 11:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leyn SA, Li X, Zheng Q, Novichkov PS, Reed S, Romine MF, Fredrickson JK, Yang C, Osterman AL, Rodionov DA. 2011. Control of proteobacterial central carbon metabolism by the HexR transcriptional regulator: a case study in Shewanella oneidensis. J. Biol. Chem. 286:35782–35794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rodionov DA, Li X, Rodionova IA, Yang C, Sorci L, Dervyn E, Martynowski D, Zhang H, Gelfand MS, Osterman AL. 2008. Transcriptional regulation of NAD metabolism in bacteria: genomic reconstruction of NiaR (YrxA) regulon. Nucleic Acids Res. 36:2032–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Earl AM, Losick R, Kolter R. 2008. Ecology and genomics of Bacillus subtilis. Trends Microbiol. 16:269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tam NK, Uyen NQ, Hong HA, Duc le H, Hoa TT, Serra CR, Henriques AO, Cutting SM. 2006. The intestinal life cycle of Bacillus subtilis and close relatives. J. Bacteriol. 188:2692–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goelzer A, Bekkal Brikci F, Martin-Verstraete I, Noirot P, Bessieres P, Aymerich S, Fromion V. 2008. Reconstruction and analysis of the genetic and metabolic regulatory networks of the central metabolism of Bacillus subtilis. BMC Syst. Biol. 2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sierro N, Makita Y, de Hoon M, Nakai K. 2008. DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 36:D93–D96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moreno-Campuzano S, Janga SC, Perez-Rueda E. 2006. Identification and analysis of DNA-binding transcription factors in Bacillus subtilis and other Firmicutes—a genomic approach. BMC Genomics 7:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Terai G, Takagi T, Nakai K. 2001. Prediction of co-regulated genes in Bacillus subtilis on the basis of upstream elements conserved across three closely related species. Genome Biol. 2:RESEARCH0048. doi:10.1186/gb-2001-2-11-research0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fadda A, Fierro AC, Lemmens K, Monsieurs P, Engelen K, Marchal K. 2009. Inferring the transcriptional network of Bacillus subtilis. Mol. Biosyst. 5:1840–1852 [DOI] [PubMed] [Google Scholar]
- 27. Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS, Dubchak IL, Alm EJ, Arkin AP. 2010. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 38:D396–D400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Felsenstein J. 1996. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 266:418–427 [DOI] [PubMed] [Google Scholar]
- 29. Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA. 2008. DBD—taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 36:D88–D92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ulrich LE, Zhulin IB. 2010. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38:D401–D407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mironov A, Vinokurova N, Gelfand M. 2000. Software for analysis of bacterial genomes. Mol. Biol. 34:222–231 [PubMed] [Google Scholar]
- 32. Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH. 2009. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37:D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ortet P, De Luca G, Whitworth DE, Barakat M. 2012. P2TF: a comprehensive resource for analysis of prokaryotic transcription factors. BMC Genomics 13:628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, Arkin AP, Mironov AA, Dubchak I. 2010. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38:W299–W307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Osterman A, Overbeek R. 2003. Missing genes in metabolic pathways: a comparative genomics approach. Curr. Opin. Chem. Biol. 7:238–251 [DOI] [PubMed] [Google Scholar]
- 37. Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eddy SR, Durbin R. 1994. RNA sequence analysis using covariance models. Nucleic Acids Res. 22:2079–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33:5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40:D109–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A. 2011. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39:D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38:D111–D118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stulke J, Hillen W. 2000. Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 54:849–880 [DOI] [PubMed] [Google Scholar]
- 45. Ochiai A, Itoh T, Kawamata A, Hashimoto W, Murata K. 2007. Plant cell wall degradation by saprophytic Bacillus subtilis strains: gene clusters responsible for rhamnogalacturonan depolymerization. Appl. Environ. Microbiol. 73:3803–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshida K, Yamaguchi M, Morinaga T, Kinehara M, Ikeuchi M, Ashida H, Fujita Y. 2008. myo-Inositol catabolism in Bacillus subtilis. J. Biol. Chem. 283:10415–10424 [DOI] [PubMed] [Google Scholar]
- 47. Setlow B, Cabrera-Hernandez A, Cabrera-Martinez RM, Setlow P. 2004. Identification of aryl-phospho-beta-d-glucosidases in Bacillus subtilis. Arch. Microbiol. 181:60–67 [DOI] [PubMed] [Google Scholar]
- 48. Zhang J, Aronson A. 1994. A Bacillus subtilis bglA gene encoding phospho-beta-glucosidase is inducible and closely linked to a NADH dehydrogenase-encoding gene. Gene 140:85–90 [DOI] [PubMed] [Google Scholar]
- 49. Schonert S, Seitz S, Krafft H, Feuerbaum EA, Andernach I, Witz G, Dahl MK. 2006. Maltose and maltodextrin utilization by Bacillus subtilis. J. Bacteriol. 188:3911–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vitreschak AG, Mironov AA, Lyubetsky VA, Gelfand MS. 2008. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA 14:717–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Akashi H, Gojobori T. 2002. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 99:3695–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tabor CW, Tabor H. 1985. Polyamines in microorganisms. Microbiol. Rev. 49:81–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Forouhar F, Lee IS, Vujcic J, Vujcic S, Shen J, Vorobiev SM, Xiao R, Acton TB, Montelione GT, Porter CW, Tong L. 2005. Structural and functional evidence for Bacillus subtilis PaiA as a novel N1-spermidine/spermine acetyltransferase. J. Biol. Chem. 280:40328–40336 [DOI] [PubMed] [Google Scholar]
- 54. Kappes RM, Kempf B, Kneip S, Boch J, Gade J, Meier-Wagner J, Bremer E. 1999. Two evolutionarily closely related ABC transporters mediate the uptake of choline for synthesis of the osmoprotectant glycine betaine in Bacillus subtilis. Mol. Microbiol. 32:203–216 [DOI] [PubMed] [Google Scholar]
- 55. Boch J, Kempf B, Schmid R, Bremer E. 1996. Synthesis of the osmoprotectant glycine betaine in Bacillus subtilis: characterization of the gbsAB genes. J. Bacteriol. 178:5121–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grundy FJ, Henkin TM. 1994. Conservation of a transcription antitermination mechanism in aminoacyl-tRNA synthetase and amino acid biosynthesis genes in gram-positive bacteria. J. Mol. Biol. 235:798–804 [DOI] [PubMed] [Google Scholar]
- 57. Zalieckas JM, Wray LV, Jr, Fisher SH. 2006. Cross-regulation of the Bacillus subtilis glnRA and tnrA genes provides evidence for DNA binding site discrimination by GlnR and TnrA. J. Bacteriol. 188:2578–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chi BK, Kobayashi K, Albrecht D, Hecker M, Antelmann H. 2010. The paralogous MarR/DUF24-family repressors YodB and CatR control expression of the catechol dioxygenase CatE in Bacillus subtilis. J. Bacteriol. 192:4571–4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hirooka K, Kunikane S, Matsuoka H, Yoshida K, Kumamoto K, Tojo S, Fujita Y. 2007. Dual regulation of the Bacillus subtilis regulon comprising the lmrAB and yxaGH operons and yxaF gene by two transcriptional repressors, LmrA and YxaF, in response to flavonoids. J. Bacteriol. 189:5170–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marciniak BC, Pabijaniak M, de Jong A, Duhring R, Seidel G, Hillen W, Kuipers OP. 2012. High- and low-affinity cre boxes for CcpA binding in Bacillus subtilis revealed by genome-wide analysis. BMC Genomics 13:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ravcheev DA, Best AA, Sernova NV, Kazanov MD, Novichkov PS, Rodionov DA. 2013. Genomic reconstruction of transcriptional regulatory networks in lactic acid bacteria. BMC Genomics 14:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.