

Abstract
Control over mRNA stability is an essential part of gene regulation that involves both endo- and exoribonucleases. RNase Y is a recently identified endoribonuclease in Gram-positive bacteria, and an RNase Y ortholog has been identified in Streptococcus pyogenes (group A streptococcus [GAS]). In this study, we used microarray and Northern blot analyses to determine the S. pyogenes mRNA half-life of the transcriptome and to understand the role of RNase Y in global mRNA degradation and processing. We demonstrated that S. pyogenes has an unusually high mRNA turnover rate, with median and mean half-lives of 0.88 min and 1.26 min, respectively. A mutation of the RNase Y-encoding gene (rny) led to a 2-fold increase in overall mRNA stability. RNase Y was also found to play a significant role in the mRNA processing of virulence-associated genes as well as in the rapid degradation of rnpB read-through transcripts. From these results, we conclude that RNase Y is a pleiotropic regulator required for mRNA stability, mRNA processing, and removal of read-through transcripts in S. pyogenes.
INTRODUCTION
RNA degradation is a strictly regulated process that involves both endo- and exoribonucleases (1). In prokaryotes, mRNA degradation is initiated by endonucleolytic cleavage and followed by exonuclease digestion (2–4). The first step is relatively slow and rate limiting, while the second step proceeds rapidly (1). In Escherichia coli, RNase E functions as the major endoribonuclease that initiates the bulk of mRNA degradation (5). Although RNase E is absent in Bacillus subtilis, the recently identified RNase Y is considered its functional analog (6). RNase E and RNase Y do not share sequence homology but are strikingly similar in function (7, 8). Both RNases are membrane-bound proteins that interact with other components to form a complex called the RNA degradosome (7, 9). These components include other RNases, an RNA helicase, and two glycolytic enzymes (10, 11). Both RNase Y and RNase E prefer 5′ monophosphorylated RNA substrates with downstream secondary structures (6, 12). The depletion of B. subtilis RNase Y results in the accumulation of about 550 mRNAs, including important transcriptional regulators for stress response and biofilm formation and metabolic operons for tryptophan biosynthesis and glycolytic enzymes (8). B. subtilis RNase Y also interacts with RNases J1 and III to control the abundance of total mRNAs (13). RNase Y of Staphylococcus aureus plays a major role in virulence gene regulation and is involved in the processing and stabilization of a global regulator system, SaePQRS (14). These observations suggest that RNase Y is the major endoribonuclease in mRNA degradation in B. subtilis and perhaps also in other Gram-positive pathogens, such as Streptococcus pyogenes.
S. pyogenes (group A streptococcus [GAS]) causes a variety of human diseases ranging from mild local infections such as pharyngitis and impetigo to life-threatening systemic diseases such as toxic shock syndrome and necrotizing fasciitis (15). GAS infections often cause serious sequelae, including acute rheumatic fever, acute glomerulonephritis, and reactive arthritis (15). Transcriptional control of GAS virulence factor production has been investigated in detail, and several specific and global gene regulators have been identified (16–18). However, regulatory control of virulence factor production is more complex and includes the interplay of several regulatory circuits (19) and a multilayer control at the posttranscriptional level (20–23). For example, the mRNAs for streptolysin S (encoded by sagA) and streptodornase (encoded by sda) are greatly stabilized during entry into the stationary phase.
An RNase Y ortholog was recently identified in S. pyogenes (24). A mutation in the S. pyogenes RNase Y-encoding gene (cvfA-rny) altered the expression of multiple GAS virulence factors in a growth-dependent manner and attenuated the virulence of the GAS strain in a murine model (24). Unlike its counterpart in B. subtilis, S. pyogenes RNase Y is not an essential enzyme (24, 25). This property makes studies of the S. pyogenes RNase Y highly amenable to global analyses of its role in mRNA degradation and, ultimately, of the regulation of virulence.
Here we used microarray and Northern blot analyses to measure global S. pyogenes mRNA half-life and to determine the role of RNase Y in mRNA metabolism on the entire transcriptome. We demonstrate that S. pyogenes has an exceptionally high mRNA turnover rate and that RNase Y plays multiple roles in mRNA degradation, processing, and read-through transcript removal.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. pyogenes NZ131 (M49) (26) and its rny gene mutant (Δrny) (25) were routinely grown in C medium (0.5% proteose peptone no. 3; 1.5% yeast extract; 10 mM K2HPO4; 0.4 mM MgSO4; 17 mM NaCl) (27) at 37°C without aeration. Erythromycin was added at a final concentration of 2 μg/ml when required. Bacterial growth curves were determined from three independent cultures with five technical replicates by using a BioScreen C machine (Piscataway, NJ).
Total RNA extraction, cDNA synthesis, and microarray analysis.
Total RNA extraction, cDNA synthesis, and microarray analysis were carried out as described previously (25, 28), with slight modifications. Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) and treated twice with Turbo DNase (Invitrogen) to remove chromosomal DNA. Fifteen micrograms of total RNA was used for cDNA synthesis with SuperScript II reverse transcriptase (Invitrogen). Two micrograms of cDNA was fragmented with DNase I (Roche, Indianapolis, IN) and 3′ end labeled with biotin-ddUTP using a BioArray terminal labeling kit (Enzo, New York, NY). A whole-genome custom GeneChip antisense expression microarray (16) was used for transcriptome analysis. On the antisense array, each gene of S. pyogenes was detected by a set of 17 25-mer oligonucleotide probes, and each intergenic region (IG) that was more than 100 bp in length was detected by a set of 25-mer probes, with gap widths ranging from 15 bp to 20 bp. (“Gap width” means the distance between the center nucleotides of two adjacent probes.) Hybridization, washing, and scanning of the custom S. pyogenes microarrays (16) were performed according to the procedures described by Affymetrix (Santa Clara, CA). Data sets were normalized based on stable RNA signals (rRNAs and tRNAs). Gene-level analysis was carried out by using the GeneChip operating software (GCOS) version 1.4 analysis program (Affymetrix). For probe-level analysis, the Affymetrix Power Tool (Affymetrx) was used to extract probe signal intensities from microarray CEL files.
mRNA decay assay and Northern blot analysis.
mRNA decay is defined as the reduction in abundance of a transcript of defined size when no new transcript is synthesized. The mRNA decay assay and Northern blot analysis were carried out as mentioned previously, with slight modifications (25). Briefly, S. pyogenes cells were grown in C medium until the late exponential phase (A600 = 0.50 to 0.55). Rifampin (1 mg/ml) and glucose (0.5% [wt/vol]) were added to the culture to inhibit new mRNA synthesis but maintain metabolic activity. Five-milliliter aliquots of the culture were withdrawn before (t = 0 min) and after (t = 0.5, 1, 2, 4, 8, 16, and 32 min) rifampin addition, rapidly chilled by mixing with 10 ml crushed ice, and harvested by centrifugation. For Northern blot analysis, total RNA was extracted with TRIzol reagent. One microgram of total RNA was separated on a denaturing agarose gel, transferred to a Hybond-N membrane (Amersham), and immobilized to the membrane by UV cross-linking (Stratalinker 1800 crosslinker; Stratagene). RNA probes were generated by T7 in vitro transcription (New England BioLabs, Ipswich, MA) of PCR fragments amplified with the primers listed in Table 1. Digoxigenin (DIG) was incorporated into RNA probes by replacing 25% standard UTP with DIG-UTP (Roche) in the in vitro transcription reaction. DIG-labeled RNA probes were hybridized to membrane-bound RNA targets at 68°C overnight. Signal visualization was achieved by using anti-DIG antibody and CDP-Star according to the manufacturer's instructions (DIG application manual; Roche Diagnostic). The abundance of gene transcripts, represented by the average pixel intensity of defined band sizes, was quantified with ImageJ software version 1.44o (29). 16S rRNA was visualized by ethidium bromide staining on an agarose gel prior to transfer and served as loading control.
Table 1.
Primers used in this study
| Primer | Sequencea |
|---|---|
| upp_1L | CGTCGTCAAACCACATCAAC |
| upp_1R(T7) | TAATACGACTCACTATAGGGGCCCAAATAAACGATCACCA |
| pepC_1L | AACCCGTCAAAGTGAAGTGG |
| pepC_1R(T7) | TAATACGACTCACTATAGGGAAACCAAACGGACTCACCAG |
| ptsI_1L | CATGAATGGCAAACCTGTTG |
| ptsI_1R(T7) | TAATACGACTCACTATAGGGTTCTGTAAGCGCACGATTTG |
| cfa_1L | TGATGCTTCAAATCCCGATA |
| cfa_1R(T7) | TAATACGACTCACTATAGGGGGTTTGATCCCCAAAACCTT |
| ska_1L | GTTGAGGGGACGAATCAAGA |
| ska_1R(T7) | TAATACGACTCACTATAGGGATTGGGCAATTGCTGCTAAC |
| ropB_1L | GGAAATTGGTGAAACCGTTG |
| ropB_1R(T7) | TAATACGACTCACTATAGGGAAACATATGATGGATCGTTTTGC |
| recA_1L | TGATCCAGCTTATGCTGCTG |
| recA_1R(T7) | TAATACGACTCACTATAGGACAAGCTCCCCTGTACGAGA |
| sodM_1L | GTTTGATGCGGAGACAATGA |
| sodM_1R(T7) | TAATACGACTCACTATAGGGGCGGCTTGGTAAAGTTCTGA |
| emm49_1L | CGTGTCTAGCGTTGCAAGAA |
| emm49_1R(T7) | TAATACGACTCACTATAGGGTCAAGGGCTTGAAGTTTGC |
| tufA_1L | CCAGGTGATGACCTTCCAGT |
| tufA_1R(T7) | TAATACGACTCACTATAGGGACCTTGTTCTACGGCGATTG |
| rnpB_F | GCAATTTTTGGATAATCGCGTA |
| rnpB_R(T7) | TAATACGACTCACTATAGGGTTTTGTTCCGGAAATAACTAGGA |
| Spy49_1274_F | TGGTGAAGCTGTACGCAAAC |
| Spy49_1274_R(T7) | TAATACGACTCACTATAGGGTTTGCTCTTTGTGGCATCTG |
| Spy49_1275_F | TCAGGCGATTACCAAAAAGG |
| Spy49_1275_R(T7) | TAATACGACTCACTATAGGGCTTGCAGACGCATTTGCTTA |
| Spy49_1276_1L | TGCGAGGGTTTGATAAGAAAG |
| Spy49_1276_1R(T7) | TAATACGACTCACTATAGGGCCAAAAACCTCTTTTTCCAATTT |
| fasX_F | CGAAGTCATGAGTTTATCGAGCA |
| fasX_R(T7) | TAATACGACTCACTATAGGGTGATATCCAAACAAAGACAACTGA |
| Anchor | TTTAGTGAGGGTTAATAAGCGGCCGCGTCGTGACTGGGAGCGC |
| T3 | GCGGCCGCTTATTAACCCTCACTAAA |
| ropB_R(RACE) | TGACCAAGGCAAAAAGGTTC |
T7 promoter sequences are underlined.
mRNA half-life calculation.
We developed a “steepest-slope” method to calculate S. pyogenes mRNA half-life. A typical mRNA decay process included a delay phase, a decay phase, and/or a stabilized phase (see Fig. S1 in the supplemental material). The most rapid mRNA reduction occurred in the decay phase, which constituted the steepest downward slope when plotted against decay time (see Fig. S1 in the supplemental material). The aim of this method was to identify the time range in which the most rapid mRNA reduction occurred. Two variables, ti and tj, were used to define the starting (ti = 0, 0.5, 1, 2, 4, or 8 min after rifampin addition) and ending (tj = 1, 2, 4, 8, 16, or 32 min after rifampin addition) points of a particular time range during an mRNA decay assay. At least one time point was included between ti and tj so that the half-life estimation was based on at least three consecutive time points. Exponential regression was carried out to determine the slope of the mRNA decay curve in each given time range defined by ti and tj, assuming that mRNA decay followed first-order kinetics, as demonstrated before (30). The time range that displayed the steepest downward slope of mRNA decay was considered the decay phase, and the mRNA half-life was calculated accordingly. Poorly expressed genes with microarray signals of less than 200 throughout the mRNA decay assay usually did not display an obvious mRNA decay phase and were excluded from half-life calculations.
5′ RACE.
To determine the transcriptional start sites (TSS) of genes, 5′ rapid amplification of cDNA ends (5′ RACE) was carried out by following the protocol described by Troutt et al. (31), with modification. Briefly, 200 ng S. pyogenes wild-type (WT) cDNA was ligated to 10 nmol 5′ phosphorylated anchor primer with T4 RNA ligase (Ambion) in a 20 μl reaction mixture. Dimethyl sulfoxide (DMSO) was added to the mixture to reach a final concentration of 10% (vol/vol) to relax secondary structures. The reaction mixture was incubated at 23°C for 18 h, heated at 65°C for 15 min to inactivate T4 RNA ligase, and subsequently used as the PCR template. PCR was carried out by using a forward primer (T3) complementary to the anchor primer and a reverse primer specific to the target gene, and the PCR product was subsequently sequenced to determine the TSS.
Statistical analyses.
One-way analysis of variance (ANOVA) and histogram analyses were carried out by using the Statplus statistical program (Analystsoft).
Microarray data accession number.
The microarray data sets were deposited in the Gene Expression Omnibus (GEO) database under platform GPL11420 with accession number GSE40198.
RESULTS
Transcriptome profiles of the WT and isogenic Δrny mutant strains.
The isogenic NZ131 Δrny mutant strain was constructed as described previously (25). The deletion of the S. pyogenes rny gene caused a slight slowdown of bacterial growth, with the generation time increasing from 50 min to 57 min (Fig. 1A). Microarray analysis of the WT and Δrny mutant strains in the late exponential phase (A600 = 0.5 to 0.55) showed very similar transcriptome profiles (Fig. 1B). Among 1,704 examined open reading frames (ORFs), 97.8% displayed a less than 2-fold difference between the two strains in transcript abundance. Only the expression of speB (streptococcal pyrogenic exotoxin B) and spi (SpeB inhibitor) was significantly reduced in the Δrny mutant, which is consistent with previous observations (24, 25). About 30 genes were significantly increased in expression in the Δrny mutant, including speG (exotoxin type G precursor), ypaA (riboflavin transporter), and two genes encoding hypothetical proteins (Spy49_1275 and Spy49_1276). Subsequent analyses showed that the mRNAs of these genes were greatly stabilized in the Δrny mutant (Table 2) (see below).
Fig 1.
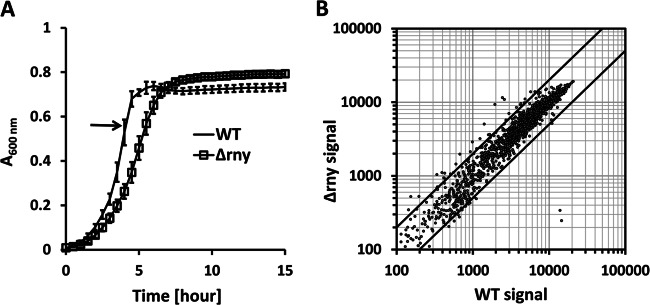
Transcriptome analysis of the WT and RNase Y mutant (Δrny). (A) Growth curves of the WT and the Δrny mutant in C medium (means ± standard deviations [SD]; n = 5). Samples were collected in the late exponential phase (optical density at 600 nm [OD600] = 0.50 to 0.55) for transcriptome analysis. (B) Microarray signals of the Δrny mutant were plotted against signals of the WT on a log-scale scatter diagram. The two diagonal lines indicate a 2-fold difference in microarray signal values between the two strains. Averages of the results of three biological replicates are presented.
Table 2.
Microarray analysis of selected genes described in this study
| Locus | Gene | Abundance (microarray signal)a |
Half-life (min) (microarray based) |
Function | ||||
|---|---|---|---|---|---|---|---|---|
| WT | Δrny mutant | Ratio | WT | Δrny mutant | Ratio | |||
| Spy49_0187 | speG | 1,946 | 9,342 | 4.8 | 0.31 | 7.44 | 23.8 | Exotoxin type G precursor |
| Spy49_0307 | ypaA | 2,442 | 11,543 | 4.7 | 0.31 | 12.79 | 41.9 | Riboflavin transporter |
| Spy49_0322 | upp | 10,747 | 8,564 | 0.8 | 1.10 | 1.39 | 1.3 | Uracil phosphoribosyltransferase |
| Spy49_0515 | tufA | 13,502 | 13,638 | 1.0 | 7.63 | 21.13 | 2.8 | Translation elongation factor Tu |
| Spy49_1010 | cfa | 10,909 | 9,374 | 0.9 | 6.89 | 6.73 | 1.0 | CAMP factor |
| Spy49_1094 | ptsI | 14,665 | 13,475 | 0.9 | 2.83 | 3.49 | 1.2 | Phosphoenolpyruvate-protein phosphotransferase of phosphotransferase system |
| Spy49_1122 | sodM | 14,655 | 14,282 | 1.0 | 7.06 | 8.57 | 1.2 | Superoxide dismutase (Mn) |
| Spy49_1274 | 6,740 | 11,315 | 1.7 | 0.85 | 9.17 | 10.8 | Hypothetical protein | |
| Spy49_ig0545 | rnpB | 15,954 | 15,035 | 0.9 | 40.33 | 34.74 | 0.9 | RNA subunit of RNase P |
| Spy49_1275 | 5,719 | 13,845 | 2.4 | 0.52 | 12.93 | 24.7 | Hypothetical protein | |
| Spy49_1276 | 6,508 | 13,878 | 2.1 | 0.25 | 13.09 | 53.0 | Conserved hypothetical protein | |
| Spy49_1280 | pepC | 9,698 | 10,397 | 1.1 | 1.48 | 3.47 | 2.3 | Aminopeptidase C |
| Spy49_1630 | ska | 10,995 | 9,202 | 0.8 | 4.52 | 3.75 | 0.8 | Streptokinase |
| Spy49_1671 | emm49 | 11175 | 11,731 | 1.0 | 2.57 | 4.94 | 1.9 | Antiphagocytic M protein |
| Spy49_1689 | spi | 14,553 | 247 | 0.0 | 8.04 | 5.59 | 0.7 | SpeB protease inhibitor |
| Spy49_1690 | speB | 13,889 | 340 | 0.0 | 8.71 | 6.18 | 0.7 | Streptococcal pyrogenic exotoxin B |
| Spy49_1691 | ropB | 5,813 | 4,807 | 0.8 | 0.87 | 0.85 | 1.0 | Rgg-like transcription regulator |
| Spy49_1753 | recA | 9,878 | 11,096 | 1.1 | 0.87 | 2.92 | 3.4 | RecA protein |
Abundance data represent average band intensities in pixels.
mRNA half-life calculation.
In order to examine global mRNA half-life, a steepest-slope method was developed based upon microarray data sets (see Materials and Methods for details) and was compared to two established methods proposed by Steglich et al. (32) and Selinger et al. (30). Specifically, Steglich's method defined the mRNA decay phase as the time range in which the exponential regression of decay displayed the minimal mean square error (32). Selinger's method defined the mRNA decay phase as the time range starting from t = 0 min to the earliest time point that displayed a decrease in mRNA abundance of more than 2-fold (30). We used the three methods to calculate S. pyogenes WT mRNA half-life and obtained consistent results (R2 = 0.78 between the steepest-slope and Steglich's methods and 0.89 between the steepest-slope and Selinger's methods) (see Fig. S2 in the supplemental material). Nevertheless, major differences were observed in highly abundant mRNAs, for which the steepest-slope method yielded smaller half-life estimations than the other two methods.
In order to evaluate the accuracy of the three methods, Northern blot analysis was carried out to determine the actual half-lives of 12 highly expressed mRNAs with a broad range of half-lives. Results showed that the steepest-slope method yielded mRNA half-lives that were closest to the actual values, while the other two methods overestimated them (see Fig. S3 in the supplemental material). These highly expressed mRNAs caused signal saturation on microarray chips, especially at the beginning of a decay assay. Selinger's and Steglich's methods were susceptible to the signal saturation and would overestimate the half-life, though the steepest-slope method was much more tolerant of signal saturation and would give a correct estimation as long as there was an obvious decay phase. An exception was observed in sodM (superoxide dismutase), the half-life of which was determined to be 0.69 min by Northern blotting (see Fig. 5 below) and was overestimated by all three microarray-based methods (7.06 min, 18.2 min, and 12.04 min by the steepest-slope, Steglich's, and Selinger's methods, respectively). Closer examination revealed that sodM-specific probe signals were saturated throughout the decay assay except for the last time point, which suggested that the sodM mRNA level was far beyond the dynamic range of the microarray. In such a case, optimizing the calculation method itself could not solve the bias problem. Thus, for our assay conditions, the steepest-slope method was the most reliable approach to estimate global mRNA half-life.
Fig 5.

Northern blot analysis of mRNAs that were differentially affected by rny gene mutation. The respective Northern blots are shown for the wild type and the Δrny mutant for the listed genes. The blots demonstrate the decay of mRNA transcripts after rifampin addition. Each blot experiment was carried out twice. The averaged half-lives in minutes are presented in the right column for the wild type and the Δrny mutant.
mRNA stability profiles in the WT and Δrny strains.
Microarray analysis and the steepest-slope method were used to estimate mRNA half-lives in the genomes of the WT and the Δrny mutant. Measurements were performed using two independent biological replicates. Results from the replicates showed high consistency for both strains (Fig. 2), which confirmed the reliability of our measurements and half-life calculation. Averaged mRNA half-lives were further used for analyses. Poorly expressed genes were excluded because of their indeterminate mRNA decay phases. Prophage genes were also excluded because of their significant heterogeneity from nonprophage genes. Specifically, the prophage mRNA abundance was 4-fold lower than that of nonprophage mRNAs, while the mean half-life was five times higher than that of the latter (7.5 min versus 1.24 min). In the end, we obtained the mRNA half-lives of 1,485 genes (87.1% of the whole genome) (see Dataset S1 in the supplemental material).
Fig 2.
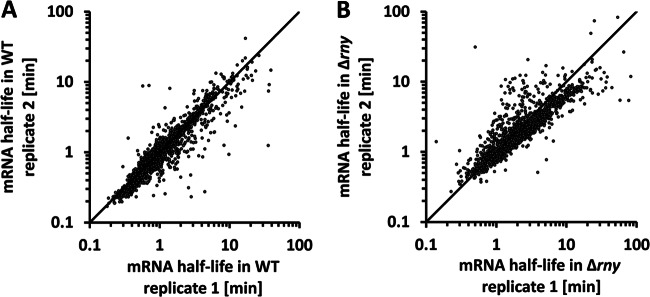
Consistency of mRNA half-life estimations between biological replicates. mRNA half-lives obtained from two biological replicates of the WT (A) and Δrny mutant (B) were plotted against each other on a log-scale scatter diagram. The diagonal line is used for visual determination of data consistency.
The median and mean half-lives of S. pyogenes mRNAs in the WT, when estimated with the steepest-slope method, were 0.89 min and 1.24 min, respectively. In the Δrny mutant, these values increased to 1.81 min and 2.79 min. In the WT, 1,267 genes had an mRNA half-life of less than 2 min (unstable mRNAs) and only 34 genes had an mRNA half-life of longer than 5 min (stable mRNAs) (Fig. 3A). In the Δrny mutant, the number of unstable mRNAs was reduced to 826 and the number of stable mRNAs increased to 178 (Fig. 3A). The effects of the rny mutation appeared to be similar for the majority of S. pyogenes mRNAs, with a 2-fold increase in overall mRNA stability (Fig. 3B). One-way ANOVA showed that the differences between the two strains in mRNA stability were statistically significant (P < 0.001).
Fig 3.
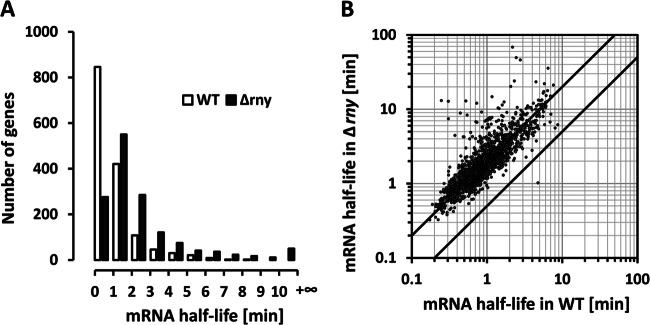
Effects of RNase Y mutation on mRNA stability. (A) Distribution of mRNA half-lives in the WT and Δrny mutant. Half-life rates averaged from two biological replicates were categorized in 1-min increments. (B) Correlation of mRNA half-lives in the WT and the Δrny mutant. The two diagonal lines indicate a 2-fold difference in mRNA half-life between the two strains.
We also used Selinger's and Steglich's methods to estimate overall S. pyogenes mRNA half-lives in the two strains. Selinger's method showed 1.02 min and 1.65 min for median and mean mRNA half-lives in the WT and 2.39 min and 3.62 min in the Δrny mutant. Steglich's method showed 1.20 min and 1.84 min for median and mean mRNA half-lives in the WT and 2.35 min and 4.18 min in the Δrny mutant. Both results showed an approximately 2-fold increase in mRNA stability in the Δrny mutant, which was in agreement with our observation using the steepest-slope method.
mRNA stability and gene function.
We hypothesized that genes belonging to different functional groups might have different mRNA stabilities. To test this hypothesis, S. pyogenes genes were categorized based on COGs (Clusters of Orthologous Groups) (33), and their mRNA half-lives in the WT were compared between functional groups (Fig. 4). One-way ANOVA indicated that significant differences in mRNA half-life existed in various functional groups (P < 0.001). Specifically, mRNAs involved in energy production and translation had a longer half-life than average (P < 0.001). For example, genes encoding ATP synthase (Spy49_0582 to Spy49_0589) and genes encoding ribosomal proteins (57 genes in total) had average mRNA half-lives of 3.9 min and 3.0 min in the WT, respectively, and those half-lives were significantly longer than the overall mRNA half-life (1.24 min). In contrast, all mRNAs involved in cell division and signal transduction were unstable mRNAs (half-life < 2 min), with average half-lives of 0.89 min and 1.01 min, respectively. However, the difference between these two groups and the whole genome was not statistically significant (P > 0.05). The vast majority of mRNA half-lives increased in the Δrny mutant, though the stability profiles among the functional groups remained similar to what was observed in the WT. These observations support our hypothesis that mRNA stability is related to the function of the gene product and are in agreement with previous findings in other species (34, 35).
Fig 4.

mRNA half-life (HL) in different functional groups. S. pyogenes genes were grouped based on COGs. (A) The numbers of unstable, intermediate, and stable mRNAs in the WT and Δrny mutant were counted for each functional group. (B) The fold changes of mRNA half-life caused by the rny gene mutation were calculated for each functional group.
Previous studies suggested the possibility that E. coli RNase E, the functional analog of RNase Y, might preferentially cleave mRNAs in certain functional groups (36). To determine whether RNase Y has a similar preferentiality in GAS, we compared the changes in mRNA half-life between the WT and Δrny mutant in each functional group (Fig. 4). In general, the rny gene mutation resulted in a 1.7- to 3.3-fold increase in mRNA half-life in all groups except for the cell division group that displayed a 6.2-fold increase in mRNA half-life. Closer examination shows that this group contained a gene locus (Spy49_1276) that was stabilized more than 30-fold in the Δrny mutant (see below for details). All other genes in this group were stabilized 2- to 3-fold, which is similar to levels seen with the other functional groups. One-way ANOVA indicated that the difference between functional groups with respect to half-life fold changes caused by the rny gene mutation was not significant (P > 0.05). Therefore, we found no evidence suggesting that RNase Y preferentially degraded mRNAs from certain functional groups.
Differential effects of RNase Y on mRNA stabilities.
While our results revealed a 2-fold increase in overall mRNA stability in the rny background (Fig. 3B), some mRNAs were unaffected or even slightly destabilized and others were greatly stabilized (Table 2). To confirm these findings, we selected a set of RNAs displaying differential stabilities and examined their half-lives with Northern blot analysis. The results showed no significant change in upp, ska, and cfa mRNA stabilities, a 2- to 3-fold increase in pepC, ptsI, and emm49 stabilities, and a 5-fold or greater increase in sodM and tufA stabilities (Fig. 5). We also examined the stability of fasX, which is a small noncoding RNA that stabilizes ska mRNA (21). Surprisingly, the fasX RNA was very stable in the WT (half-life > 6 min) but was considerably destabilized in the Δrny mutant (half-life < 2 min). Meanwhile, the abundance of fasX RNA in the WT was more than 2-fold higher than in the Δrny mutant (Fig. 5A and data not shown). The decreased fasX transcript abundance in the Δrny may directly result from the high RNA degradation rate, though other regulatory mechanisms may also be involved. The whole S. pyogenes transcriptome, including ORFs and intergenic regions, was screened for destabilized RNAs in the Δrny mutant, and only fasX has been identified so far (data not shown). These findings suggest that RNase Y plays different roles in degrading different RNA targets.
RNase Y functions in mRNA processing.
Northern blot analysis revealed that RNase Y is involved in the processing of ropB (rgg) mRNA (Fig. 6A). The ropB gene encodes a transcriptional regulator that activates the streptococcus exotoxin B-encoding gene (speB) (27, 37). The two genes locate next to each other on the chromosome in opposite orientations (38). Northern blot analysis showed that, in the WT, the ropB locus specifies three transcripts of different sizes (1.2 kb, 1.1 kb, and 1.0 kb, respectively) (Fig. 6A). The 1.2-kb ropB transcript was less abundant and degraded faster than the two shorter transcripts. In the Δrny mutant, the 1.2-kb transcript became the most prominent form of ropB mRNA, while the 1.0-kb transcript was less abundant and the 1.1-kb transcript was undetectable (Fig. 6A). The 1.2-kb transcript was stabilized only slightly in the Δrny mutant, which suggested that its increased abundance was not due to increased stability (Fig. 6A). Rather, our results suggest that the 1.2-kb transcript could be the primary ropB transcript that underwent endonucleolytic cleavages to generate the 1.1-kb and 1.0-kb transcripts. These results also suggested that RNase Y was very likely the enzyme responsible for ropB mRNA processing.
Fig 6.

Northern blot and probe-level analyses of the ropB mRNA. (A) Estimation of the ropB transcript half-lives in the WT and the Δrny mutant with Northern blot analysis. Each blot experiment was carried out twice. The averaged half-lives (in minutes) are presented in the right column. (B) Arrangements of the ropB and speB genes on the chromosome and the ropB mRNA structure. The ropB and speB promoters are indicated as bent arrows. Two pairs of inverted repeats in the ropB 5′ UTR are indicated as stem-loop structures. Two putative cleavage sites are indicated with scissor symbols. The predicted ropB TTS and the first putative cleavage site are marked with asterisks. (C) Probe-level analysis of the ropB mRNA. The average probe signal intensities upstream and downstream of the predicted ropB TTS are shown as solid lines (dashes indicate individual results). The average probe signal half-lives upstream and downstream of the first putative cleavage site are also shown as solid lines (open circles indicate individual results). The left and right dotted vertical lines indicate the shifts of the probe signal intensities and probe signal half-life, which correspond to the ropB TTS and the first putative cleavage site, respectively.
We next used 5′ RACE to determine the 5′ ends of three ropB mRNA transcripts. The ropB transcripts were expected to have identical 3′ ends but different 5′ ends, since the ropB gene had a strong Rho-independent terminator immediately downstream of the coding region (data not shown). The 5′ end of the 1.0-kb ropB transcript was determined to be 139 nucleotides (nt) upstream of the ropB translational start site. We were unable to determine the 5′ ends of the other two transcripts, presumably because of the complicated secondary structures in that region. Based on transcript sizes, the 5′ ends of the 1.2-kb and 1.1-kb transcripts were predicted to be 300 to 400 nt and 200 to 300 nt upstream of the translational start site, respectively (Fig. 6B). The speB gene TSS had been previously determined to be 244 bp upstream of the ropB translational start site (38), which was confirmed in this study (Fig. 6B). These findings suggested that the ropB and speB promoters overlapped in opposite directions on the chromosome.
The ropB mRNA was further analyzed using our microarray data. Probe-level analysis showed an abrupt upshift of probe signal intensity at approximately 350 nt upstream of the ropB translational start site (Fig. 6B), which strongly suggests that the ropB TSS was in that region. It also showed an upshift of probe signal half-life at approximately 210 nt upstream of the ropB translational start site, with average probe half-life increasing from 0.50 min to 1.16 min (Fig. 6B). The upshift was likely due to the differential stabilities of primary (1.2-kb) and processed (1.1-kb and 1.0-kb) ropB transcripts (Fig. 6B).
RNase Y degrades rnpB read-through transcripts.
Microarray analysis showed that the rny mutation significantly stabilized the mRNAs of three consecutive hypothetical gene open reading frames, Spy49_1274, Spy49_1275, and Spy49_1276 (Table 2). A BLASTP search indicated that Spy49_1274 encodes a Holiday junction-specific endonuclease, Spy49_1275 encodes a putative RNA methyltransferase, and Spy49_1276 encodes a cell division initiation protein that is highly homologous to the GpsB protein in B. subtilis (39). The Spy49_1274 and Spy49_1275 genes are organized as a bicistronic operon, while Spy49_1276 is located 491 bp upstream of the operon (Fig. 7A). Sequence analysis showed that the intergenic region between Spy49_1276 and the Spy49_1275-Spy49_1274 operon contained an rnpB gene encoding the RNA component of RNase P. RNase P is an essential ribozyme that catalyzes the 5′ processing of pre-tRNAs (40).
Fig 7.

The Spy49_1274-Spy49_1276 region. (A) Chromosomal arrangement of the Spy49_1274-Spy49_1276 region and Northern blot analysis of their transcripts. (B) The mRNA stabilities of Spy49_1274 and rnpB read-through transcripts were determined by Northern blotting. Each blot experiment was carried out twice. The averaged half-lives (in minutes) are presented in the right column.
The rnpB transcript itself was extremely stable (with a half-life of longer than 30 min) and highly abundant in S. pyogenes, though the transcripts of its surrounding genes (Spy49_1274, Spy49_1275, and Spy49_1276) were less abundant and very unstable in the WT, with an mRNA half-life of less than 1 min (Table 2). Their mRNA stabilities were greatly enhanced in the Δrny mutant, with half-life increasing to longer than 10 min, making them the mRNAs most profoundly affected by RNase Y in the S. pyogenes genome. The steady-state levels of these mRNAs also increased dramatically in the Δrny mutant compared to the WT (Table 2). Our subsequent Northern blot analyses showed that it was the accumulation of rnpB read-through transcripts, rather than the stabilization of primary transcripts, that caused the increased expression of the genes surrounding rnpB. The rnpB upstream gene (Spy49_1276) did not have a strong terminator at its 3′ end and was cotranscribed with rnpB to form a 0.7-kb transcript (Fig. 7A). This transcript was poorly expressed and very unstable in the WT but became highly abundant and extremely stable in the Δrny mutant (Fig. 7). We were unable to detect the full-length Spy49_1275-Spy49_1274 transcript and could detect only the Spy49_1274 single-gene transcript (Fig. 7A). It was possible that the full-length Spy49_1275-Spy49_1274 transcript underwent processing to generate two single-gene transcripts and that the Spy49_1274 transcript was more stable than the Spy49_1275 transcript. The rny mutation stabilized the Spy49_1274 single-gene mRNA only slightly, with the half-life increasing from 0.28 min to 0.56 min (Fig. 7B). Meanwhile, two long read-through transcripts that were transcribed from the Spy49_1276 and rnpB promoters emerged in the Δrny mutant. The read-through transcripts were not detectable in the WT but were highly abundant and very stable in the Δrny mutant (Fig. 7A). These observations suggested that RNase Y was involved in the rapid degradation of rnpB read-through transcripts.
DISCUSSION
In this study, we examined S. pyogenes mRNA stability and the role of RNase Y in mRNA degradation on a transcriptome-wide scale. To the best of our knowledge, this is the first attempt to determine precisely the global mRNA half-life in an obligate human pathogen. Our results showed that S. pyogenes had a higher mRNA turnover rate than other bacteria with known global half-lives. With our newly developed steepest-slope method, the median half-life of S. pyogenes mRNAs was estimated to be 0.89 min, with about 85% of mRNA half-lives being less than 2 min. Similar short mRNA half-life estimations were made by using two established half-life calculation methods (see Results), which suggested that the observed high S. pyogenes mRNA turnover rate was not a method bias. In contrast, the median half-life of Bacillus cereus mRNAs was estimated by different groups to be 2.4 min (35) and 5 min (41). In E. coli, about 80% of mRNAs had a half-life of between 3 and 8 min (34). In Staphylococcus aureus, about 90% of mRNAs had a half-life of less than 5 min, though the exact values were not determined (14).
The rny gene mutation led to a 2-fold increase in overall mRNA half-life without significantly affecting the steady-state levels of most mRNA species. Similar observations were made by Kang et al., who reported that the differential gene transcription caused by an rny mutation was not obvious in the exponential phase (24). These observations seem to contradict each other, since one would expect that increased mRNA stability would cause increased mRNA abundance. However, this expectation is based on the assumption that the mRNA synthesis rate remains unaltered in the Δrny mutant, which may not be the case. We believe that these seemingly contradicting observations may be explained by the following observations. (i) Gene transcription, but not mRNA degradation, is the dominant factor that determines the mRNA steady-state level. This notion was suggested by Bernstein et al., as they observed increased global mRNA stability but unaltered mRNA abundance in the E. coli RNase E mutant (34). (ii) Decreased mRNA turnover rate may reduce the free nucleotide pool, which in turn may slow down new mRNA synthesis. (iii) The organism may use other strategies to detect mRNA abundance and adjust mRNA synthesis accordingly. The rapid mRNA turnover rate of S. pyogenes may have alternative roles, perhaps including facilitation of adaptation of the organism to fast-changing environments at different stages of bacterial infection. It would be intriguing to understand whether the rapid mRNA turnover is a unique feature of S. pyogenes or a common theme in other streptococci and/or in other human pathogens when such data sets become available.
fasX, a small regulatory RNA, was severely destabilized in the Δrny mutant. This small RNA enhances streptokinase activity by base pairing to the 5′ end of ska mRNA and stabilizing the transcript (21). However, the ska mRNA stability did not change significantly in the Δrny mutant (this study and reference 21). This finding may be attributable to the stabilizing effect of rny mutation on ska mRNA being counteracted by reduced fasX abundance, the net outcome of which is unchanged ska mRNA stability. A thorough screen of the S. pyogenes genome suggests that fasX is the only RNA that is destabilized by an RNase Y mutation (data not shown). The mechanism to explain this observation remains unclear and is worthy of further investigation.
RNase Y-mediated mRNA processing in the well-studied gapA operon of B. subtilis has been previously described (42). RNase Y mediates the mRNA cleavage between the cggR and gapA protein-coding regions, which leads to decreased stability of the cggR-containing transcript and increased stability of the gapA-containing transcript (10). We found in this and a previous study (25) that RNase Y was involved in the processing of two functionally related mRNAs, speB and ropB. Both mRNAs were processed in the 5′ untranslated region (UTR) and were stabilized after processing. These findings support the idea that RNA processing plays an important role in posttranscriptional regulation. The identification of processed RNAs on a genome-wide scale has been a challenge for traditional microarray analysis because RNA processing does not usually lead to an obvious change in mRNA abundance (35). However, since RNA processing changes the stability of mRNAs, those mRNAs that display segmental stability are very likely processed by some endoribonuclease(s). We are currently using this approach to search for processed RNAs in the S. pyogenes genome.
The finding that the speB and ropB mRNAs overlap is in agreement with a previous study by Neely et al. (38). Two possible regulatory mechanisms may underlie this unique chromosomal arrangement. Overlapping promoters can function as a repression-activation switch, where one promoter is turned on and the other is simultaneously turned off by the same regulatory protein (43). We have observed that the speB gene is highly expressed only in the late exponential and stationary phases, while the ropB gene is constitutively expressed in the exponential phase but rapidly diminished in expression after entry into the stationary phase (25). The distinct expression profiles of the two genes imply that such a regulatory mechanism might exist for the speB-ropB gene pair. Another possibility is that the 5′ UTR of the ropB mRNA, after RNase Y processing, may form a small antisense RNA that binds to the speB mRNA and modulates its stability. These two possibilities are under investigation in our laboratory.
RNase P is an essential endoribonuclease that catalyzes the maturation of tRNAs. Its catalytic function is achieved by an RNA component encoded by rnpB (40). We found in this study that RNase Y is responsible for the rapid degradation of rnpB read-through transcripts. A similar observation has been made in E. coli, where RNase E mediates the degradation of rnpB read-through transcripts (44). The RNase P RNA is extremely stable in the WT of both species, where all RNases are present. The presence of extra sequences upstream and/or downstream of the rnpB sequence makes the read-through transcript much more susceptible to RNase attack. It is unlikely that the extra nucleotides alter the secondary structure of the endogenous rnpB transcript, since Ko et al. have observed identical secondary structures in the rnpB mRNAs with and without extra nucleotides (44). It is possible that RNase E/Y cleaves the extended sequences with high efficiency and that the cleaved RNAs bearing monophosphates at the 5′ end are the preferred substrates for further degradation. Although the exact mechanism of the RNase Y action remains to be elucidated, our results clearly show that RNase Y is crucial for the rapid degradation of read-through transcripts containing highly structured sequences.
Supplementary Material
ACKNOWLEDGMENTS
J.K. was supported by NIH/NIDCR grant R00DE018400.
We thank Justin Merritt (University of Oklahoma Health Sciences Center, Microbiology and Immunology) for helpful discussions.
Footnotes
Published ahead of print 29 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00097-13.
REFERENCES
- 1. Andrade JM, Pobre V, Silva IJ, Domingues S, Arraiano CM. 2009. The role of 3′-5′ exoribonucleases in RNA degradation. Prog. Mol. Biol. Transl. Sci. 85:187–229 [DOI] [PubMed] [Google Scholar]
- 2. Mudd EA, Krisch HM, Higgins CF. 1990. RNase E, an endoribonuclease, has a general role in the chemical decay of Escherichia coli mRNA: evidence that rne and ams are the same genetic locus. Mol. Microbiol. 4:2127–2135 [DOI] [PubMed] [Google Scholar]
- 3. Condon C. 2003. RNA processing and degradation in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 67:157–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lehnik-Habrink M, Lewis RJ, Mäder U, Stülke J. 2012. RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol. Microbiol. 84:1005–1017 [DOI] [PubMed] [Google Scholar]
- 5. Carpousis AJ, Luisi BF, McDowall KJ. 2009. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl Sci. 85:91–135 [DOI] [PubMed] [Google Scholar]
- 6. Shahbabian K, Jamalli A, Zig L, Putzer H. 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 28:3523–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lehnik-Habrink M, Newman J, Rothe FM, Solovyova AS, Rodrigues C, Herzberg C, Commichau FM, Lewis RJ, Stülke J. 2011. RNase Y in Bacillus subtilis: a natively disordered protein that is the functional equivalent of RNase E from Escherichia coli. J. Bacteriol. 193:5431–5441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lehnik-Habrink M, Schaffer M, Mäder U, Diethmaier C, Herzberg C, Stülke J. 2011. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol. 81:1459–1473 [DOI] [PubMed] [Google Scholar]
- 9. Liou GG, Jane WN, Cohen SN, Lin NS, Lin-Chao S. 2001. RNA degradosomes exist in vivo in Escherichia coli as multicomponent complexes associated with the cytoplasmic membrane via the N-terminal region of ribonuclease E. Proc. Natl. Acad. Sci. U. S. A. 98:63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Völker U, Stülke J. 2009. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell Proteomics 8:1350–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carpousis AJ. 2007. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 61:71–87 [DOI] [PubMed] [Google Scholar]
- 12. Mackie GA. 1998. Ribonuclease E is a 5′-end-dependent endonuclease. Nature 395:720–723 [DOI] [PubMed] [Google Scholar]
- 13. Durand S, Gilet L, Bessières P, Nicolas P, Condon C. 2012. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 8:e1002520 doi:10.1371/journal.pgen.1002520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marincola G, Schäfer T, Behler J, Bernhardt J, Ohlsen K, Goerke C, Wolz C. 2012. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol. Microbiol. 85:817–832 [DOI] [PubMed] [Google Scholar]
- 15. Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kreth J, Chen Z, Ferretti J, Malke H. 2011. Counteractive balancing of transcriptome expression involving CodY and CovRS in Streptococcus pyogenes. J. Bacteriol. 193:4153–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steiner K, Malke H. 2002. Dual control of streptokinase and streptolysin S production by the covRS and fasCAX two-component regulators in Streptococcus dysgalactiae subsp. equisimilis. Infect. Immun. 70:3627–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA, III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect. Immun. 77:3141–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carroll RK, Musser JM. 2011. From transcription to activation: how group A streptococcus, the flesh-eating pathogen, regulates SpeB cysteine protease production. Mol. Microbiol. 81:588–601 [DOI] [PubMed] [Google Scholar]
- 20. Le Rhun A, Charpentier E. 2012. Small RNAs in streptococci. RNA Biol. 9:414–426 [DOI] [PubMed] [Google Scholar]
- 21. Ramirez-Peña E, Treviño J, Liu Z, Perez N, Sumby P. 2010. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol. Microbiol. 78:1332–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barnett TC, Bugrysheva JV, Scott JR. 2007. Role of mRNA stability in growth phase regulation of gene expression in the group A streptococcus. J. Bacteriol. 189:1866–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang SO, Caparon MG, Cho KH. 2010. Virulence gene regulation by CvfA, a putative RNase: the CvfA-enolase complex in Streptococcus pyogenes links nutritional stress, growth-phase control, and virulence gene expression. Infect. Immun. 78:2754–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z, Itzek A, Malke H, Ferretti JJ, Kreth J. 2012. Dynamics of speB mRNA transcripts in Streptococcus pyogenes. J. Bacteriol. 194:1417–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McShan WM, Ferretti JJ, Karasawa T, Suvorov AN, Lin S, Qin B, Jia H, Kenton S, Najar F, Wu H, Scott J, Roe BA, Savic DJ. 2008. Genome sequence of a nephritogenic and highly transformable M49 strain of Streptococcus pyogenes. J. Bacteriol. 190:7773–7785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lyon WR, Gibson CM, Caparon MG. 1998. A role for trigger factor and an rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J. 17:6263–6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ajdić D, Pham VT. 2007. Global transcriptional analysis of Streptococcus mutans sugar transporters using microarrays. J. Bacteriol. 189:5049–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abramoff MD, Magelhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Intl. 11:36–42 [Google Scholar]
- 30. Selinger DW, Saxena RM, Cheung KJ, Church GM, Rosenow C. 2003. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res. 13:216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Troutt AB, McHeyzer-Williams MG, Pulendran B, Nossal GJ. 1992. Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proc. Natl. Acad. Sci. U. S. A. 89:9823–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steglich C, Lindell D, Futschik M, Rector T, Steen R, Chisholm SW. 2010. Short RNA half-lives in the slow-growing marine cyanobacterium Prochlorococcus. Genome Biol. 11:R54 doi:10.1186/gb-2010-11-5-r54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tatusov RL, Koonin EV, Lipman DJ. 1997. A genomic perspective on protein families. Science 278:631–637 [DOI] [PubMed] [Google Scholar]
- 34. Bernstein JA, Khodursky AB, Lin PH, Lin-Chao S, Cohen SN. 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U. S. A. 99:9697–9702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kristoffersen SM, Haase C, Weil MR, Passalacqua KD, Niazi F, Hutchison SK, Desany B, Kolstø AB, Tourasse NJ, Read TD, Økstad OA. 2012. Global mRNA decay analysis at single nucleotide resolution reveals segmental and positional degradation patterns in a Gram-positive bacterium. Genome Biol. 13:R30 doi:10.1186/gb-2012-13-4-r30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee K, Bernstein JA, Cohen SN. 2002. RNase G complementation of rne null mutation identifies functional interrelationships with RNase E in Escherichia coli. Mol. Microbiol. 43:1445–1456 [DOI] [PubMed] [Google Scholar]
- 37. Chaussee MS, Ajdic D, Ferretti JJ. 1999. The rgg gene of Streptococcus pyogenes NZ131 positively influences extracellular SPE B production. Infect. Immun. 67:1715–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neely MN, Lyon WR, Runft DL, Caparon M. 2003. Role of RopB in growth phase expression of the SpeB cysteine protease of Streptococcus pyogenes. J. Bacteriol. 185:5166–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, Edwards DH. 2008. Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Mol. Microbiol. 68:1029–1046 [DOI] [PubMed] [Google Scholar]
- 40. Kazantsev AV, Pace NR. 2006. Bacterial RNase P: a new view of an ancient enzyme. Nat. Rev. Microbiol. 4:729–740 [DOI] [PubMed] [Google Scholar]
- 41. Hambraeus G, von Wachenfeldt C, Hederstedt L. 2003. Genome-wide survey of mRNA half-lives in Bacillus subtilis identifies extremely stable mRNAs. Mol. Genet. Genomics 269:706–714 [DOI] [PubMed] [Google Scholar]
- 42. Meinken C, Blencke HM, Ludwig H, Stülke J. 2003. Expression of the glycolytic gapA operon in Bacillus subtilis: differential syntheses of proteins encoded by the operon. Microbiology 149(Pt 3):751–761 [DOI] [PubMed] [Google Scholar]
- 43. Bendtsen KM, Erdossy J, Csiszovszki Z, Svenningsen SL, Sneppen K, Krishna S, Semsey S. 2011. Direct and indirect effects in the regulation of overlapping promoters. Nucleic Acids Res. 39:6879–6885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ko JH, Han K, Kim Y, Sim S, Kim KS, Lee SJ, Cho B, Lee K, Lee Y. 2008. Dual function of RNase E for control of M1 RNA biosynthesis in Escherichia coli. Biochemistry 47:762–770 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.