

Abstract
Although increased lymphocyte turnover in chronic human immunodeficiency virus and simian immunodeficiency virus (SIV) infection has been reported in blood, there is little information on cell turnover in tissues, particularly in primary SIV infection. Here we examined the levels of proliferating T cell subsets in mucosal and peripheral lymphoid tissues of adult macaques throughout SIV infection. To specifically label cells in S-phase division, all animals were inoculated with bromodeoxyuridine 24 h prior to sampling. In healthy macaques, the highest levels of proliferating CD4+ and CD8+ T cells were in blood and, to a lesser extent, in spleen. Substantial percentages of proliferating cells were also found in intestinal tissues, including the jejunum, ileum, and colon, but very few proliferating cells were detected in lymph nodes (axillary and mesenteric). Moreover, essentially all proliferating T cells in uninfected animals coexpressed CD95 and many coexpressed CCR5 in the tissues examined. Confocal microscopy also demonstrated that proliferating cells were substantial viral target cells for SIV infection and viral replication. After acute SIV infection, percentages of proliferating CD4+ and CD8+ T cells were significantly higher in tissues of chronically infected macaques and macaques with AIDS than in those of the controls. Surprisingly, however, we found that proliferating CD4+ T cells were selectively decreased in very early infection (8 to 10 days postinoculation [dpi]). In contrast, levels of proliferating CD8+ T cells rapidly increased after SIV infection, peaked by 13 to 21 dpi, and thereafter remained significantly higher than those in the controls. Taken together, these findings suggest that SIV selectively infects and destroys dividing, nonspecific CD4+ T cells in acute infection, resulting in homeostatic changes and perhaps continuing loss of replication capacity to respond to nonspecific and, later, SIV-specific antigens.
INTRODUCTION
Early profound loss of memory CD4+ T cells, particularly in the intestine, is a hallmark of both human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) infection, and understanding the mechanisms of this loss remains a central issue in our understanding of the pathogenesis of AIDS (1). Reduced production of central memory CD4+ T cells has been proposed to be responsible for CD4+ T cell loss in rapidly progressing macaques (2). Others have suggested exhaustion of the immune system during HIV/SIV infection as a result of accelerated T cell turnover (3); therefore, the information on T cell turnover might have important implications for understanding T lymphocyte homeostasis and AIDS pathogenesis.
During HIV infection, CD4 depletion and the various immune defects associated with infection could affect the capacity of the immune system to develop effector-memory CD4+ T cells. Under normal, homeostatic conditions, there are baseline levels of proliferating CD4+ and CD8+ cells continuously replenishing cells lost in the body through attrition, subclinical infections, or other immunologic processes. It is clear that HIV and SIV induce proliferation and regeneration of peripheral T cells in acute and chronic infection (4–9), and massive production of HIV particles in blood, paralleled by a rapid turnover of CD4+ T lymphocytes, has been demonstrated after withdrawal of antiretroviral therapy (10–12). Increases in proliferating CD4+ and CD8+ T lymphocytes in blood have been described in HIV infection (8, 9), and studies in macaques demonstrate that SIV infection accelerates lymphocyte turnover in all lymphocyte subsets (5–7). However, studies analyzing changes in telomere length suggest that CD8+ T cell proliferation increases, whereas CD4+ T cell proliferation does not (13, 14). Still other studies show distinct cycling profiles of CD4+ and CD8+ T cells in blood during chronic SIV infection in macaques (5, 15). This suggests either differential regulation of CD4+ and CD8+ T cell proliferation, selective viral targeting and elimination of specific cell subsets, or differential regeneration of T cell subsets occurring in other tissues. Most information on T cell turnover rates has been limited to the rates in peripheral blood, and few studies have examined proliferation and T cell turnover in tissues, particularly in the intestine, which is a primary target for acute SIV and HIV infection. Further, it is increasingly clear that the immunologic and virologic events that occur during the earliest stages of infection may have a strong impact on disease progression. Moreover, studies on T cell turnover have focused on chronic infection, and little is known regarding very early events in SIV infection. Examining the earliest changes in proliferating T cell subsets in blood is more likely to detect selective viral targeting and elimination of specific cell subsets.
To examine the proliferation of T cell subsets in tissues, we administered bromodeoxyuridine (BrdU) to healthy and SIV-infected rhesus macaques as a single pulse 24 h prior to tissue collection in all animals. Since BrdU is a thymidine analog incorporated only by cells synthesizing DNA, this protocol specifically allows detection of cells in the synthesis (S) phase of cell division. Thus, this is a more specific method for distinguishing cells in S-phase division than other markers, such as Ki67, which persists throughout all active phases of the cell cycle (G1, S, G2, and mitosis) (16), and some data suggest that Ki67 expression may be artificially increased in chronic HIV infection, as memory CD4+ T cells appear to be increasingly Ki67 positive (Ki67+) when arrested in G1 phase of the cycle (5, 17). Thus, we selected a single pulse-label administration of BrdU 24 h prior to sacrifice to detect and quantify only those cells in S-phase cell division. This method reliably labeled proliferating T cell subsets in the blood, lymph nodes, spleen, and intestines of uninfected and SIV-infected macaques.
MATERIALS AND METHODS
Animals, SIV, and BrdU inoculation.
Tissues from 50 rhesus macaques (Macaca mulatta) from 4 to 13 years old were obtained from the Tulane National Primate Research Center. All monkeys were housed and maintained in accordance with the standards of the Association for Assessment and Accreditation of Laboratory Animal Care, and all studies were reviewed and approved by the Tulane Institutional Animal Care and Use Committee. Of these, 10 uninfected animals were euthanized for tissue collection as controls, and another 40 were infected with SIVmac251 or SIVmac239 and euthanized for tissue collection at various time points, including during very early (acute) infection at 8 (n = 6), 10 (n = 4), 13 (n = 5), and 21 (n = 2) days postinoculation (dpi) or during chronic infection (defined here as infection over 60 days) either with no overt signs of disease (chronic asymptomatic; n = 7) or with illness that could not be definitively attributed to AIDS (e.g., nonresponsive diarrhea, weight loss; n = 7). An additional 9 animals were defined as having AIDS by the presence of either classic opportunistic infections or absolute CD4 counts in blood below 200 cells/μl. All animals examined during acute infection (infection for 21 days or less) were intravenously infected with SIVmac251 to reduce the variation that may occur with mucosal inoculations, but macaques examined during chronic infection were either intravenously or intravaginally inoculated and grouped together irrespective of the route of inoculation. For in vivo BrdU pulse-labeling, BrdU (Sigma) was dissolved in saline (20 mg/ml), filter sterilized, and intraperitoneally inoculated into macaques (60 mg/kg of body weight) 24 h prior to tissue collection.
Cell isolation and flow cytometry.
Tissues were collected from the intestine (jejunum, ileum, and colon), spleen, and mesenteric and axillary lymph nodes within minutes of euthanasia and transported to the lab on ice for immediate processing, and duplicate samples were fixed in formalin and embedded in paraffin for immunohistochemistry. Viable intestinal lymphocytes were isolated and stained for flow cytometry as previously described (18, 19). Briefly, intestinal segments were subjected to serial incubations with EDTA to remove the epithelium, followed by digestion with collagenase to extract lamina propria lymphocytes. Cell suspensions were adjusted to 107 cells/ml, and 100-μl aliquots (106 cells) were stained for 30 min at 4°C with appropriately diluted concentrations of monoclonal antibodies. Peripheral blood and spleen cells were stained using a whole-blood lysis technique. Cells from all tissues were stained with either BrdU-fluorescein isothiocyanate (FITC) or Ki67-FITC and CD20-phycoerythrin (PE) or CCR5-PE (3A9) with CD4-allophycocyanin (APC) (L200) and CD8-peridinin chlorophyll protein (PerCP) (SK1; BD Biosciences) for 4-color flow cytometry; cells from 5 healthy and 12 SIV-infected macaques were stained with BrdU-FITC, CD8-PerCP, CD4-APC, CD95-PE-Cy5, HLADR-PE-Cy7, CD20-APC, CD69-APC-Cy7, CD3-Pacific Blue (BD Biosciences), CD8-PE-Texas Red (Caltag Laboratories), and CD4-Qdot655 (NIH) for 10-color flow cytometry. For intracellular BrdU staining, surface-stained cells were washed in Dulbecco phosphate-buffered saline (PBS)–bovine serum albumin, fixed, and permeabilized with BD Cytofix/Cytoperm buffer, followed by staining for BrdU, according to the manufacturer's instructions, including a 1-h incubation with DNase, followed by washing with BD Perm/Wash buffer and staining with fluorescent anti-BrdU-FITC for 20 min at room temperature. Cells were washed again (BD Perm/Wash buffer), and all samples were resuspended with BD stabilizing fixative buffer (BD Biosciences) and acquired on a FACSCalibur flow cytometer (Becton, Dickinson) for 4-color samples or a FACSAria flow cytometer (Becton, Dickinson) for 10-color flow cytometry samples within 24 h of fixation. Data were analyzed with FlowJo software (Tree Star, Inc.). At least 10,000 lymphocytes were collected for analysis from each sample.
Phenotyping SIV-infected cells in tissues by in situ hybridization and multilabel confocal microscopy.
A combination of 3-color immunofluorescence for CD3 and BrdU by immunohistochemistry and in situ hybridization for SIV mRNA was performed on formalin-fixed, paraffin-embedded sections of specimens from 4 animals early after SIV infection (2 animals each at 8 dpi and 10 dpi). Briefly, 5-μm sections were cut and adhered to charged glass slides. After deparaffinization in xylene, rehydration in PBS, and antigen retrieval with steam (citrate buffer), sections were incubated with antisense SIV riboprobes (Lofstrand Labs Ltd.) encompassing essentially the entire SIV genome, as previously described (19, 20). Labeled cells were visualized using horseradish alkaline phosphatase-conjugated sheep antidigoxigenin antibodies. SIV mRNA-positive cells were detected by a 2-hydroxy-3-naphthoic acid-2 phenylanilide phosphate (HNPP) fluorescence detection set (Roche Diagnostics Corporation), with cells appearing red. Sections were also colabeled using unconjugated primary antibodies for BrdU and CD3 and then with secondary antibodies conjugated to either Alexa 488 or Alexa 633 (Molecular Probes, Eugene, OR). After staining, slides were washed, mounted with fluorescent mounting medium (DakoCytomation), and visualized using a confocal microscope. Confocal microscopy was performed using a Leica TCS SP2 confocal microscope equipped with three lasers (Leica Microsystems, Exton, PA). Individual optical slices representing 0.2 μm and 32 to 62 optical slices were collected at a resolution of 512 by 512 pixels. NIH Image software (version 1.62) and Adobe Photoshop software (version 7.0) were used to assign colors to the channels collected: HNPP-Fast Red, which fluoresces when exposed to a 568-nm-wavelength laser, appears red; Alexa 488 (Molecular Probes) appears green; Alexa 633 (Molecular Probes) appears blue; and the differential interference contrast (DIC) image is grayscale. Results for the four channels were collected simultaneously.
Statistics.
Graphical presentation and statistical analysis of the data were performed using GraphPad Prism (version 4.0) software (GraphPad Software Inc., San Diego, CA). Comparisons of BrdU expression in tissues throughout SIV infection were analyzed by a one-way analysis of variance (ANOVA). A Mann-Whitney U test was used for comparison of BrdU incorporation by T cell subsets in cells from healthy and infected macaques and between CD4+ and CD8+ T cells at specific time points. Correlations between samples were calculated and expressed using the Spearman coefficient of correlation. P values of <0.05 were considered significant.
RESULTS
Distribution of BrdU+ CD4+/CD8+ T cells in tissues of healthy rhesus macaques.
In uninfected adult animals, S-phase proliferating cells were rare but easily detectable in most tissues, with the highest levels of BrdU-positive (BrdU+) CD4+ and CD8+ T cells being found in blood and, to a lesser extent, in spleen. Substantial percentages of ‘proliferating cells were also found in intestinal tissues, including the jejunum, ileum, and colon, but very few proliferating cells were detected in lymph nodes (axillary and mesenteric) (Fig. 1A). Notably, the baseline percentages of CD4+ T cells in S-phase division (BrdU+) were similar to those of BrdU+ CD8+ T cells in all tissues of healthy, uninfected animals (Fig. 1A). This was confirmed by Ki67 staining of uninfected animals (Fig. 1B). As expected, the percentages of Ki67+ T cells were consistently much higher than those of 24-h pulse-labeled BrdU+ T cells, but the ratios and proportions of Ki67+ T cells were consistent with those of BrdU+ cells between the tissues examined (Fig. 1).
Fig 1.

Percentages and distribution of proliferating CD4+ and CD8+ T cells in tissues of healthy (noninfected) adult rhesus macaques detected by BrdU incorporation (A) and Ki67 staining (B). Note for both CD4+ and CD8+ T cells more proliferating cells in blood and spleen than in other tissues (P < 0.05 by Mann-Whitney U test), but there were no significant differences between proliferating CD4+ and CD8+ T cells, and the ratios and proportions of BrdU+ T cells are consistent with those of Ki67+ cells in all tissues examined. Graphs represent mean percentages of BrdU+/Ki67+ cells when gating through either CD4+ or CD8+ lymphocytes ± SEM. Axi LN, axillary lymph node; Mes LN, mesenteric lymph node; Jej, jejunum; LPL, lamina propria lymphocytes.
To phenotype proliferating cells, coexpression of CD95 (a memory marker) and CCR5 (a coreceptor for HIV/SIV infection) on BrdU+ CD4+ and CD8+ T cell subsets was examined in selected animals. As shown in Fig. 2A, almost all proliferating (BrdU+) CD4+ T cells coexpressed CD95, suggesting that only memory cells were in S-phase division. Notably, many BrdU+ T cells also coexpressed CCR5 regardless of the tissue examined (Fig. 2B).
Fig 2.

Phenotyping of proliferating (BrdU+) T cells in various tissues of rhesus macaques. (A and B) Flow cytometry plots show that BrdU+ T cells mostly coexpress CD95 (memory marker), suggesting that they are memory cells (A), and substantial numbers of BrdU+ cells coexpress CCR5 (HIV/SIV coreceptor) (B) in all tissues from healthy rhesus macaques examined.
Dynamics of BrdU+ CD4+ and BrdU+ CD8+ T cells in vivo during primary SIV infection.
To evaluate the effects of SIV infection on proliferating T cells in vivo, we also compared the levels of BrdU+ T lymphocytes from the blood, spleen, mesenteric lymph nodes, and intestines of animals at various stages of SIV infection. ANOVA indicated that primary SIV infection had profound effects on both proliferating CD4+ T cells (P = 0.003 in blood, P = 0.008 in spleen, P = 0.07 in mesenteric lymph nodes, P = 0.03 in jejunal lamina propria lymphocytes) and CD8+ T cells (P < 0.0001 in blood, P < 0.0001 in spleen, P = 0.00032 in mesenteric lymph nodes, P = 0.06 in jejunal lamina propria lymphocytes) in rhesus macaques (Fig. 3). Remarkably, there were also different rates of CD4+ and CD8+ T cell turnover between different regions of the intestine, suggesting differential regulation or homing and/or T cell turnover between the small (jejunum, ileum) and large (colon) intestine (Fig. 3).
Fig 3.

Comparison of proliferating (BrdU+) CD4+ T cells and CD8+ T cells from various tissues in uninfected and SIV-infected macaques throughout SIV infection. Data represent mean percentages of CD4+ or CD8+ T cells labeled with BrdU (mean ± SEM). *, P < 0.05; **, P < 0.01.
Increased lymphocyte turnover has been previously reported in blood in late acute and chronic HIV and SIV infection (2, 5, 8, 21–24). Consistent with this, here we show increased T cell turnover in all tissues, including spleen, lymph node, jejunum, and blood, both in chronic infection and in macaques with AIDS. Surprisingly, however, although increased turnover of CD4+ T cells was found in most tissues by day 21 and through chronic infection and AIDS (Fig. 3), the ratio and percentage of proliferating CD4+ T cells were significantly decreased in early SIV infection, reaching their nadirs at 10 dpi, and then levels gradually increased (Fig. 3 and 4). These findings suggest that selective depletion of homeostatic, non-virus-specific (baseline) proliferating CD4+ T cells is an early feature of SIV infection. Moreover, we found that SIV infection resulted in progressively increasing percentages of BrdU+ CD8+ cells in all tissues examined during primary infection. As shown in Fig. 3 and 4, increased levels of BrdU+ CD8+ T cells were detected at 8 dpi. These levels peaked at 13 dpi in mesenteric lymph nodes and jejunum, and they peaked at 21 dpi in blood and spleen and then declined after 21 dpi, yet increased levels compared to those at baseline were sustained throughout infection in all tissues examined. This is consistent with the hypothesis that chronic and persistent immune activation due to SIV infection is associated with progression to AIDS.
Fig 4.

Distinct changes in proliferating CD4+ (top) and CD8+ (bottom) T cells in early stages of SIV infection in tissues of rhesus macaques. Data represent mean percentages of BrdU+ cells in gated CD4+ or CD8+ T cells (mean ± SEM).
Disparate levels of proliferating CD8+ and CD4+ T cells occur in very early SIV infection.
Although increased lymphocyte turnover was detected in both CD4+ and CD8+ T cells in chronically infected animals, the profiles were quite different in very early infection. In uninfected macaques, there were no significant differences between BrdU+ CD4+ T and BrdU+ CD8+ T cells (Fig. 1). However, rates of proliferation of CD4+ T cells differed markedly from those of CD8+ T cells in very early infection (Fig. 3 and 4). Although the rates of proliferating CD4+ and CD8+ T cells were very similar in uninfected macaques, the percentages of CD4+ BrdU+ cells were significantly lower than those of CD8+ BrdU+ cells within 8 days after SIV infection in blood and spleen (Fig. 4). Further, higher levels of BrdU+ CD8+ cells were also found through 21 dpi in tissues (Fig. 3). Combined, these results reflect the differential effects of SIV infection on CD4+ and CD8+ T cell subsets in very early infection, as previously described (5), but by examining the percentages of cells at multiple time points in the first 13 days, a significant, yet transient loss of proliferating CD4+ T cells was detected 8 to 10 days after infection (Fig. 4).
Selective loss of proliferating CD4+ T cells occurs in very acute SIV infection.
A rapid decrease in proliferating CD4+ T cells was detected in early SIV infection. Considering that essentially all proliferating CD4+ T cells were memory cells and many coexpressed CCR5 (Fig. 2), we hypothesized that activated CD4+ T cells could be direct targets for SIV infection and lysis, resulting in their rapid loss in early infection. Unfortunately, we were not able to sort and quantify proliferating CD4+ cell subsets due to technical issues associated with intracellular staining techniques for BrdU and Ki67, which is a limitation of this study. However, we phenotyped SIV-infected cells in tissues from early infection by multilabel confocal microscopy and showed substantial numbers of SIV-infected cells in sections costained with BrdU (Fig. 5). This suggests that, proportionally, CD4+ T cells in S-phase division are more susceptible to SIV infection and replication than other memory CD4+ T cells. These findings combined support the hypothesis that SIV directly infects and rapidly destroys homeostatic (nonspecific) proliferating cells and, by inference, many of the earliest proliferating virus-specific CD4+ T cells responding to primary infection.
Fig 5.
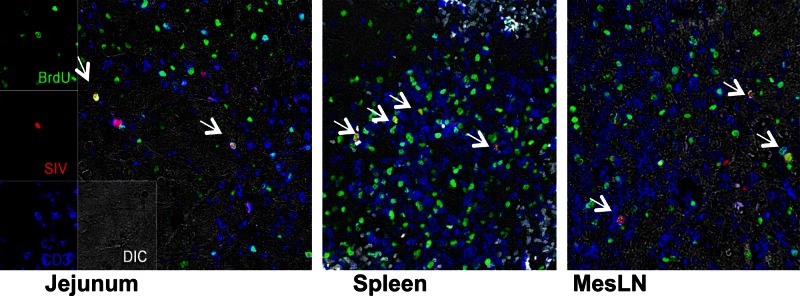
Phenotyping of SIV-infected T cells in tissues of infected macaques early in the course of infection by confocal microscopy. As indicated by arrows, several SIV-infected cells (SIV mRNA, red) in tissues appearing white or yellow when dually labeled are also BrdU positive. These results suggest that proliferating cells are major early targets for SIV infection.
Plasma viral loads and their association with proliferating CD4+ T cells in blood during primary SIV infection.
As previously reported, we found peak viral loads of 107 to 108 SIV RNA copies/ml in plasma at about 10 days of infection, which then declined by day 21 (Fig. 6). Further, we found that plasma viral loads indirectly correlated with both total CD4+ T cells (r = 0.47, P = 0.002) and BrdU+ CD4+ T cells (r = −0.53, P = 0.0005) in blood throughout SIV infection. However, in very early SIV infection (8 through 21 days postinfection), plasma viral loads indirectly correlated with the levels of BrdU+ CD4+ T cells (r = −0.5, P = 0.03) but not total CD4+ T cells (r = 0.1, P = 0.59) (Fig. 6). Taken together, these results suggest that proliferating CD4+ T cells are disproportionally being infected and eliminated in all tissues, whereas CD8+ T cells are proliferating (and increasing) at a markedly accelerated pace in tissues in response to infection.
Fig 6.

Changes in plasma viral load (solid line), CD4+ T cells (dotted line, left), and proliferating CD4+ T cells (dashed line, right) in blood during early SIV infection (P = 0.036, Spearman r = −0.5).
DISCUSSION
Although lymphocyte turnover has been extensively studied in chronic HIV/SIV infection, there is little information on turnover in acute infection, especially in tissues. Here we used a single pulse-labeling of BrdU to compare the distribution and frequency of proliferating T cells in various tissues of healthy rhesus macaques and compared the kinetics of T lymphocyte turnover in acute SIV infection through progression to AIDS. Consistent with previous reports (5), we found increased turnover in both CD4+ and CD8+ T cells during chronic infection in both asymptomatic and symptomatic SIV-infected macaques as well as in SIV-infected animals with AIDS. Interestingly, however, when examining blood and tissue samples from very early time points, we found that the levels of proliferating CD4+ T cells rapidly decreased in very early SIV infection, whereas we found that the levels of proliferating CD8+ T cells in the same tissues significantly increased at these time points (Fig. 3 and 4). Although it is difficult to definitively determine the dynamics of T cell turnover in vivo, this suggests that the early transmitted founder SIV has a propensity to infect and replicate in proliferating memory CD4+ T cells in blood and tissues.
Both HIV and SIV cause rapid, persistent depletion of memory CD4+ T cells throughout the body after infection, which may play a critical role in determining the course of disease. However, the mechanism of this early CD4+ T cell loss remains one of the most critical questions in HIV pathogenesis (1). We and others have previously shown that the rapid and selective depletion of intestinal CD4+ T cells was associated with a combination of their state of activation and expression of high levels of CCR5 (25–27). In this study, detectable yet variable levels of proliferating T cells were found in different tissues of uninfected macaques (Fig. 1). Further, all dividing T cells (BrdU+) coexpressed CD95, regardless of tissue site, as shown in Fig. 2, indicating that these are activated memory T cells, a known target for SIV. Moreover, substantially higher percentages of BrdU+ CD4+ T cells than all CD4+ T cells were CCR5+ (data not shown), suggesting that they are potential targets for direct infection with transmitted founder HIV/SIV. We also found that SIV-infected cells in tissues coexpressed BrdU in tissue sections (Fig. 5), clearly indicating that BrdU+ T cells are targets for direct viral infection. In further support of this, we found rapidly decreased levels of proliferating CD4+ T cells at between 8 and 10 days of infection, whereas markedly increased levels of proliferating CD8+ T cells were detected in the same tissues and at the same time points (Fig. 3 and 4). By focusing on early time points when samples are usually not obtained (most studies examine blood on a weekly basis, at days 7, 14, 21, etc.), we detected a proportional loss of proliferating CD4+ T cells that was rapidly overcome by increased proliferative CD4+ T cell responses (presumably to SIV antigens), which were first detected by day 13. These differential changes in proliferating CD4+ and CD8+ T cells indicate that primary SIV infection has early differential impacts on CD4+ and CD8+ T cell subsets. These results combined are strongly supportive of a selective infection and depletion of proliferating CD4+ T cells, particularly since proliferating CD8+ T cells were increased in primary SIV infection. Moreover, these results suggest that direct infection and destruction of the earliest-responding CD4+ T cells represent another mechanism by which SIV/HIV subvert the earliest attempts to mount an immune response.
Unfortunately, the inability to sort BrdU+ cells for quantitative viral quantification is a limitation of this study. Nonetheless, it has previously been shown that HIV selectively infects and destroys HIV-specific CD4+ cells, but the mechanism of selective infection has remained obscure (28). Here we show that there is a selective propensity for SIV to also infect dividing cells that are obviously proliferating under conditions of normal homeostasis or possibly in response to subclinical (nonspecific) infections. By inference, proliferating cells that will soon be responding to SIV antigens will also be selectively targeted. Thus, we propose the hypothesis that for SIV, transmitted founder viruses infect and deplete both virus-specific and non-virus-specific proliferating CD4+ T cells in very early infection in vivo, resulting in immediate impairments in the ability to effectively respond to new antigens and to the SIV or HIV infection itself. At about 12 to 13 days of infection, the emergence of virus-specific T cell responses to SIV infection overwhelms the ability to accurately track the dynamics of homeostatic and infectious T cell clones, and a selective destruction of S-phase CD4+ T cells in the face of marked proliferative responses to virus apparently results in a net increase in proliferative CD4+ and CD8+ T cells. Notably, the depletion of BrdU+ CD4+ T cells was found in animals only at 8 to 10 dpi, and by day 13, the levels of BrdU+ CD4+ T cells were restored, and this population persisted and increased in all chronically infected animals. This suggests that ongoing activation, proliferation, destruction, and turnover of CD4+ T cells begins at as early as 8 days and occurs in secondary lymphoid tissues throughout SIV infection.
Definitively determining the mechanism of CD4+ T cell depletion remains a central unresolved issue in AIDS research, and our data do not definitively prove the mechanisms of the targeting of CD4+ BrdU+ cells, yet it is logical that at least their capacity to support viral integration and replication may be facilitated within an actively dividing cell. Nonetheless, this rapid infection and destruction of proliferating CD4+ T cells detected in the earliest stages of infection likely continue throughout infection but are masked by marked yet insufficient attempts to restore T cell homeostasis through activation and proliferation of new target cells for infection. This undoubtedly impairs the earliest attempts to induce effective immune responses to SIV/HIV infection and likely affects homeostatic regulation of virus-specific and non-virus-specific CD4+ T cell turnover from very early in infection and may set the stage for viral persistence and the establishment of opportunistic infections. Thus, the selective loss of proliferating CD4+ T cells represents another very early alteration in immune homeostasis that may be directly attributed to primary SIV/HIV infection.
ACKNOWLEDGMENTS
We thank Julie Bruhn and Calvin Lanclos for flow cytometry support and Megan Gardner, Meagan Watkins, Terri Rasmussen, and Maury Duplantis for technical support.
We declare that we have no conflicts of interest.
This work was supported by NIH grant R01 AI084793 and the National Center for Research Resources and the Office of Research Infrastructure Programs (ORIP) of the National Institutes of Health through grant OD011104-51.
Footnotes
Published ahead of print 17 April 2013
REFERENCES
- 1. Chase A, Zhou Y, Siliciano RF. 2006. HIV-1-induced depletion of CD4+ T cells in the gut: mechanism and therapeutic implications. Trends Pharmacol. Sci. 27:4–7 [DOI] [PubMed] [Google Scholar]
- 2. Picker LJ, Hagen SI, Lum R, Reed-Inderbitzin EF, Daly LM, Sylwester AW, Walker JM, Siess DC, Piatak M, Jr, Wang C, Allison DB, Maino VC, Lifson JD, Kodama T, Axthelm MK. 2004. Insufficient production and tissue delivery of CD4+ memory T cells in rapidly progressive simian immunodeficiency virus infection. J. Exp. Med. 200:1299–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hellerstein MK, McCune JM. 1997. T cell turnover in HIV-1 disease. Immunity 7:583–589 [DOI] [PubMed] [Google Scholar]
- 4. Hellerstein MK. 1999. Measurement of T-cell kinetics: recent methodologic advances. Immunol. Today 20:438–441 [DOI] [PubMed] [Google Scholar]
- 5. Kaur A, Hale CL, Ramanujan S, Jain RK, Johnson RP. 2000. Differential dynamics of CD4(+) and CD8(+) T-lymphocyte proliferation and activation in acute simian immunodeficiency virus infection. J. Virol. 74:8413–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mohri H, Bonhoeffer S, Monard S, Perelson AS, Ho DD. 1998. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science 279:1223–1227 [DOI] [PubMed] [Google Scholar]
- 7. Rosenzweig M, DeMaria MA, Harper DM, Friedrich S, Jain RK, Johnson RP. 1998. Increased rates of CD4(+) and CD8(+) T lymphocyte turnover in simian immunodeficiency virus-infected macaques. Proc. Natl. Acad. Sci. U. S. A. 95:6388–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sachsenberg N, Perelson AS, Yerly S, Schockmel GA, Leduc D, Hirschel B, Perrin L. 1998. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 187:1295–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang ZQ, Notermans DW, Sedgewick G, Cavert W, Wietgrefe S, Zupancic M, Gebhard K, Henry K, Boies L, Chen Z, Jenkins M, Mills R, McDade H, Goodwin C, Schuwirth CM, Danner SA, Haase AT. 1998. Kinetics of CD4+ T cell repopulation of lymphoid tissues after treatment of HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 95:1154–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126 [DOI] [PubMed] [Google Scholar]
- 11. Perelson AS, Essunger P, Ho DD. 1997. Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS 11(Suppl A):S17–S24 [PubMed] [Google Scholar]
- 12. Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, Saag MS, Shaw GM. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117–122 [DOI] [PubMed] [Google Scholar]
- 13. Palmer LD, Weng N, Levine BL, June CH, Lane HC, Hodes RJ. 1997. Telomere length, telomerase activity, and replicative potential in HIV infection: analysis of CD4+ and CD8+ T cells from HIV-discordant monozygotic twins. J. Exp. Med. 185:1381–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolthers KC, Bea G, Wisman A, Otto SA, de Roda Husman AM, Schaft N, de Wolf F, Goudsmit J, Coutinho RA, van der Zee AG, Meyaard L, Miedema F. 1996. T cell telomere length in HIV-1 infection: no evidence for increased CD4+ T cell turnover. Science 274:1543–1547 [DOI] [PubMed] [Google Scholar]
- 15. Monceaux V, Ho Tsong Fang R, Cumont MC, Hurtrel B, Estaquier J. 2003. Distinct cycling CD4(+)- and CD8(+)-T-cell profiles during the asymptomatic phase of simian immunodeficiency virus SIVmac251 infection in rhesus macaques. J. Virol. 77:10047–10059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scholzen T, Gerdes J. 2000. The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 182:311–322 [DOI] [PubMed] [Google Scholar]
- 17. Combadere B, Blanc C, Li T, Carcelain G, Delaugerre C, Calvez V, Tubiana R, Debre P, Katlama C, Autran B. 2000. CD4+Ki67+ lymphocytes in HIV-infected patients are effector T cells accumulated in the G1 phase of the cell cycle. Eur. J. Immunol. 30:3598–3603 [DOI] [PubMed] [Google Scholar]
- 18. Veazey RS, Rosenzweig M, Shvetz DE, Pauley DR, DeMaria M, Chalifoux LV, Johnson RP, Lackner AA. 1997. Characterization of gut-associated lymphoid tissue (GALT) of normal rhesus macaques. Clin. Immunol. Immunopathol. 82:230–242 [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Xu H, Pahar B, Alvarez X, Green LC, Dufour J, Moroney-Rasmussen T, Lackner AA, Veazey RS. 2010. Simian immunodeficiency virus selectively infects proliferating CD4+ T cells in neonatal rhesus macaques. Blood 116:4168–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Borda JT, Alvarez X, Kondova I, Aye P, Simon MA, Desrosiers RC, Lackner AA. 2004. Cell tropism of simian immunodeficiency virus in culture is not predictive of in vivo tropism or pathogenesis. Am. J. Pathol. 165:2111–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clark DR, de Boer RJ, Wolthers KC, Miedema F. 1999. T cell dynamics in HIV-1 infection. Adv. Immunol. 73:301–327 [DOI] [PubMed] [Google Scholar]
- 22. De Boer RJ, Mohri H, Ho DD, Perelson AS. 2003. Turnover rates of B cells, T cells, and NK cells in simian immunodeficiency virus-infected and uninfected rhesus macaques. J. Immunol. 170:2479–2487 [DOI] [PubMed] [Google Scholar]
- 23. Hazenberg MD, Stuart JW, Otto SA, Borleffs JC, Boucher CA, de Boer RJ, Miedema F, Hamann D. 2000. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95:249–255 [PubMed] [Google Scholar]
- 24. Lempicki RA, Kovacs JA, Baseler MW, Adelsberger JW, Dewar RL, Natarajan V, Bosche MC, Metcalf JA, Stevens RA, Lambert LA, Alvord WG, Polis MA, Davey RT, Dimitrov DS, Lane HC. 2000. Impact of HIV-1 infection and highly active antiretroviral therapy on the kinetics of CD4+ and CD8+ T cell turnover in HIV-infected patients. Proc. Natl. Acad. Sci. U. S. A. 97:13778–13783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093–1097 [DOI] [PubMed] [Google Scholar]
- 26. Picker LJ. 2006. Immunopathogenesis of acute AIDS virus infection. Curr. Opin. Immunol. 18:399–405 [DOI] [PubMed] [Google Scholar]
- 27. Veazey R, Lackner A. 2003. The mucosal immune system and HIV-1 infection. AIDS Rev. 5:245–252 [PubMed] [Google Scholar]
- 28. Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. 2002. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417:95–98 [DOI] [PubMed] [Google Scholar]