

Abstract
Metastatic malignant melanoma remains one of the most therapeutically challenging forms of cancer. Here we test replication-competent vesicular stomatitis viruses (VSV) on 19 primary human melanoma samples and compare these infections with those of normal human melanocyte control cells. Even at a low viral concentration, we found a strong susceptibility to viral oncolysis in over 70% of melanomas. In contrast, melanocytes displayed strong resistance to virus infection and showed complete protection by interferon. Several recombinant VSVs were compared, and all infected and killed most melanomas with differences in the time course with increasing rates of melanoma infection, as follows: VSV-CT9-M51 < VSV-M51 < VSV-G/GFP < VSV-rp30. VSV-rp30 sequencing revealed 2 nonsynonymous mutations at codon positions P126 and L223, both of which appear to be required for the enhanced phenotype. VSV-rp30 showed effective targeting and infection of multiple subcutaneous and intracranial melanoma xenografts in SCID mice after tail vein virus application. Sequence analysis of mutations in the melanomas used revealed that BRAF but not NRAS gene mutation status was predictive for enhanced susceptibility to infection. In mouse melanoma models with specific induced gene mutations including mutations of the Braf, Pten, and Cdkn2a genes, viral infection correlated with the extent of malignant transformation. Similar to human melanocytes, mouse melanocytes resisted VSV-rp30 infection. This study confirms the general susceptibility of the majority of human melanoma types for VSV-mediated oncolysis.
INTRODUCTION
Of all skin cancers, melanoma has the highest mortality rate, and it accounts for approximately 75% of skin cancer-related deaths (1); the incidence rate has tripled over the last 3 decades (2). One contributing factor to the poor prognosis of advanced stage malignant melanoma is the high predisposition to develop brain metastases. It is the third most common solid tumor to metastasize to the brain, and stage IV melanoma patients have the highest risk (30 to 50%) of all cancer patients of tumor spread to the central nervous system (CNS) (3, 4). FDA-approved drugs such as dacarbazine and interleukin-2 have a limited impact on overall survival due to responses restricted to subsets of patients; the recently approved drug ipilimumab shows benefits in some patients (5). Response rates of newer drugs like BRAF inhibitors are higher (6, 7), but activity of these molecular-target agents depends on the presence of the corresponding mutation. In addition, response duration is often limited due to the development of secondary resistance (8). Key oncogenic driver mutations have been recognized delineating molecular pathways as possible targets for therapeutic intervention (9, 10). Among those, BRAF mutation, PTEN mutation, CDKN2A mutation, and AKT amplification are the most common events. The BRAFV600 mutation stands out as the most common melanoma mutation and is found in 50 to 65% of patients with melanoma (9).
A number of viruses have been studied using in vitro and in vivo models of melanoma tumors (11–21). A promising oncolytic virus with broad tropism against many cancers (22), vesicular stomatitis virus (VSV), has also been studied using melanoma models (23–25). Most of these melanoma studies have focused on mouse melanoma models and have facilitated substantially our current understanding of the role of the immune response in oncolytic virotherapy. However, the most commonly used mouse melanoma cell line B16 may be partially resistant to VSV infection (26). In addition, systemic application of potentially oncolytic virus, which was successfully shown in animal studies on other cancers (22, 27) and which is a prerequisite to target a metastatic tumor, remains to be studied with human melanoma.
As an oncolytic virus, VSV displays a strong affinity to tumors with defects in their interferon (IFN) signaling pathway (28, 29), and up to 50% of human melanomas carry defects in this pathway (30). Patients not responding to IFN treatment might ultimately benefit from agents exploiting a defective interferon pathway in these cancer cells. A number of recombinant variants of VSV have been introduced as potential oncolytic agents with modified biological properties mostly focusing on attenuation (22, 31–34). In contrast, the VSV variant used in many experiments in the current study, VSV-rp30, shows a more rapid infection and lysis of human glioma (35) and sarcoma (36) but had not been studied in melanoma. We also tested three other VSV variants including a wild-type-like phenotype (VSV-G/GFP) and attenuated phenotypes VSV-M51 and VSV-CT9-M51. We tested 19 primary human melanoma samples and normal human melanocytes for their susceptibility to VSV-rp30 infection and lysis. We find that the majority of melanomas are completely infected and killed by VSV even in the presence of IFN. This infection appears to be independent from NRAS mutation status; mutations BRAFV600E and BRAFV600K correlated with higher VSV susceptibility. Systemic application of VSV successfully targeted multiple subcutaneous as well as intracranial melanoma xenografts.
MATERIALS AND METHODS
Cells.
Human melanoma tissue from primary and metastatic sites was obtained from surgical cases at Yale New Haven Hospital, as previously described in detail (37). Most human melanoma samples were isolated and obtained from the Specimen Resource Core of the Yale SPORE in Skin Cancer; the YU prefix indicates tissue generated at Yale University. Tissue collection and processing were in accordance with the Health Insurance Portability and Accountability Act (HIPAA) and the institutional Human Investigative Committee protocol. Normal human melanocytes were derived from newborn foreskin tissue as described previously (38) and maintained as primary culture in Opti-MEM medium, described below, with the additional supplements 12-O-tetradecanoyl phorbol-13-acetate (16 nmol/liter), 3-isobutyl-1-methylxanthine (0.1 mmol/liter), cholera toxin (2.5 nmol/liter), Na3VO4 (1 μmol/liter), and N6,2′-O-dibutyryladenosine 3:5-cyclic monophosphate (0.1 mmol/liter) (Sigma-Aldrich).
Stably transfected rYUMAC cells expressing dsRed red fluorescent protein (RFP) were generated in a manner similar to that described previously (27). Briefly, YUMAC cells were transfected with plasmid pDsRed monomer-C1 coding for monomeric red fluorescent protein under the control of a cytomegalovirus (CMV) ie1 promoter and with a G418 selection construct, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Stable expression was achieved after 3 weeks of maintenance in medium enriched with G418 (Sigma, St. Louis, MO).
Mouse melanoma lines with defined mutation patterns were derived from transgenic mouse models expressing combinations of melanoma-associated mutations in the form of BrafV600E activation, Pten and/or Cdkn2a inactivation, and β-catenin stabilization as described in detail previously (39–41). In short, mouse models were generated by conditional melanocyte-specific expression of a Tyr::CreERT2 construct leading to Cre-mediated conversion of Braf, PTEN, and other genes. Tumor-associated genes were recombined after release of Cre recombinase from Hsp90 initiated by topical application of 4-hydroxytamoxifen (4-HT) and subsequent nuclear translocation of Cre recombinase. Tumor tissues were triturated through a 20-gauge needle and filtered through a 100-μm nylon cell strainer.
All human and mouse melanoma cells were maintained in Opti-MEM supplemented with 5% fetal bovine serum (FBS) and were kept in a humidified atmosphere containing 5% CO2 at 37°C.
Viruses.
Six recombinant VSV variants were used in this study: wild-type-based VSV-G/GFP, VSV-rp30, VSV-M51, VSV-CT9-M51, VSV-P126, and VSV-L223. Virus construction and characteristics of the first four have been detailed previously (34). VSV-G/GFP is based on the VSV Indiana serotype with an additional copy of the viral G protein gene fused to the GFP (green fluorescent protein) gene located at the 5th gene position. VSV-rp30 evolved from VSV-G/GFP through repeated passage on and adaptation to U87 human glioblastoma cells (35). VSV-M51 contains a deletion at amino acid position 51 of the VSV M protein; VSV-CT9-M51 is additionally modified by having a truncated cytoplasmic tail of the viral G protein, to 9 amino acids (42). VSV-M51 and VSV-CT9-M51 include a GFP reporter gene at the 5th position coding for cytosolic GFP. VSV-P126 andVSV-L223 are described below.
All viruses were plaque purified and propagated, their titers were determined on BHK-21 cells, and they were stored in aliquots at −80°C. For titer determinations, a standard plaque assay technique with 0.5% agar overlay was employed.
VSV genome sequencing.
Viral genomic RNA was extracted from VSV-G/GFP and VSV-rp30 using the QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA). Reverse transcription (RT) of isolated VSV genomic RNA was performed using Thermo-X Reverse Transcriptase (Invitrogen, Carlsbad, CA) at a reaction temperature of 53.9°C and an RT oligonucleotide with the sequence 5′-ACG AAG ACA AAC AAA CCA TTA TTA TC-3′, designed to anneal to the initial 26 bases of the 3′-end of the VSV genome. Full-length genomic VSV-G/GFP cDNA (13.5 kb) was used as the template in a series of 6 PCRs that generated overlapping products covering the entire genome and ranging in size from 2.0 to 2.7 kb. Primers were designed on the basis of the VSV-G/GFP sequence (GenBank accession number FJ478454). PCR was performed using the Expand High Fidelity PCR kit (Roche Diagnostics, Indianapolis, IN), and the reaction products were purified using the QIAquick PCR Purification kit (Qiagen). Purified products were sequenced by the W. M. Keck DNA Sequencing Facility (Yale University). Sequencing of each PCR product was performed on both the sense and antisense DNA strands. Potential sequencing ambiguities that arose in one strand were resolved by inspection of sequence results from the complementary strand.
Restriction digestion using BsrDI (New England BioLabs, Beverly, MA) was performed to assess homogeneity of the mutation throughout the VSV-rp30 population. A 2,126-bp PCR product was amplified from VSV-G/GFP and VSV-rp30 cDNA using the same methods as described above and primers 960F, 5′-AAA ACC CTG CCT TCC ACT TC-3′, and 3047R, 5′-AAC AGA TCG ATC TCT GTT AG-3′, at an annealing temperature of 52°C for 30 cycles. The presence of the C1772T mutation in the gene for the P protein of VSV-rp30 creates a unique BsrDI restriction site that yields two smaller species of 1,298 bp and 828 bp. PCR products were purified and quantified as described above, and 350 ng of each was included in restriction digestion reactions and incubated according to the manufacturer's instructions. Digestion products were run on 1.2% agarose gels stained with ethidium bromide.
VSV-P126 and VSV-L223 plasmid construction, virus recovery, and verification.
The plasmid pVSV-G/GFP encoding the parent virus was constructed from pVSV-XN2 (43) and pBS-G/GFP as described previously (44). To construct the genomic VSV plasmid encoding the P126 mutation, a 1-kb BstZ17I-XbaI fragment spanning the N and P genes and including the mutation site was first digested from pVSV-XN2 and then subcloned into pEBX, a modified pCR-Blunt II-TOPO plasmid (Invitrogen). The pEBX plasmid was generated by inserting a short linker encoding a BstZ17I site into the multiple-cloning site (MCS) of pCR-Blunt II-TOPO between the EcoRI and XbaI sites. This linker was prepared by digesting an annealed pair of complementary oligonucleotides and, after insertion, yielded the following modified sequence between the EcoRI and XbaI sites: 5′-GAATTC CA GTATAC TAT TCTAGA-3′ with the EcoRI, BstZ17I, and XbaI sites (in that order) underlined. Insertion of the 1-kb N/P gene BstZ17I-XbaI fragment digested from pVSV-XN2 into pEBX yielded the subclone pN/Psub. Next, a 0.4-kb BlpI-XbaI fragment of pN/Psub was removed and replaced with a comparable gene-synthesized fragment (Genscript, Piscataway, NJ) encoding the P126 mutation to create the plasmid pN/P-P126. This newly mutagenized BstZ17I-XbaI fragment was then digested from pN/P-P126 and used to replace the comparable sequence of pVSV-G/GFP to create pVSV-P126.
To construct the VSV plasmid bearing the L223 mutation, a 4.4-kb NheI-FseI fragment encoding the N-terminal half of the L protein gene was digested from pVSV-XN2 and then subcloned into pENFP, another modified version of pCR-Blunt II-TOPO. The pENFP plasmid was generated by first digesting pCR-Blunt II-TOPO with FseI and DraIII, blunting the ends, and then ligating to destroy the FseI and DraIII restriction sites located in an unused Zeocin resistance gene in the plasmid backbone. This procedure resulted in the deletion of 77 bp between the FseI and DraIII sites to create pCR-Blunt II-TOPO(−)FseI/DraIII. Next, a short linker encoding an NheI and FseI site was inserted into the MCS of pCR-Blunt II-TOPO(−)FseI/DraIII between the EcoRI and PstI sites to create pENFP. This linker was prepared by digesting an annealed pair of complementary oligonucleotides and, after insertion, yielded the following modified sequence between the EcoRI and PstI sites: 5′-GAATTC GCTAGC ATA GGCCGGCC CTGCAG-3′ with the EcoRI, NheI, FseI, and PstI sites (in that order) underlined. Insertion of the 4.4-kb L gene NheI-FseI fragment digested from pVSV-XN2 into pENFP yielded the subclone pLsub. Next, a 0.3-kb SacII-BstAPI fragment of pLsub was removed and replaced with a comparable gene-synthesized fragment (Genscript) with the L223 mutation to create the plasmid pL-L223. This newly mutagenized NheI-FseI fragment was then digested from pL-L223 and used to replace the comparable sequence of pVSV-G/GFP to create pVSV-L223.
Recombinant VSVs were recovered from pVSV-P126 and pVSV-L223 plasmid DNA through transfection of BHK-21 cells and infection with vaccinia virus-T7 (45, 46). Seed stocks of the additional plasmids used in recombinant VSV recovery (pBS-N, pBS-P, and pBS-L), along with the plasmids pVSV-XN2 and pBS-G/GFP, were kindly provided by J. Rose (Yale University). Verification of the successful recovery of VSV-P126 and VSV-L223 was performed by sequencing RT-PCR products amplified from genomic RNA harvested from infected BHK-21 cells. VSV-P126, VSV-L223, VSV-rp30, and VSV-G/GFP were compared for infection effects, replication rates, and plaque sizes in several human melanomas.
Quantification of infection and cytotoxicity.
All VSV variants express the GFP reporter gene. Cells were seeded in either 24- or 12-well dishes at 5 × 104 or 1 × 105 cells per well, respectively, and incubated overnight. VSV variants were added by changing the medium to medium containing the indicated viral concentration for a final multiplicity of infection (MOI) of 0.1 or 10 PFU per cell, respectively. At the indicated time points, ranging from 24 to 48 h, infectivity rates of melanoma cultures were assessed as the number of GFP-positive cells compared to the total amount of cells, using an Olympus IX71 fluorescence microscope connected to a SPOT RT digital camera (Diagnostic Instruments, Sterling Heights, MI). A minimum of 10 microscopic fields were quantified per condition. Phase-contrast imaging was used for assessment of cytopathic effects (CPE) (rounding, blebbing, detachment, shrinkage, and cell fusion). Cytotoxicity was quantified using ethidium homodimer (EthD-1; Molecular Probes, Eugene, OR), which labels dead cells with a red nuclear fluorescence. EthD-1 stock solution was added to infected cultures in 24-well dishes at the indicated time points for a final concentration of 4 μM. Dead cells were quantified in experimental culture dishes after virus infection using a fluorescence microscope.
Interferon treatment.
Interferon pretreatment was performed by incubating cultures with medium containing 100 IU/ml human IFN-αA/D (catalog no. I4401; Sigma-Aldrich) for 3 h before adding virus at an MOI of 10. IFN-αA/D is a hybrid molecule based on recombinant human IFN-αA and -αD and acts on both mouse and human IFN-α receptors, as explained in the Sigma I 4401 product sheet.
Quantitative RT-PCR for analysis of interferon-stimulated genes (ISGs) in human melanoma.
RNA from human melanoma cultures with or without 6-h IFN-α treatment was isolated from cell lysates using the RNeasy Plus kit (Qiagen, Valencia, CA). DNase-treated RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). TaqMan gene expression assays were used for quantitative PCR of β-actin and interferon-stimulated genes OAS-1 and Mx1 using an ICycler iQ Real Time PCR system (Bio-Rad, Hercules, CA, USA). Assays were purchased from Applied Biosystems (Foster City, CA). Each sample was measured using triplicate PCRs. OAS-1 and Mx1 quantities were normalized to corresponding β-actin expression levels for each cell type.
Animal procedures.
For human melanoma xenografts, 4- to 6-week-old immunodeficient homozygous CB-17-SCID mice (Taconic, Inc.) were used. For subcutaneous flank tumor xenografting, mice were anesthetized using a 20% (vol/vol) isoflurane-propylene glycol mix. Melanoma cells (5 × 105) tested for pathogen-free status were suspended in phosphate-buffered saline (PBS), and 100-μl suspensions were injected into a skin fold on the flanks of the mice. Mice received subcutaneous flank xenografts at up to 4 different sites. Palpable tumors formed within 3 weeks. For intracranial melanoma xenografts, mice were anesthetized by intraperitoneal (i.p.) injection of a ketamine-xylazine mixture (100 and 10 mg/kg body weight, respectively) and placed into a stereotactic frame. A Hamilton syringe controlled by a Stoelting stereotactic injector (Stoelting, Wood Dale, IL) was used to inject 1 μl of melanoma cell suspension containing 5 × 104 cells into the striatum (2 mm lateral, 0.4 mm rostral to bregma, at a 3-mm depth). The following human melanoma cells were used for intracranial tumor grafts: red fluorescent rYUMAC, melanotic YUSIK, or amelanotic YUSIT. For systemic virus application, a single bolus of VSV-rp30 (1 × 108 PFU in 100 μl PBS) was injected via tail vein. Animals were monitored daily for body weight, grooming, and overall health. Mice were euthanized with a pentobarbital overdose and perfused transcardially with 4% paraformaldehyde at the indicated time points ranging from 2 to 5 days after virus inoculation. Subcutaneous and intracranial tumors were examined via fluorescent stereomicroscope (Olympus SZX12; Olympus Optical, Tokyo, Japan) before microtome sectioning for subsequent histological analysis. All experiments were performed in accordance with institutional guidelines of the Yale University Animal Care and Use Committee.
RESULTS
VSV-rp30 selectively infects a broad range of human melanoma cells.
Little is known about VSV targeting of human melanoma cells freshly derived from patients undergoing resective surgery and potential correlations between melanoma mutation profiles and the susceptibility to viral oncolysis. Here, we present for the first time a large panel of different human melanomas with distinct growth characteristics and mutational defects. Of the 19 human patient-derived melanoma samples tested, 5 originated from primary tumor sites; all other melanomas were collected from metastatic or recurrent sites (Table 1).
Table 1.
Summary of susceptibility of human melanoma cells for VSV infection and genetic and clinical characteristicsa
| Melanoma sample ID | Low MOI infection | VSV replication | IFN insensitivity | OS score | Gene mutation status |
Clinical characteristic/site(s) | Gender/age (yr) | |
|---|---|---|---|---|---|---|---|---|
| BRAF | NRAS | |||||||
| YUSIK | +++ | +++ | +++ | 9 | V600E | WT | Metastasis/lymph node | F/49 |
| 501MEL | +++ | +++ | +++ | 9 | V600E | WT | Metastasis/lymph node | M/71 |
| YUMAC | +++ | +++ | +++ | 9 | V600K | WT | Metastasis/back | M/52 |
| YUSIT | +++ | +++ | +++ | 9 | V600K | WT | Metastasis | M/67 |
| YUTICA | +++ | +++ | + | 7 | WT | Q61R | Metastasis/pleural fluid | M/65 |
| YUSIV | +++ | +++ | + | 7 | WT | WT | Metastasis/chest | F/61 |
| YURIF | +++ | +++ | − | 6 | V600K | WT | Recurrence/thigh | M/52 |
| YUFIC | +++ | + | 4 | WT | Q61R | Metastasis/lymph node | M/65 | |
| YUCYLO | +++ | + | 4 | V600E | WT | Primary/upper arm | F/62 | |
| YUPEET | +++ | ++ | − | 5 | V600E | WT | Primary/back | M/54 |
| YUAME | +++ | ++ | − | 5 | WT | G13R | Metastasis/lymph node | M/61 |
| YUGEN8 | +++ | ++ | 5 | V600E | WT | Metastasis/brain | F/44 | |
| YUHEF | ++ | +++ | +++ | 8 | WT | WT | Metastasis/lung | M/52 |
| YUDOSO | ++ | + | + | 4 | WT | Q61K | Primary/abdomen | M/84 |
| YUNIGE | + | ++ | ++ | 5 | WT | WT | Metastasis/brain, CSF | M/59 |
| YUFULO | + | + | 2 | WT | Q61L | Primary/ocular uveal | M/86 | |
| YUROL | + | + | + | 3 | WT | WT | Recurrence/calf | M/85 |
| YUKIM | − | ++ | − | 2 | WT | Q61R | Metastasis/lymph node | F/71 |
| YULOVY | − | − | − | 0 | WT | Q61L | Primary/calf | F/83 |
Human melanoma cells were ranked for their susceptibility to VSV infection at low MOI (0.1), as follows: +++, 80 to 100%; ++, 50 to 79%; +, 10 to 49%; −, less than 10%. The OS (oncolytic susceptibility) score reflects the sum of individual scores for infectivity (0 to 3), interferon resistance (0 to 3), and viral replication capacity (0 to 3), with 9 being the highest OS score and 0 being the lowest. WT, wild type; CSF, cerebrospinal fluid.
Compared to the parent wild-type-based VSV-G/GFP, VSV-rp30 showed somewhat enhanced infectivity toward human glioblastoma (35) and sarcomas (36). To determine the general susceptibility of human melanomas for VSV-rp30 infection, semiconfluent cultures of 17 human melanoma samples were infected at a high (10) and low (0.1) MOI. Fluorescence microscopy was used to detect GFP expression to quantify infection rates at 36 h postinoculation (hpi). The ratio of the number of infected cells to total cells is shown in Fig. 1A.
Fig 1.

VSV-rp30 infection of 17 short-term human melanoma cultures. Cultures of human melanoma from primary and metastatic sites were infected with VSV-rp30 at a low (0.1) or high (10) MOI with VSV-rp30 and analyzed 36 h later for viral infection (A) displayed as percentage of GFP-expressing cells and for rates of cytopathic effects (B). Viral infection was correlated with cytopathic effects.
Fourteen of 17 melanomas showed complete or nearly complete infection by 36 hpi with a high MOI. Three melanoma types, YUKIM, YULOVY, and YUROL, showed less than 50% infection, even with a high MOI of VSV-rp30, indicating a partial resistance. In contrast, normal human melanocytes were remarkably resistant to VSV-rp30 even at a high MOI, with only 3.8% ± 0.6% cells infected after 36 h.
Low MOI (0.1) conditions are useful to test for the oncolytic potency of a virus since it factors in viral replication as a prerequisite for complete infection of a culture. At a low MOI, the majority of melanoma derivations (11 of 17) showed complete infection by 36 hpi. These include YUAME, YUFIC, YUGen8, YUMAC, YUPEET, YURIF, YUSIK, YUSIT, YUSIV, YUTICA, and 501Mel. Four melanomas, YUKIM, YUNIGE, YUROL, and YULOVY, displayed significantly reduced infection rates, showing fewer than 30% of cells infected. YUDOSO and YUHEF showed substantial infection, with 60 to 80% of cells infected. VSV susceptibility of melanomas with BRAF mutation V600E or V600K was significantly higher than that of non-BRAF-mutated cells (P = 0.02; Mann-Whitney test) (see Fig. 6A, left box). In contrast, NRAS mutation status did not have a significant effect on infection rates (see Fig. 6A, center box) compared to infection rates in cells with wild-type status for BRAF and NRAS (see Fig. 6A, right box).
Fig 6.
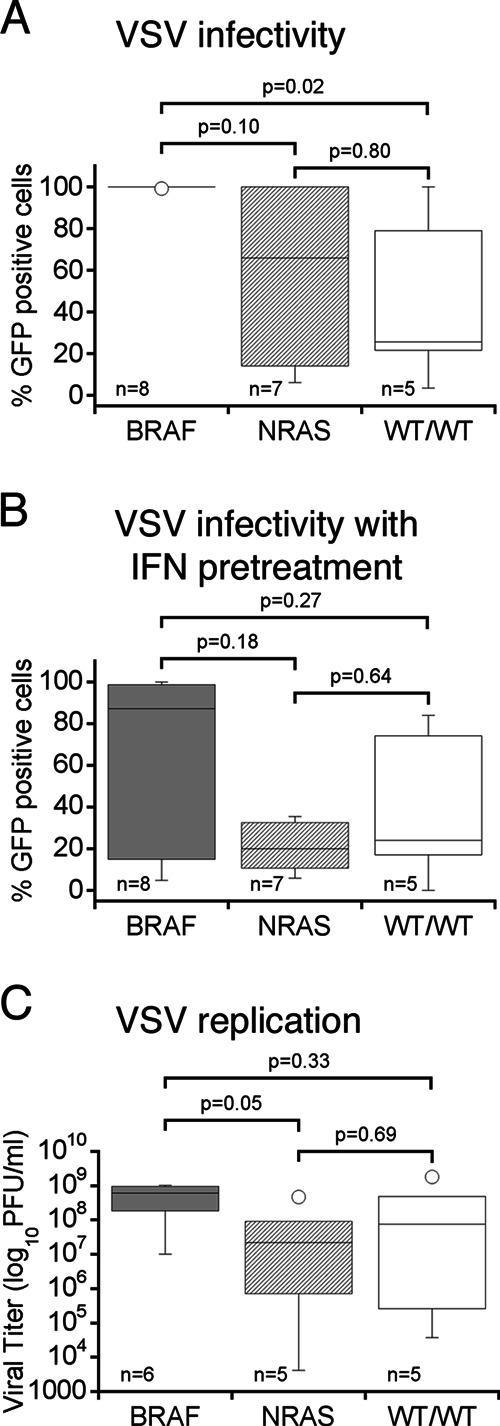
Relationship between BRAF and NRAS mutation status and VSV susceptibility. Box plots of human melanoma cells are grouped according to their melanoma-associated BRAF (dark gray panels) and NRAS (light gray panels) mutation status. WT/WT (white panels) indicates wild-type gene status for both BRAF and NRAS. Parameters assessed for correlation were (i) infectivity at low MOI (0.1) (A), (ii) infectivity after IFN pretreatment (B), and (iii) viral replication (C). All melanoma cells with BRAF mutation show a strong susceptibility for VSV infection, whereas NRAS-mutated cells and wild-type cells vary in their susceptibility. No association was found for RAS mutation and VSV susceptibility. Horizontal bars within a box indicate group means; statistically significant difference is defined at P values of <0.05 using the Mann-Whitney test.
To corroborate the view that infection as determined by reporter gene expression correlates with cell death, we studied cytopathic effects using phase-contrast microscopy. This analysis revealed infected cells displaying robust cytopathic effects (rounding, blebbing, membrane fusion, and detachment) (Fig. 2A, column 2). The ratios of the number of cytopathic cells to the total number of cells paralleled the infection ratios and are shown in Fig. 1B (correlation coefficient for all GFP expression rates versus cell death rates, R, was 0.94 for low MOI and 0.93 for high MOI). Consistent with the small number of cells showing reporter gene expression, normal human melanocytes showed very few signs of cytopathic effects (see Fig. 3C).
Fig 2.

Oncolytic action of VSV on human melanoma. (A) Representative collection of 5 human melanoma cultures infected with VSV-rp30 at an MOI of 0.1 for 36 h. EthD-1 (red) labels the nuclei of dead or dying cells. Fluorescence imaging reveals a strong correlation between the number of infected cells (green) and dead cells (red) and the appearance of cytopathic effects (phase-contrast images). (B) Bar graph showing quantification of cytotoxicity of 7 human melanoma cultures infected with VSV-rp30 at an MOI of 0.1 using EthD-1 cell death staining.
Fig 3.

IFN pretreatment retards VSV-rp30 infection and cytolytic rates on some human melanomas. (A) The presence of IFN reduced infection in about one half of melanoma samples tested at 36 hpi. (B) The other half of the melanoma samples displayed cytopathic effects after VSV-rp30 infection even in the presence of IFN. (C) Panel of representative images of YUMAC melanoma cells and normal human melanocytes (control cells) taken 24 h after VSV-rp30 inoculation at high (10) or low (0.1) MOI and in the presence of IFN-α. Phase-contrast images show normal cell morphology or cytopathic effects. GFP expression indicates viral infection.
Ethidium homodimer (EthD-1) stains dead cells only after the integrity of the plasma membrane has deteriorated. To corroborate our morphological analysis, an EthD-1 cell death assay was used to assess a representative selection of human melanoma to confirm the correlation between morphological CPE of virus and subsequent cell death (Fig. 2A, right column). Quantification of ratios of EthD-1-indicated that cell death rates were comparable to CPE (Fig. 2B) at 36 hpi with VSV-rp30 (0.1 MOI), yielding a correlation coefficient, R, of 0.87. These data are consistent with the notion that CPEs observed after VSV infection are indicative of subsequent cell death. YULOVY cells were highly resistant to VSV-rp30 infection as indicated by low GFP and EthD-1 ratios, whereas the other melanomas showed a high level of dead cell staining. In addition to the 17 human melanoma cultures listed above that were systematically compared in parallel experiments, two additional primary melanoma cultures (YUCYLO and YUFULO) were assessed independently for GFP expression and lytic effects after VSV infection in separate experiments (data not shown). A summary of infection susceptibility and other characteristics for all tested 19 melanomas is shown in Table 1.
Effect of interferon administration on VSV infection of human melanoma.
The selectivity of the oncolytic action of VSV is based in part on its sensitivity to interferon-mediated antiviral defenses that protect normal cells from infection. This IFN response is often compromised in tumor cells of various derivations (29, 47). On the other hand, IFN-α has been used as a therapeutic agent against melanoma and displays a relapse-free survival advantage (48).
Tumor responses to IFN vary substantially, possibly indicating that defects in IFN signaling are present in a substantial number of melanomas. We therefore addressed to what extent IFN-α inhibits VSV infection and cell death of human melanoma cells. Cultures of human melanomas were incubated with 100 IU IFN-αA/D for 3 h prior to application of VSV-rp30 at an MOI of 10. Thirty-six hours later, fluorescence microscopy was used to quantify GFP expression, indicative of infection rates (Fig. 3A). Approximately 37% of the human melanomas tested (7 of 19, i.e., YUHEF, YUMAC, YUNIGE, YUSIK, YUSIT, YUGEN8, and 501Mel) were unresponsive to IFN-mediated protection from VSV-rp30 infection. In contrast, IFN almost completely blocked infection of YUAME, YULOVY, YUPEET, and YURIF cells, all of which except YULOVY were completely infected by VSV under control conditions (Fig. 1A). In addition, IFN reduced infection rates in YUDOSO, YUFIC, YUKIM, YUROL, YUSIV, and YUTICA. The effect of IFN pretreatment on VSV-rp30 infection of human melanoma cells was independent from either BRAF or NRAS mutation status (see Fig. 6B).
Interestingly, cytopathic effect rates noticeably exceeded infection rates in the presence of IFN (Fig. 3B) compared to control conditions, in which the extent of cytopathic effects paralleled infection rates (R = 0.81). Even in cells displaying low infection rates, widespread cell death was seen (YUDOSO, YUFIC, YUKIM, YURIF, and YUTICA), suggesting a possible synergistic effect of VSV infection and interferon. We did not, however, observe direct cytotoxicity of IFN-α on these melanomas in the absence of VSV-rp30, with cell numbers in IFN-treated dishes not significantly different from control cell numbers (data not shown). In contrast to the modest attenuation of infection rates of VSV-rp30 by IFN in melanoma, normal human melanocytes were completely protected by IFN (Fig. 3).
Expression of interferon-stimulated genes (ISGs) in melanoma cells compared to normal melanocytes.
In order to assess the responses of human melanoma and normal melanocytes to interferon application, we used quantitative reverse transcription-PCR (qRT-PCR) to examine the expression of two well-characterized interferon-stimulated genes, Mx1 and OAS (49, 50). After 6 h of IFN-α treatment (100 U/ml), RNA harvested from normal human melanocytes showed a substantial increase in levels of Mx1 (1,900-fold) and OAS (2,930-fold) compared to untreated controls. In contrast, 11 melanomas varied substantially in their IFN response and displayed significantly less induction of ISGs than did control cells. Three melanoma cultures (YUTICA, YUMAC, and YUSIT) showed a considerable induction of ISGs in the range of several hundredfold for Mx1 and OAS. The remaining 8 melanomas showed relatively little induction of ISGs compared to normal melanocytes, with particularly low values seen in YUSIK, 501mel, YULOVY, YUNIGE, and YUDOSO, which responded to IFN treatment with Mx1 induction of only 1.2- to 5-fold and YUDOSO and YULOVY, which showed an OAS induction of only 5- to 6-fold (Fig. 4). Interestingly, with some melanoma cultures, such as YUSIK and 501mel, the strongly attenuated ISG induction correlated with a lack of protection by IFN from VSV-rp30 infection, whereas in other melanomas it does not. Examples for the latter include YULOVY, YUAME, and YURIF, which when treated with IFN show little induction of the tested ISGs but are completely protected from VSV-rp30 infection. Conversely, the melanomas with the strongest upregulation of ISGs, YUMAC and YUSIT, paradoxically showed no protection of these cells by IFN from VSV infection (Fig. 3 and Table 1). The finding that interferon-induced expression of antiviral effectors Mx1 and OAS is not highly predictive of the level of IFN-mediated viral resistance in melanoma cells suggests that ISG expression may not consistently correlate with virus susceptibility.
Fig 4.
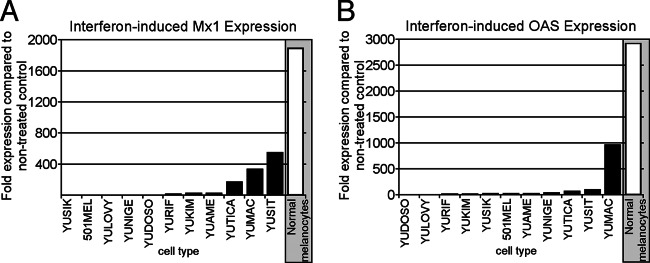
Reduced induction of interferon-stimulated genes in melanoma compared to normal melanocytes. Quantitative RT-PCR was applied for gene expression studies on control and IFN-αA/D-treated cultures of 11 human melanoma samples and normal human melanocytes. Two specific interferon-stimulated genes (ISGs), Mx1 (A) and OAS (B), were analyzed. β-Actin was used as a reference for each cell type to normalize expression levels. Data reflect the fold induction relative to nontreated control cultures. Normal melanocytes showed induction levels of several thousandfold for both ISGs. In contrast, melanoma tumor cells showed significantly reduced induction of ISGs.
Comparison of recombinant VSVs in infecting melanoma.
An important factor determining the potency of an oncolytic viral agent is the speed with which it can replicate and locally amplify the cytolytic effect before the adaptive immune system mounts an effective response. From this perspective, a virus with faster replication may present a favorable phenotype. Employing standard plaque assay techniques, we quantified the replication rates of VSV-rp30 and wild-type-based VSV-G/GFP on 15 melanoma cultures and normal human melanocytes. Eighty percent confluent duplicate cultures were infected with each virus at an MOI of 0.1. Samples of supernatant were collected at 7 hpi and at a later time point, 24 hpi. At 7 hpi, virus progeny production varied widely between different melanoma cultures, ranging from 102 to 106 PFU/ml. VSV-rp30 produced significantly higher titers of progeny than VSV-G/GFP in 13 of 15 melanoma cultures (Fig. 5A). For 12 of these cultures (excluding outlier YUROL) the mean (and standard error of the mean [SEM]) difference of viral replication was 3.8 ± 0.6 in favor of VSV-rp30. One melanoma culture, YUROL, had an unusually high replication advantage of VSV-rp30 over VSV-G/GFP; however, replication in YUROL was also unusually low and thus showed a relatively large error in the measurement. One melanoma culture, YUDOSO, showed higher titers for VSV-G/GFP than for VSV-rp30 (64-fold). Viral titers were higher at 24 hpi, ranging from 105 to 3 × 109 PFU/ml. Differences between total virus propagation of VSV-rp30 and VSV-G/GFP were less apparent, suggesting that VSV-rp30's advantage is a factor of speed more than total progeny produced/cell (Fig. 5B). In contrast, measurable viral replication in normal melanocytes was absent at 7 hpi and was highly reduced compared to 14 of 15 melanomas after 24 h. Although not statistically significant, melanoma cells with BRAF mutations showed a clear trend toward greater replication than cells without these mutations (P = 0.07; Mann-Whitney test) (Fig. 6C). No such trend was observed in NRAS-mutated cells (P = 0.14).
Fig 5.
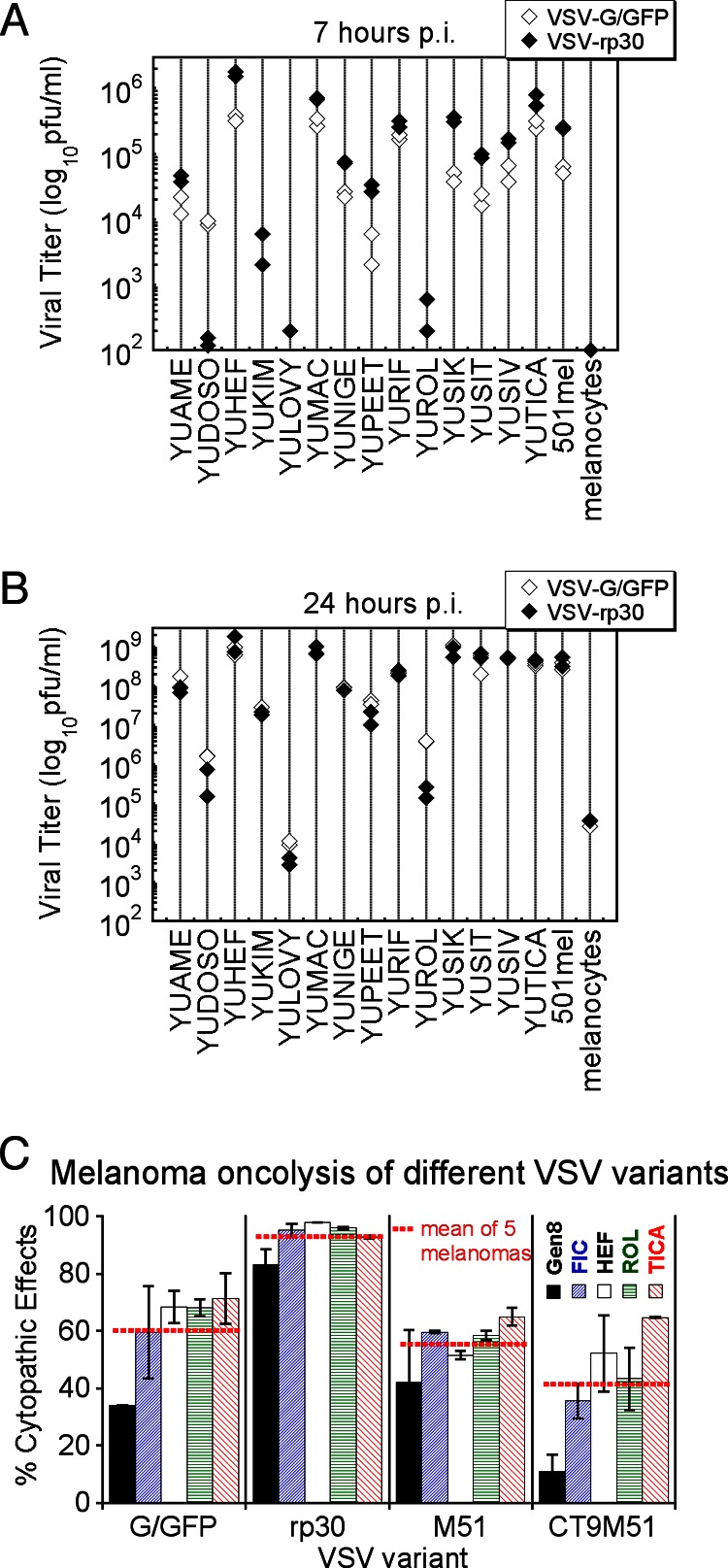
Replication and oncolytic effects of different VSVs in melanoma cells. Viral replication was quantified at 7 (A) and 24 (B) hours following virus inoculation at an MOI of 0.1. For most melanoma cells, infection of VSV-rp30 (black diamonds) resulted in higher titers than VSV-G/GFP (white diamonds), with a particularly significant difference at the early time point. Viral replication in normal melanocytes yielded significantly smaller amounts of VSV progeny. (C) Comparison of low MOI (0.1) infection of different VSV (VSV-G/GFP, VSV-rp30, VSV-M51, and VSV-CT9-M51) on 5 different human melanoma cultures at 24 hpi. VSVrp30 showed the strongest cytopathic effect on melanoma cells. Displayed is the percentage of cytopathic effects compared to noninfected controls. This was based on analysis of duplicate cultures and 10 microscopic fields per cell type and condition. Horizontal red bars indicate the mean values of 5 melanoma cultures combined.
We also compared VSV-rp30 and VSV-G/GFP with two attenuated VSV-variants, VSV-M51 and VSV-CT9-M51. A representative panel of 5 melanoma cultures were infected with VSV at a low MOI of 0.1 and assessed for CPE rates at 24 hpi. All 5 melanomas, YUFIC, YUHEF, YUROL, YUTICA, and YUGen8, were nearly dead or dying after VSV-rp30 infection, with a mean of 93.0% ± 2.6% for all 5 samples. In contrast, infection with VSV-G/GFP (60.3% ± 6.9%), VSV-M51 (55.4% ± 4.0%), and VSV-CT9-M51 (41.4% ± 9.0%) resulted in significantly less cell lysis at 24 hpi (Fig. 5C). Importantly, at later time points, even the most attenuated VSV variants resulted in widespread cell death of the melanoma tested (data not shown), indicating that all four VSVs successfully killed multiple melanoma cultures.
VSV-rp30 genome sequencing reveals two nonsynonymous mutations.
VSV-rp30 was originally generated through repeated passage of VSV-G/GFP on human glioblastoma U87 cultures to select viral progeny with faster growth kinetics on multiple human gliomas (35). To identify the mutation(s) associated with this phenotype, we sequenced the entire genome of VSV-rp30 and the parent strain VSV-G/GFP (Fig. 7). The sequences of both viruses were found to be identical except for four point mutations located in the genes for the P protein (C1772T) and L protein (G7693T, A11712G, and C11736T), with nucleotide numbering according to GenBank accession number FJ478454 (Fig. 7B). Inspection of the sequence chromatograph data indicated that all of the detected VSV-rp30 mutations were represented by a single unambiguous chromatographic peak (Fig. 7B), suggesting that these mutations were present throughout the VSV-rp30 viral population. The C1772T mutation appears in the second position of codon 126 of the P protein gene and results in a replacement of the serine (S) in VSV-G/GFP with a leucine (L) in VSV-rp30 (S126L). The G7693T mutation appears in the first position of codon 223 of the gene for L protein and results in a replacement of the asparagine (D) in VSV-G/GFP with a tyrosine (Y) in VSV-rp30 (D223Y). The A11712G and C11736T mutations were both silent base substitutions appearing in the third position of codons 1562 and 1570 of the L protein gene. Since the C1772T mutation results in the creation of a Bsr DI restriction site in VSV-rp30, we were able to further assess the apparent homogeneity of this mutation within the viral population by means of restriction digestion. Using a 2.1-kb PCR product encompassing the C1772T mutation site, Bsr DI yielded a complete digestion of the VSV-rp30-amplified PCR product and no detectable digestion of VSV-G/GFP-derived product (Fig. 7C). These results indicate an absence of any detectable sequence heterogeneity within the VSV-rp30 population at the identified mutation site.
Fig 7.

VSV-rp30 sequencing and mutation identification. Sequencing of the entire genome for both VSV-G/GFP and VSV-rp30 was performed using genomic-length cDNAs. (A) Diagram illustrating the parent VSV-G/GFP genome and the positions of the four mutations (C1772T, G7693T, A11712G, and C11736T) identified in the glioblastoma cell-adapted variant VSV-rp30. (B) Sections of the sequencing chromatographs showing both the parent VSV-G/GFP (above) and the mutated VSV-rp30 codons (below). Altered nucleotides are shown in bold type. The affected protein and corresponding amino acid positions are shown below each pair of chromatographs. Nucleotide numbering is based on the VSV-G/GFP sequence (GenBank accession number FJ478454). (C) BsrDI restriction digestion of a 2.1-kb PCR product encompassing the C1772T mutation site identified in the P gene of VSV-rp30. VSV-rp30 PCR product incubated in the presence (+) of BsrDI resulting in the complete digestion of the product into the predicted 1.3-kb and 0.8-kb fragments (lane 5), indicating that all of the VSV-rp30 product contained the C1772T mutation recognized by BsrDI. (D) Representative panel of micrographs showing phase-contrast and corresponding GFP fluorescence of YUSIK melanoma tissue infected with VSV-G/GFP, VSV-rp30, VSV-P126, and VSV-L223 at an MOI of 0.1 at 15 hpi. (E) Graph depicting infection rates using GFP expression analysis for each VSV variant on three different human melanomas (mean ± SEM; n = 10 microscopic fields per condition). (F) Comparison of replication of VSV variants on 3 different human melanoma samples. Horizontal lines show means of duplicate titer determinations. (G) Comparison of plaque sizes. Black circles indicate mean (± SEM) relative plaque sizes compared to control VSV-G/GFP plaque diameters (n = 60 per condition). The “mean” row shows the combined mean relative plaque sizes from all three melanoma cells tested.
Because sequencing VSV-rp30 revealed two nonsynonymous mutations, we asked whether both mutations are required for the oncolytically enhanced phenotype of VSV-rp30. We generated single mutant variants VSV-P126 and VSV-L223 by inserting the corresponding mutations into the parental wild-type-based strain VSV-G/GFP. Plaque-purified and expanded stocks of VSV-G/GFP, VSV-rp30, VSV-P126, and VSV-L223 were generated at the same time and compared for infectivity, spread, and propagation in three human melanoma cultures. Cells were infected at an MOI of 0.1 and analyzed at 15 hpi (Fig. 7D). Infection rates were higher in VSV-rp30-infected cultures than in VSV-G/GFP-infected cultures for all three melanomas tested. Similarly, single mutant VSV-P126 showed GFP expression rates greater than that of VSV-G/GFP but lower than that of VSV-rp30. VSV-L223, on the other hand, showed little or no increased infectivity compared to VSV-G/GFP (Fig. 7E). For quantification of viral progeny generation of the 3 VSV variants, we used plaque assays of serial dilutions of supernatants of infected melanoma cells collected at 15 hpi. VSV-rp30 reached titers 5.2-fold higher than those of VSV-G/GFP. The single mutant VSV-P126 increased titers 2.2-fold compared to VSV-G/GFP. VSV-L223 did not increase viral titers significantly (Fig. 7F). The inability of a single mutant to recapitulate the full effect of VSV-rp30 and the absence of a substantive effect of the L-protein mutation alone suggest the possibility for a synergistic relationship between the two active mutations.
We next used viral plaque size measurements on human melanoma cultures. Virus dilutions were plated onto melanoma monolayers, covered with an agar overlay, and analyzed a day later (Fig. 7G). VSV-rp30 (mean ± SEM, 131% ± 2.4%) and VSV-P126 (120% ± 2.5%) generated larger plaques than the parent virus VSV-G/GFP (100% ± 2.1%). VSV-L223 showed little to no difference compared to VSV-G/GFP (101% ± 2.2%).
Defined mutation pattern in oncogenically transformed mouse melanocytes correlates with VSV susceptibility.
Most cancers are caused by a combination of mutations that lead to unrestricted growth. Although the status of two melanoma-associated mutations (in BRAF and NRAS) was determined for most of the human melanomas tested in our study, the complete set of mutations was not. We therefore employed a mouse melanoma model with only one to three defined mutations. The number of mutations required for cancer formation can be relatively small. In mice, induction of changes in as few as two genes can transform melanocytes into melanoma cells with high penetrance in vivo (40). We asked whether an increased level of VSV infection correlated with particular mutations driving melanoma formation. Mouse melanomas were formed by induction of specific mutations in Braf, Pten, Cdkn2a, and beta catenin (Ctnnb1) in various combinations (40, 41). In order to investigate whether there is a correlation between transforming mutations and VSV oncolysis, 7 mouse melanoma cell lines with different induced mutations and normal mouse melanocytes were infected at a low MOI (0.1) and analyzed for GFP expression and cytopathic effects at 24 hpi.
Although no clear correlation between any single mutation and the extent of VSV infection was observed, we noted a trend toward increased infection rates in melanoma cells with a greater number of mutations compared to melanomas with fewer mutations or melanocytes with no mutations (Fig. 8). For example, melanoma designation 1111, with only two mutations (Pten and Cdkn2a), showed an infection rate 7-fold lower than those of melanoma type 2901 or 0888, with three mutations (Braf, Pten, and Cdkn2a). Comparing the infectivity of three-mutation melanoma cells with that of two-mutation melanoma cells revealed a trend toward higher susceptibility of cells with more mutations (P = 0.09; one-tailed Mann-Whitney-Wilcoxon test) for VSV infection.
Fig 8.

VSV-rp30 infection and cytopathic rates on mouse melanoma with defined mutation pattern. (A) Oncogenically transformed mouse melanocytes expressing combinations of Braf, Pten, Cdkn2a, or beta catenin and normal mouse melanocytes were infected by VSV-rp30 at an MOI of 0.1. Infection rates and cytopathic effect rates were quantified at 24 h postinoculation. Cells with more mutations showed a trend for greater susceptibility to VSV-rp30 infection than cells with fewer mutations, with normal mouse melanocytes showing very limited signs of infection. Gray shading in boxes indicates cells with three mutations. (B) Representative micrographs showing phase-contrast and GFP fluorescence images of infected cultures from cell type 2901, with three mutations, compared to cell type 1111, with two mutations, and normal mouse melanocytes.
Targeting single and multiple intra- and extracranial human melanoma xenografts by intravenously administered VSV.
We previously showed that systemically applied VSV successfully targets human glioma and sarcoma in intracranial and subcutaneous in vivo tumor models (27, 36). Here, we address the potential of VSV-rp30 to target human melanoma in vivo after tail vein injection; immunocompromised CB17 SCID mice received either subcutaneous or intracranial xenografts or a combination of both. To emphasize the multisite characteristic of metastatic melanoma, mice received up to 4 subcutaneous flank tumors (Fig. 9A, displaying two tumors). To facilitate the identification of tumor cells, human YUMAC cells were stably transfected with a red fluorescent protein (RFP) reporter gene (in the following referred to as rYUMAC) (Fig. 9B and C). Three weeks after tumor seeding, mice received an intravenous application of 100 μl VSV-rp30 containing 108 PFU. Tumors were harvested at 3 or 5 days postinfection (dpi). Fluorescence microscopy analysis of the subcutaneous rYUMAC melanoma masses revealed substantial (3 dpi) or complete (5 dpi) infection of the tumor mass, which could easily be distinguished from surrounding normal tissue due to its red fluorescence (Fig. 9E and H). GFP expression in VSV-infected tumors indicated selective and complete targeting of the red tumor mass. No viral infection was noted in the surrounding tissue or in sections of liver, spleen, or heart (not shown).
Fig 9.

Intravenous VSV-rp30 targets multiple subcutaneous melanoma xenografts. SCID mice bearing multiple subcutaneous rYUMAC melanoma xenografts were given a single tail vein injection of VSV-rp30 (1 × 108 PFU/100 μl). (A) Skin of the back and flank section with bilateral xenografts, with the subcutaneous (s.c.) side facing up. Human YUMAC melanoma cells modified to express red fluorescent protein (RFP) allow tracing of tumor masses surrounded by normal tissue, as illustrated by stereomicroscope images (B and C). Green filter images confirm VSV-rp30 infection restricted to red tumor masses at 5 dpi. Microsections of individual tumors reveal details of close adherence of viral intratumoral spread to the tumor border, as seen in green-red merged low-magnification images (D and G) and high-magnification details (E, F, H, I).
To assess the long-term safety of VSV-rp30, adult Swiss-Webster mice (n = 18) were given a strong dose of intravenous virus (108 PFU) into the tail vein. Over the course of >30 days, we found no negative consequences. All mice remained healthy and showed no weight loss and no neurological symptoms. We found no VSV reporter gene expression in any of the major organs after the mice were euthanized. These data suggest that widespread virus inoculation does not generate health problems and that the virus is eliminated over time.
For intracranial melanoma xenografts using stereotactic injection of 1 μl of melanoma cell suspension containing 5 × 104 cells, three different melanoma cultures were chosen for CNS placement: rYUMAC (n = 5), melanotic YUSIK (n = 6), and amelanotic YUSIT (n = 6). Of the mice receiving intracranial rYUMAC, three also carried subcutaneous rYUMAC tumors and two mice received 4 intracranial injections each. Of the mice transplanted with YUSIK or YUSIT, three of each group received a single striatal injection and three mice received intracranial injections at three different locations plus subcutaneous tumor grafts. Four to 6 weeks after melanoma inoculation, most mice received 100 μl of VSV-rp30 containing 108 PFU. Additionally, a short-term tumor growth protocol was used for two rYUMAC-carrying mice, in which VSV-rp30 was applied 5 days after intracranial tumor injection. Brains of mice were harvested between 2 and 5 days and analyzed using fluorescence microscopy for signs of VSV targeting of the tumor masses. YUSIK tumors (Fig. 10A) and single tumor cells (Fig. 10C) were highly melanotic and could be microscopically detected via bright-field imaging. GFP expression patterns indicating virus infection were found to be exclusively limited to melanin granule-containing cells (Fig. 10B and C). Of note, even cell clusters composed of few cells distant from the main tumor were successfully targeted by VSV. Importantly, no signs of infection were seen outside the tumor in the surrounding brain parenchyma at the times examined. DAPI (4′,6-diamidino-2-phenylindole) nuclear stain was used to assess cells in the tumor and surrounding normal brain tissue (Fig. 10E). YUSIT tumor xenografts were found to grow amelanotic without melanin granules. As with YUSIK xenografts, the whitish YUSIT tumor mass showed complete infection by VSV without GFP-reported signs of infection outside the tumor mass (data not shown).
Fig 10.

Targeting of intracranial human melanoma xenografts by intravenous VSV-rp30. (A to E) Melanin pigment-filled human melanoma YUSIK cells were injected stereotactically in the striatum of SCID mice. Border of tumor mass (A) and isolated cell clusters surrounded by brain parenchyma (C) were easily traceable in bright-field observation of brain microsections. A single injection of 108 PFU VSV-rp30 led to complete infection of large tumor masses (B) as well as isolated tumor cells (D) within 2 days, with limited infection of surrounding brain tissue; the cellularity of normal brain tissue is indicated by DAPI nuclear stain (E). Alternatively, the amelanotic human melanoma YUMAC tissue was stably transfected to express red fluorescent protein for easy tracing of tumor cells injected into the brain of SCID mice (F to J). Short-term brain melanoma xenografts (7 days) were established through stereotactic injection into the striatum of SCID mice (F, H). Tail vein injection of VSV-rp30 led to complete infection of the red tumor cells within 2 days (G, H). DAPI stain illustrating the cellularity of the brain parenchyma. Red YUMAC tumor-bearing mice not receiving VSV-rp30 via tail vein were monitored for an additional 3 weeks, and brains were harvested and analyzed for progressed tumor growth (K). Detail sections (L) display high cellularity via DAPI stain (M) and negative signal for GFP fluorescence in the absence of virus (N).
Two mice intracranially injected with rYUMAC melanoma cells were used in an alternate setting with a shortened time between tumor inoculation and virus injection. Five days after tumor inoculation, mice received the same intravenous dose as above. The rYUMAC short-term tumor xenografts featured cell dispersion that was easily traceable due to the red fluorescence of rYUMAC cells and reduced tumor organization with few signs of tumor vascularization. Three days after systemic VSV-rp30 application, brains were harvested, and fluorescence microscopic analysis revealed successful targeting of rYUMAC short-term grafts by VSV-rp30 (Fig. 10F to J). This is particularly noteworthy because the tumors at this stage are considerably smaller and lack the tumor organization shown in tumors grown for weeks. Two additional mice carrying intracranial rYUMAC tumors were left untreated; the mice were sacrificed after 3 weeks, and brains were analyzed for tumor expansion. Tumors remained strongly positive for RFP expression; no green fluorescence was detected in the absence of virus (Fig. 10K to N).
DISCUSSION
In light of the genetic heterogeneity of advanced-stage melanoma, we tested a large panel of 19 human melanoma samples in which some of the driver mutations had been identified. As listed in Table 1, they comprise a wide selection of phenotypes of primary and metastatic tumors. The majority of the human melanoma samples tested were highly susceptible to VSV oncolysis; in contrast, normal melanocytes were resistant to infection.
Human melanoma samples.
Most previous reports on VSV and melanoma focused on mouse melanoma tumors in syngeneic models (25, 26, 51, 52). The most commonly used B16 mouse melanoma model has been referred to in some studies as VSV resistant or partially resistant (24), whereas in others it was shown to be VSV susceptible (53). VSV activates an immune response against mouse melanoma that can reduce tumor mass (54, 55). Our data showed that VSV is very efficient at infecting and killing the majority of melanomas tested but was less efficient in targeting a minority of melanomas. Our samples were directly derived from excised human melanoma samples with minimal cell passaging. Recent work on melanoma cell lines that have been long established shows a similar refractoriness to virus infection in a minority of lines (56). Established cancer cell lines often accumulate additional mutations that promote long-term propagation in vitro. The human melanoma cells used here were taken directly from patients' tissue with minimal passaging and present a closer approximation of the endogenous genetic and pathological situation. Thus, our data based on freshly prepared melanoma are consistent with findings based on the long-term cultures of established cell lines. In our work with primary human melanoma in vitro, only 5 of the 19 melanoma samples showed a low rate of infection after low-titer virus application; the remaining 14 melanoma cultures were very susceptible to VSV. This ratio (26% resistance) is consistent with a previous report of VSV infection resistance rates (22) of established cancer lines from the NCI 60 panel, which included some melanomas, and with a recent report on 6 established melanoma lines (56). In our hands, the observed cytopathic effect (CPE) rates and EthD-1-staining of dead cells across the panel correlated very highly with infection rates (R = 0.94 and R = 0.87, respectively), consistent with the rapid cell death-inducing consequences of VSV infection (57) and supportive of the potential efficacy of VSV as an oncolytic agent in melanoma. Importantly, normal human melanocytes showed a remarkable inherent resistance to infection. To our knowledge, a direct comparison of VSV susceptibility of human melanoma and normal human melanocytes has not been described before, nor has there been an analysis of VSV infection in a broad panel of partially genotyped melanomas.
Melanoma infection and interferon.
One mechanism of VSV oncoselection is based on defects of the interferon pathway (47, 58). Here, in the context of VSV oncolysis of advanced melanoma, the role of the interferon response is fundamental in two aspects: (i) IFN is an approved drug for melanoma therapy, and (ii) IFN is a key element underlying VSV oncoselectivity. IFN has been FDA approved for adjuvant advanced-stage melanoma therapy (2). IFN shows a direct growth-arresting effect on a subset of malignancies (59, 60). On the other hand, resistance to the antitumor action of IFN is common (30, 61). Melanomas less responsive to IFN might be expected to be impaired in their antiviral responses. When melanoma cultures were preincubated with IFN, we found VSV infection to be greatly attenuated in 5 samples, whereas infection was not or only mildly affected in 12 other samples.
Type I IFN receptor activation is followed by an upregulation of several hundred IFN-stimulated genes (62). The ISG (OAS and Mx1) response of normal human melanocytes exposed to IFN was orders of magnitude greater than that of the 11 melanoma cultures tested. Although YUSIT and YUMAC had the strongest Mx1 response among tumors, and despite the known association of Mx1 expression with VSV resistance, YUSIT and YUMAC were among the melanoma samples that appeared to be unprotected by IFN. Conversely, several melanomas that showed protection from VSV infection by IFN (YUAME, YUDOSO, and YURIF) did not demonstrate a strong ISG response for Mx1 or OAS. That the Mx1 and OAS response to IFN does not correlate with the ability of IFN to protect melanoma from VSV suggests that other ISGs might underlie IFN-mediated protection. Although many ISGs are upregulated by IFN, in tumor cells with multiple mutations this may not be the case, and expression of the ISG(s) most responsible for inhibiting VSV growth (whose identity is an open question) may not correlate with expression of the two ISGs measured here (Mx1 and OAS).
VSVs effectively infect melanoma cells.
All recombinant VSVs tested were effective in killing multiple melanoma cultures with somewhat different rates of infection. VSV-G/GFP and VSV-rp30 displayed a faster course of melanoma infection and cell killing, but over time, the more attenuated variants VSV-M51 and VSV-CT9-M51 also killed melanoma cells. Building upon our previous report of the effectiveness of VSV-rp30 on human sarcoma tumors (36), the current data support the view that although VSV-rp30 was originally adapted to human glioblastoma cells, its enhanced oncolytic profile is not restricted to glioblastoma cells (35). Our viral replication studies revealed that VSV-rp30 propagated faster than VSV-G/GFP in the majority of melanomas, particularly at early collecting points. Some studies have suggested that replication speed is crucial for the success of oncolytic virus treatment because fast-replicating viruses would spread further through tumor tissue before the antiviral immune response reaches its full potential (63). Furthermore, faster oncolytic action consequently leads to larger amounts of tumor cell debris and tumor cell antigens being exposed in the initial round of infection, which can enhance an antitumor immune response. In contrast, others have argued that the therapeutic effect of oncolytic viruses might in large part depend on virus-induced stimulation of antitumor immune response and that rapidly replicating viruses impair the overall effect by diverting immune response elements from the tumor to the virus (54). Of note, the latter studies focused on multiple virus injections into single tumors; in contrast, systemic application of oncolytic VSV in a setting in which tumor metastases have spread throughout the body may confer some advantage in attenuating melanoma. In this setting, fast viral replication would be desirable to ensure widespread early tumor infection before the systemic immune system is upregulated to control further virus spread.
Sequencing and comparing the genome of VSV-rp30 revealed the presence of four mutations in VSV-rp30: C1772T (S126L) in the viral P protein and G7693T (D223Y), A11712G, and C11736T in the viral L protein, with the last two being silent mutations. Because the P and L proteins of VSV form an RNA polymerase complex that is responsible for both viral mRNA transcription and genome replication, mutations in these proteins can affect viral propagation. Whereas the P protein itself may not display enzymatic activity, it may modulate L protein activity, with phosphorylation of serine/threonine residues on P being important for this function (64). Interestingly, the mutation at position P126 falls within the active domain 1 of the P protein, which has been suggested to function as a binding domain for the L protein (65), potentially underlying a synergistic action between the two mutations in VSV-rp30. The hypothesis about the two mutations affecting the direct interplay between the P and L proteins is supported by our findings that single mutant recombinant VSVs expressing either P126 or L223 do not fully recapitulate the enhanced phenotype of VSV-rp30; this suggests either an additive or synergistic effect of the two active mutations.
In vivo systemic virus application to tumor xenografts.
A key challenge of our study was to address not only the multisite tumor location of metastatic melanoma but also the common CNS involvement of this disease. Advanced melanoma has a high propensity to spread to the CNS. Systemic application of VSV as a cotreatment modality for melanoma was described recently (23, 66) in studies that centered on VSV as an adjuvant immunotherapeutic agent in a murine B16 melanoma model. To our knowledge, systemic application of VSV against human melanoma has not been previously shown. We demonstrate that a single intravenous application of VSV is sufficient for targeting and productive spread throughout multiple tumors; this stands in contrast to findings with in vivo applications in mouse B16 melanoma models (23, 54), although part of the difference may relate to the status of the systemic immune system. Importantly, single cells that had spread from the site of tumor placement in the brain were still targeted by intravenous VSV, indicating that the virus can find single tumor cells. VSV was able to infect large portions of the tumor within a few days. A sterile needle stab wound or xenotransplantation of nontumor control cells into the brain did not lead to CNS entry and infection after systemic injection of VSV (27). This supports the notion that VSV-rp30 targets both established and developing tumors. VSV-M51 mutants have been described as safe in mice after intratumoral injection (56). Although VSV-rp30 infected melanoma tumors faster than the two M51 mutants (VSV-M51 and VSV-CT9/M51) that we tested, control immunocompetent mice given a high intravenous dose of VSV-rp30 showed no adverse health consequences.
At the time of tumor analysis, no viral infection was observed beyond the tumor border or in any control tissue samples (liver, spleen, or lung), supporting a tumor-selective targeting of infection. However, since we examined human melanomas in SCID mice, in the long term such immunocompromised mice may not fully clear virus infections (27). On the other hand, attenuation of VSV variants and systemic immune targeting of VSV enhance the safety profile of VSV application in the brain (34, 67, 68). Future work will need to address the balance between high replication speed and strategies to limit the potential for neurotoxicity.
Melanoma mutations and VSV susceptibility.
Analysis of a large panel of human melanoma provided the opportunity to compare melanoma driver mutation patterns with susceptibility for viral infection and oncolytic control by VSV. One of the most frequent driver mutations in melanoma formation is BRAF V600E, found in approximately 50% of cases, and associated with a higher risk for developing brain metastases (3). Other common mutations include NRAS mutations, found in 15 to 20% of melanomas (9). We generated a cumulative scoring system for VSV oncolytic susceptibility (OS score), which combined infection rates, replication potency, and IFN insensitivity for each melanoma type. Table 1 lists all melanoma cells used in this study ranked by OS score from high to low (OS score 9 being the highest). Cells with BRAF mutation showed a significantly higher OS score than cells with wild-type BRAF or cells with RAS mutation. When dissecting individual susceptibility parameters, all human melanomas with BRAF mutations showed complete infection, in contrast to cells with normal BRAF, which displayed various degrees of infection. This suggests that BRAF mutations might be predictive for successful VSV targeting. Nonetheless, about one-half of the non-BRAF-mutated cells also show significant VSV susceptibility, suggesting that BRAF mutation alone is not a prerequisite for VSV infection. Corroborating the notion that BRAF-mutated cells are likely to be successfully targeted by VSV oncolysis are the results from our viral replication analysis, in which all tested BRAF-mutated cells produced substantial amounts of viral progeny. In contrast, we did not find strong evidence for a clear correlation between RAS mutations and VSV infection. Importantly, the absence of either the BRAF or the RAS mutation did not exclude enhanced VSV infection. If confirmed with larger samples, this might lead to the possibility that unlike novel specifically targeted therapeutic agents like the BRAF inhibitor vemurafenib (PLX4032), which shows activity in patients with the corresponding mutation, oncolytic VSV might potentially serve as a potential therapy option for a broader range of patients.
Although the status of selected melanoma-associated mutations was determined for all our tested human melanoma cells, the global genetic alteration status was not. We therefore employed a mouse melanoma model in which tumors from transgenic mice expressing combinations of oncogenic mutations in Braf, Pten, Cdkn2a, and beta catenin were used to study VSV infection rates. In human melanoma, we found that BRAF-mutated cells were highly susceptible to VSV oncolysis. In mouse melanoma, BRAF-mutated cells 2901 and 0888 showed complete infection, compared to partial infection in other BRAF-mutated cells, i.e., 0753 and 2697. Therefore, unlike human melanoma, BRAF status alone does not appear to be predictive for VSV oncolysis in mouse melanoma. Similarly, neither inactivation of Cdkn2a nor beta catenin activation determined VSV susceptibility in mouse melanoma. However, despite the lack of single gene association, cells with a greater number of mutations appeared to show greater infection than cells with fewer mutations, suggesting a trend for an association between the number of genetic defects and VSV infection. In summary, we show that human melanomas are an attractive selective target for VSV-mediated direct oncolysis. The high degree of direct oncolysis in human primary melanoma complements previous work showing that the VSV can initiate or enhance an immune attack on mouse melanoma (26, 51, 54, 55, 66).
ACKNOWLEDGMENTS
We thank Yang Yang, Vitaliy Rogulin, and Antonella Bacchiocchi for technical facilitation, R. Halaban and the Yale SPORE in Skin Cancer funded by the National Cancer Institute (1 P50 CA121974, R. Halaban, PI) for the generous contribution of melanomas, and H. Kluger, J. Paglino, and M. Robek for helpful suggestions.
Support was provided by NIH grants CA124737, CA161048, and CA175577 (to A.N.V.D.P.) and CA112054 (to M.W.B.).
Footnotes
Published ahead of print 3 April 2013
REFERENCES
- 1. Mocellin S, Pasquali S, Rossi CR, Nitti D. 2010. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J. Natl. Cancer Inst. 102:493–501 [DOI] [PubMed] [Google Scholar]
- 2. Lee B, Mukhi N, Liu D. 2012. Current management and novel agents for malignant melanoma. J. Hematol. Oncol. 5:3 doi: 10.1186/1756-8722-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carlino MS, Fogarty GB, Long GV. 2012. Treatment of melanoma brain metastases: a new paradigm. Cancer J. 18:208–212 [DOI] [PubMed] [Google Scholar]
- 4. Sloan AE, Nock CJ, Einstein DB. 2009. Diagnosis and treatment of melanoma brain metastasis: a literature review. Cancer Control 16:248–255 [DOI] [PubMed] [Google Scholar]
- 5. Trinh VA, Hwu WJ. 2012. Ipilimumab in the treatment of melanoma. Expert Opin. Biol. Ther. 12:773–782 [DOI] [PubMed] [Google Scholar]
- 6. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. 2010. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363:809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363:711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dummer R, Flaherty KT. 2012. Resistance patterns with tyrosine kinase inhibitors in melanoma: new insights. Curr. Opin. Oncol. 24:150–154 [DOI] [PubMed] [Google Scholar]
- 9. Flaherty KT, Hodi FS, Fisher DE. 2012. From genes to drugs: targeted strategies for melanoma. Nat. Rev. Cancer 12:349–361 [DOI] [PubMed] [Google Scholar]
- 10. Swick JM, Maize JC, Sr 2012. Molecular biology of melanoma. J. Am. Acad. Dermatol. 67:1049–1054 [DOI] [PubMed] [Google Scholar]
- 11. Au GG, Beagley LG, Haley ES, Barry RD, Shafren DR. 2011. Oncolysis of malignant human melanoma tumors by Coxsackieviruses A13, A15 and A18. Virol. J. 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, Peach H, Phillips RM, Bond J, Pandha H, Harrington K, Vile R, Russell S, Selby P, Melcher AA. 2013. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 20:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eberle J, Fecker LF, Hossini AM, Kurbanov BM, Fechner H. 2008. Apoptosis pathways and oncolytic adenoviral vectors: promising targets and tools to overcome therapy resistance of malignant melanoma. Exp. Dermatol. 17:1–11 [DOI] [PubMed] [Google Scholar]
- 14. Hwang TH, Moon A, Burke J, Ribas A, Stephenson J, Breitbach CJ, Daneshmand M, De Silva N, Parato K, Diallo JS, Lee YS, Liu TC, Bell JC, Kirn DH. 2011. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol. Ther. 19:1913–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. MacKie RM, Stewart B, Brown SM. 2001. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet 357:525–526 [DOI] [PubMed] [Google Scholar]
- 16. Moehler M, Sieben M, Roth S, Springsguth F, Leuchs B, Zeidler M, Dinsart C, Rommelaere J, Galle PR. 2011. Activation of the human immune system by chemotherapeutic or targeted agents combined with the oncolytic parvovirus H-1. BMC Cancer 11:464 doi: 10.1186/1471-2407-11-464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muster T, Rajtarova J, Sachet M, Unger H, Fleischhacker R, Romirer I, Grassauer A, Url A, Garcia-Sastre A, Wolff K, Pehamberger H, Bergmann M. 2004. Interferon resistance promotes oncolysis by influenza virus NS1-deletion mutants. Int. J. Cancer 110:15–21 [DOI] [PubMed] [Google Scholar]
- 18. Nettelbeck DM, Rivera AA, Balague C, Alemany R, Curiel DT. 2002. Novel oncolytic adenoviruses targeted to melanoma: specific viral replication and cytolysis by expression of E1A mutants from the tyrosinase enhancer/promoter. Cancer Res. 62:4663–4670 [PubMed] [Google Scholar]
- 19. Shmulevitz M, Gujar SA, Ahn DG, Mohamed A, Lee PW. 2012. Reovirus variants with mutations in S1 and L2 genome segments exhibit enhanced virion infectivity and superior oncolysis. J. Virol. 86:7403–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomas DL, Doty R, Tosic V, Liu J, Kranz DM, McFadden G, Macneill AL, Roy EJ. 2011. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol. Immunother. 60:1461–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zamarin D, Vigil A, Kelly K, Garcia-Sastre A, Fong Y. 2009. Genetically engineered Newcastle disease virus for malignant melanoma therapy. Gene Ther. 16:796–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stojdl DF, Lichty BD, BRtenOever Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275 [DOI] [PubMed] [Google Scholar]
- 23. Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, Brunelliere J, Bramson JL, Lichty BD, Wan Y. 2010. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 18:1430–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leveille S, Samuel S, Goulet ML, Hiscott J. 2011. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 18:435–443 [DOI] [PubMed] [Google Scholar]
- 25. Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, Vile R. 2011. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol. Ther. 19:150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leveille S, Goulet ML, Lichty BD, Hiscott J. 2011. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J. Virol. 85:12160–12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 28:1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balachandran S, Barber GN. 2000. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life 50:135–138 [DOI] [PubMed] [Google Scholar]
- 29. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6:821–825 [DOI] [PubMed] [Google Scholar]
- 30. Wong LH, Krauer KG, Hatzinisiriou I, Estcourt MJ, Hersey P, Tam ND, Edmondson S, Devenish RJ, Ralph SJ. 1997. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J. Biol. Chem. 272:28779–28785 [DOI] [PubMed] [Google Scholar]
- 31. Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J. Virol. 81:2056–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, Udem SA. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 360:36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van den Pol AN, Davis JN. 2013. Highly attenuated recombinant vesicular stomatitis virus VSV-12′GFP displays immunogenic and oncolytic activity. J. Virol. 87:1019–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wollmann G, Tattersall P, van den Pol AN. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paglino JC, van den Pol AN. 2011. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J. Virol. 85:9346–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng E, Trombetta SE, Kovacs D, Beech RD, Ariyan S, Reyes-Mugica M, McNiff JM, Narayan D, Kluger HM, Picardo M, Halaban R. 2006. Rab33A: characterization, expression, and suppression by epigenetic modification. J. Invest. Dermatol. 126:2257–2271 [DOI] [PubMed] [Google Scholar]
- 38. Tworkoski K, Singhal G, Szpakowski S, Zito CI, Bacchiocchi A, Muthusamy V, Bosenberg M, Krauthammer M, Halaban R, Stern DF. 2011. Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Mol. Cancer Res. 9:801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D, Rimm DL, McMahon M, Bosenberg M. 2011. beta-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 20:741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. 2009. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 41:544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Held MA, Curley DP, Dankort D, McMahon M, Muthusamy V, Bosenberg MW. 2010. Characterization of melanoma cells capable of propagating tumors from a single cell. Cancer Res. 70:388–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Publicover J, Ramsburg E, Robek M, Rose JK. 2006. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J. Virol. 80:7028–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schnell MJ, Buonocore L, Whitt MA, Rose JK. 1996. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol. 70:2318–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dalton KP, Rose JK. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279:414–421 [DOI] [PubMed] [Google Scholar]
- 45. Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. U. S. A. 92:4477–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitt MA. 2010. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 169:365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wollmann G, Robek MD, van den Pol AN. 2007. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J. Virol. 81:1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kirkwood JM, Manola J, Ibrahim J, Sondak V, Ernstoff MS, Rao U. 2004. A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin. Cancer Res. 10:1670–1677 [DOI] [PubMed] [Google Scholar]
- 49. Haller O, Kochs G, Weber F. 2007. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 18:425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Staeheli P, Pavlovic J. 1991. Inhibition of vesicular stomatitis virus mRNA synthesis by human MxA protein. J. Virol. 65:4498–4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. 2007. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 67:2840–2848 [DOI] [PubMed] [Google Scholar]
- 52. Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 77:8843–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fernandez M, Porosnicu M, Markovic D, Barber GN. 2002. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J. Virol. 76:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, Melcher A, Vile R. 2010. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 17:158–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Pulido J, Pavelko K, Pease L, Melcher A, Vile R. 2010. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 70:4539–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blackham AU, Northrup SA, Willingham M, D'Agostino RB, Jr, Lyles DS, Stewart JH., 4th 2013. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery 153:333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gaddy DF, Lyles DS. 2005. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 79:4170–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barber GN. 2005. VSV-tumor selective replication and protein translation. Oncogene 24:7710–7719 [DOI] [PubMed] [Google Scholar]
- 59. Ferrantini M, Capone I, Belardelli F. 2007. Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie 89:884–893 [DOI] [PubMed] [Google Scholar]
- 60. Kalvakolanu DV. 2004. The GRIMs: a new interface between cell death regulation and interferon/retinoid induced growth suppression. Cytokine Growth Factor Rev. 15:169–194 [DOI] [PubMed] [Google Scholar]
- 61. Davar D, Tarhini AA, Kirkwood JM. 2012. Adjuvant therapy for melanoma. Cancer J. 18:192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Conzelmann KK. 2005. Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. J. Virol. 79:5241–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ebert O, Shinozaki K, Huang TG, Savontaus MJ, Garcia-Sastre A, Woo SL. 2003. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 63:3605–3611 [PubMed] [Google Scholar]
- 64. Rose JK, Whitt MA. 2001. Rhabdoviridae: the viruses and their replication, p 1221–1244 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 4th ed Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 65. Banerjee AK, Barik S. 1992. Gene expression of vesicular stomatitis virus genome RNA. Virology 188:417–428 [DOI] [PubMed] [Google Scholar]
- 66. Rommelfanger DM, Wongthida P, Diaz RM, Kaluza KM, Thompson JM, Kottke TJ, Vile RG. 2012. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res. 72:4753–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D, Lichty B, Power A, Johnston RN, Hamilton M, Parney I, Bell JC, Forsyth PA. 2006. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J. Natl. Cancer Inst. 98:1546–1557 [DOI] [PubMed] [Google Scholar]
- 68. Ozduman K, Wollmann G, Ahmadi SA, van den Pol AN. 2009. Peripheral immunization blocks lethal actions of vesicular stomatitis virus within the brain. J. Virol. 83:11540–11549 [DOI] [PMC free article] [PubMed] [Google Scholar]