

Abstract
T helper 17 (Th17) cells play an important role in mucosal immune homeostasis and maintaining the integrity of the mucosal epithelial barrier. Loss of Th17 cells has been extensively documented during human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) infections. The lack of effective repopulation of Th17 cells has been associated with chronic immune activation mediated by the translocation of microbial products. Using ex vivo analysis of purified peripheral blood CD4 T cells from SIV-infected rhesus macaques, we show that the suppression of interleukin-17 (IL-17) expression correlated with upregulated expression of negative regulatory genes PIAS3, SHP2, and SOCS3 in CD4 T cells. Suppressed Th17 expression was accompanied by elevated levels of soluble CD14 (sCD14) and lipopolysaccharide binding protein (LBP) in the plasma during early stages of infection. Plasma viral loads rather than sCD14 or LBP levels correlated with acute immune activation. Additionally, we observed a significant increase in the expression of CD14 on peripheral blood monocytes that correlated with IL-23 expression and markers of microbial translocation. Taken together, our results provide new insights into the early events associated with acute SIV pathogenesis and suggest additional mechanisms playing a role in suppression of Th17 cells.
INTRODUCTION
Progressive human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) infections are characterized by chronic immune activation (1) that is partially mediated by translocated microbial products (2). Microbial translocation has been correlated with the loss of T helper 17 (Th17) cells, and numerous studies have documented the loss of Th17 cells during early SIV infection (3, 4). Loss of Th17 cells was associated with translocation of Salmonella into the systemic circulation (5). Others (3, 4, 6, 7) have shown that the loss of interleukin-17 (IL-17)-producing cells accompanied mucosal damage and translocation of bacterial products during pathogenic SIV infections. Favre et al. (4) demonstrated that the loss of Th17 cells was accompanied by systemic T cell activation. In contrast, nonpathogenic infection of sooty mangabeys was associated with preservation of the Th17 cells and mucosal barrier integrity (6, 8), whereas Ciccone et al. (9) showed that Th17 cells were preserved in HIV-infected long-term nonprogressors.
Similar to chronic infection, a high level of immune activation has also been observed during acute SIV infection (10–13). It is not clear if microbial products translocate early in infection and if these translocated products contribute to immune activation during acute infection. We sought to address this question by examining the markers of microbial translocation and immune activation in rhesus macaques during acute SIV infection and correlated these with Th17 expression. Our results showed that markers of microbial translocation were significantly upregulated in the plasma of SIV-infected animals very early during the course of infection and that this was accompanied by a loss of IL-17 expression in CD4 T cells. Interestingly, plasma viral loads rather than IL-17 expression or markers of microbial translocation positively correlated with CD8+ KI-67+ T cells, suggesting that acute viral replication rather than translocated bacterial products likely plays a primary role in acute immune activation. Suppressed IL-17 expression correlated with upregulated expression of various negative regulatory genes, such as PIAS3, SHP2, and SOCS3, indicating a potential role for these genes in suppressing Th17 responses during SIV infection.
MATERIALS AND METHODS
Animals, infection, and samples.
Rhesus macaques (Macaca mulatta) of Indian origin (n = 6) were used for this study. The animals were housed in accordance with the American Association for Accreditation of Laboratory Animal Care guidelines and were seronegative for SIV, simian retrovirus (SRV), and simian T-cell leukemia virus (STLV) type 1. Animals were infected intravenously with uncloned pathogenic SIVmac251.
Peripheral blood was collected at various time points and used for isolation of plasma and peripheral blood mononuclear cells (PBMCs). PBMCs were isolated by density gradient centrifugation and cryopreserved. Plasma viral loads were determined by real-time PCR using reverse-transcribed viral RNA as the template, as previously described (14). Commercial enzyme-linked immunosorbent assay kits were used to measure plasma soluble CD14 (sCD14; R&D Systems) and lipopolysaccharide (LPS) binding protein (LBP; Cell Sciences) levels as per the manufacturer's instruction.
Antibodies and flow cytometry.
Isolated cells were labeled with specific panels of antibodies containing different combinations of CD3-Cy7-allophycocyanin (APC), CD4-Pacific Blue (PB), CD28-Cy5-phycoerythrin (PE), CD95-fluorescein isothiocyanate (FITC), and CD8-Alexa Fluor 700 or CD3-Cy7-APC, CD4-PB, CD28-Cy5-PE, CD95-APC, CD8-Alexa Fluor 700, and Ki-67–FITC and analyzed by flow cytometry as described previously (15–21). All the antibodies were obtained from BD Biosciences (San Diego, CA) and titrated using rhesus macaque PBMCs. Ki-67 expression in CD8 memory T cells was determined by intracellular staining using a Cytofix/Cytoperm kit from BD Biosciences. Labeled cells were fixed with 0.5% paraformaldehyde and analyzed using a Becton, Dickinson LSR II flow cytometer.
For analysis of IL-17, STAT3 (signal transducer and activator of transcription 3), PIAS3 (protein inhibitor of activated STAT3), SHP2 (Src homology region 2 domain-containing phosphatase 2), and SOCS3 (suppressor of cytokine signaling 3) mRNA expression, total viable peripheral blood CD4 T cells from days 7 and 35 postinfection (p.i.) were sorted by negative selection using a BD FACSAria sorter. We did not have sufficient samples from the other time points; hence, we were unable to include these samples in our analysis. To avoid signaling and activation through CD3 and CD4, cells were labeled only with anti-CD14, anti-CD8, anti-CD20, and the live/dead amine-reactive dye VIVID. VIVID-positive dead cells were excluded along with CD14+, CD8+, and CD20+ cells. An aliquot of the negatively sorted cells was labeled with anti-CD3 and anti-CD4 to determine purity and enrichment of the sorted subset. Approximately 5 × 105 CD4+ T cells/well were seeded in 96-well tissue culture plates and stimulated with 40 units/ml of recombinant human interleukin-6 (rIL-6; R&D Systems) in serum-free RPMI 1640 for 15 min at 37°C, as previously described for rhesus macaques (22). Unstimulated cells were set up simultaneously as controls. After 15 min, cells were harvested, washed, and used for RNA isolation. For ex vivo analysis of Toll-like receptor 4 (TLR4) and IL-23 mRNA in monocytes, CD14+ monocytes were positively sorted from PBMCs at days 7 and 35 p.i. and used ex vivo for RNA isolation.
Relative qPCR assay.
RNA was isolated using an RNeasy kit (Qiagen Sciences, Gaithersburg, MD) and treated with Ambion Turbo DNase (Applied Biosystems, Austin, TX) to remove contaminating DNA. Each DNase-treated RNA sample was tested using a quantitative PCR (qPCR) assay with the β-actin primers described below to confirm that RNA was free from DNA contamination. Purified RNA was reverse transcribed with a SuperScript III first-strand synthesis kit (Invitrogen, Carlsbad, CA) to synthesize cDNA that was used to determine the expression of genes using an ABI 7500 instrument (Applied Biosystems). TaqMan qPCR was performed using high-fidelity Platinum Taq polymerase (Invitrogen) as described previously with Macaca mulatta-specific (i) IL-17 primers IL-17-F (ACCAATCCCAAAAGGTCCTC) and IL-17-R (TCTCTCAGGGTCCTCATTGC) and probe IL-17-P (FAM-CAACCGATCCACCTCACCTTGG-BHQ1, where FAM is 6-carboxyfluoresein and BHQ1 is black hole quencher 1), (ii) STAT3 primers STAT3-F (GGAGGAGTTGCAGCAAAAAG) and STAT3-R (GATTCTCTCCTCCAGCATCG) and probe STAT3-P (FAM-CCCCATTGTACAGCACCGGC-BHQ1), (iii) PIAS3 primers PIAS3-F (ACCATTGCCCTTCTATGAAGTC) and PIAS3-R (AGGTAAAGTGCGCTTCCTCA) and probe PIAS3-P (FAM-ACCACCCTTGCATCCACTTCTA-BHQ1), (iv) SHP2 primers SHP2-F (ATATGGCGTCATGCGTGTTA) and SHP2-R (TCCGTATTCCCTTGTCCAAC) and probe SHP2-P (FAM-TGTCAAAGAAAGTGCTGCTCATGA-BHQ1), (v) SOCS3 primers SOCS3-F (TTCTACTGGAGCGCAGTGAC) and SOCS3-R (CTGTCGCGAATCAGAAAGGT) and probe SOCS3-P (FAM-AGGCGAACCTGCTGCTCAGC-BHQ1) (vi) IL-6 primers IL-6-F (ATGCAATAACCACCCCTGAA) and IL-6-R (AAGAGCCCTCAGGTTGGACT) and probe IL-6-P (FAM-TGCTGACGAAGCTGCAGGCA-BHQ1), (vii) IL-21 primers IL-21-F (TGTGAATGACTTGGACCCTGAA) and IL-21-R (AAACAGGAAATAGCTGACCACTCA) and probe IL-21-P (FAM-TCTGCCAGCTCCAGAAGATGTAGAGACAAACT-BHQ1), (viii) IL-23p19 primers IL-23-F (CCAGCAGCTTTCACAGAAGC) and IL-23-R (TCTTAGATCCATGTGTCCCACT) and probe IL-23-P (FAM-TGGCCTGGAGTGCACATCCA-BHQ1), (ix) transforming growth factor β (TGF-β) primers TGFb-F (TGTCATAGATTTCGTTGTGGGTTT) and TGFb-R (GTACAACAGCACCCGCGAC) and probe TGFb-P (FAM-ACCATTAGCACGCGGGTGACCTCC-BHQ1), and (x) TLR4 primers TLR4-F (CCTTTCAGCTCTGCCTTCAC) and TLR4-R (CACCTTTCGGCTTTTATGGA) and probe TLR4-P (FAM-ATTCCCGGTGTGGCCATTGC-BHQ1); and gene expression was normalized to Macaca mulatta β-actin housekeeping gene expression using β-actin-specific primers β-Actin-F (ATGCTTCTAGGCGGACTGTG) and β-Actin-R (AAAGCCATGCCAATCTCATC) and probe β-Actin-P (FAM-TGCGTTACACCCTTTCTTGACAAAACC-BHQ1). Primers and probes were designed using Primer-3 software (23). Collected data were analyzed using the 2−ΔΔCT (ddCT, where CT is the threshold cycle) method with ABI 7500 software, and fold changes in gene expression levels were calculated as previously described (24).
Data analysis.
Flow cytometric data were analyzed using FlowJo (version 9.2) software (Tree Star, Inc., Ashland, OR). Statistical analysis was performed using the t test with GraphPad Prism (version 4.0) software (GraphPad Prism Software, Inc. San Diego, CA). A P value of <0.05 was considered significant. Error bars represent standard errors. Linear regression analysis was performed to determine line of fit, correlations were derived using the Spearman correlation, and a P value of <0.05 was considered significant.
RESULTS
Plasma sCD14 and LBP levels are significantly elevated during acute SIV infection.
Acute SIV infection has been associated with the loss of memory CD4 T cells in peripheral blood (10, 19, 25). To confirm these findings, we first evaluated plasma viral loads (Fig. 1a) and the levels of memory CD4 T cells (Fig. 1b) at days 0, 7, 14, and 35 p.i. Plasma viremia peaked at day 14 p.i., followed by a significant decline to a set point. In line with previous reports, the decline in plasma viral loads was accompanied by a significant loss of memory CD4 T cells at day 35 p.i. compared to the levels at days 0 and 7 p.i. (10, 19, 25).
Fig 1.
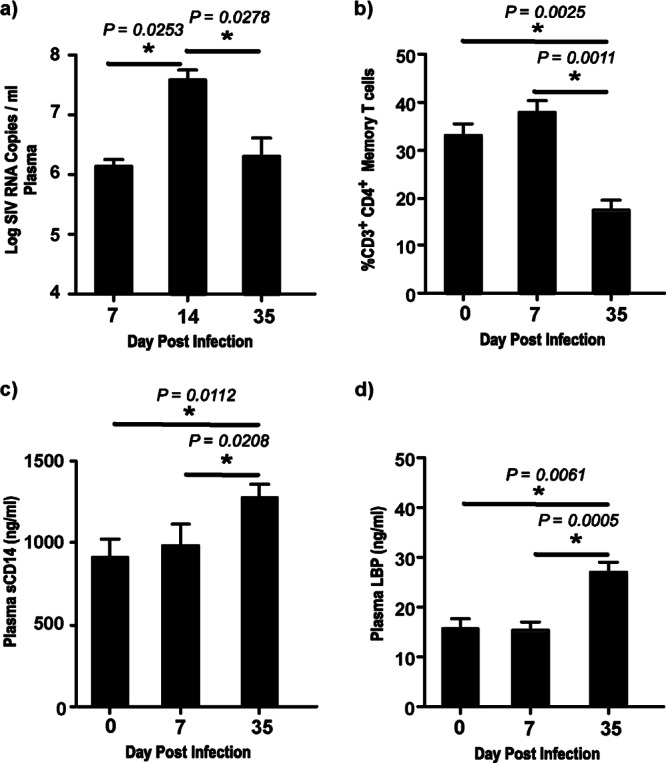
Plasma sCD14 and LBP levels are significantly elevated during acute SIV infection. (a) Plasma viral loads at days 7, 14, and 35 p.i. The limit of detection was 30 copies/ml of plasma. (b) Percentage of total CD3+ CD4+ memory T cells in peripheral blood at days 0, 7, and 35 p.i. Memory CD4 T cells were identified on the basis of the expression of CD28 and CD95, with all memory CD4 T cells expressing CD95. (c and d) Plasma sCD14 (c) and plasma LBP (d) levels at days 0, 7, and 35 p.i.
Next, we examined the plasma levels of sCD14 (Fig. 1c) and LBP (Fig. 1d) at day 35 p.i. and compared them to those at days 0 and 7 p.i. Soluble CD14 and LBP have been extensively used as markers of microbial translocation (2, 26–28). We observed a significant increase in the plasma levels of both sCD14 and LBP at day 35 p.i. compared to those at the earlier time points, suggesting that early viral replication and CD4 T cell loss were accompanied by an increase in the markers of microbial translocation.
IL-17 expression is significantly suppressed during acute SIV infection and negatively correlates with sCD14 and LBP levels.
To determine if the increased levels of markers of microbial translocation were accompanied by a loss of Th17 cells, we evaluated the ex vivo expression of IL-17 in purified populations of negatively sorted CD4 T cells (Fig. 2a) following short-term stimulation with IL-6 at days 7 and 35 p.i. Previous studies have shown that both the Th17 compartment (19) and the total memory CD4 T cell compartment (10, 25) were largely intact during the first 7 to 10 days after infection. Our results showed that IL-17 expression was significantly suppressed at day 35 p.i. compared to that at day 7 p.i. (Fig. 2b).
Fig 2.
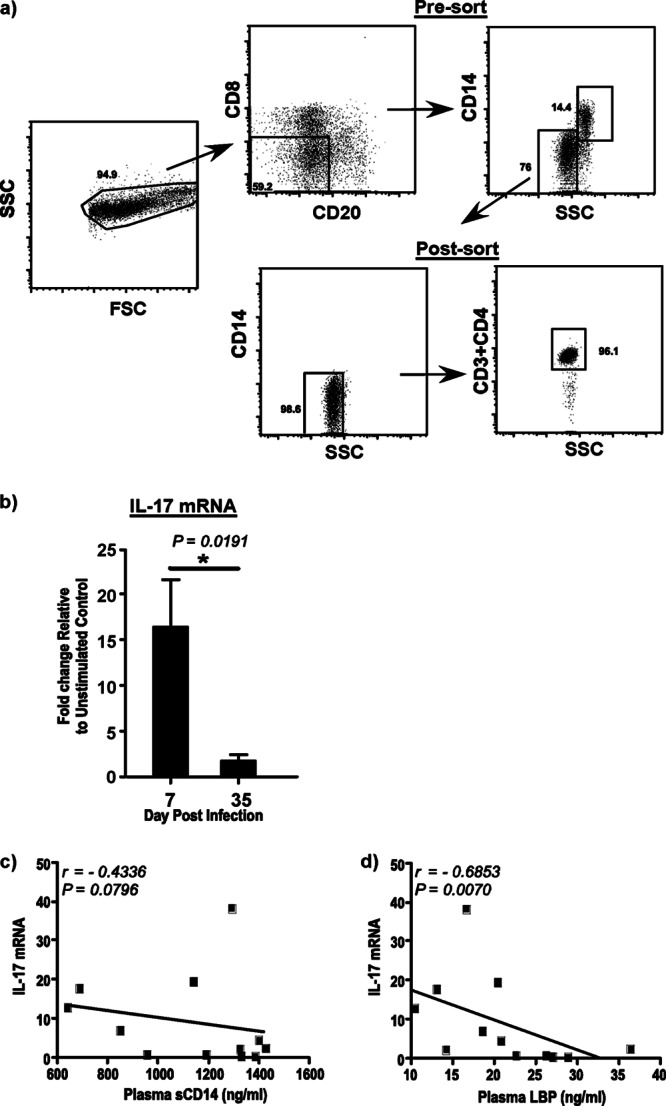
IL-17 expression in CD4 T cells is significantly suppressed during acute SIV infection. (a) Gating strategy used to sort pure populations of peripheral blood CD4 T cells by fluorescence-activated cell sorting. PBMCs were labeled with anti-CD20, anti-CD8, and anti-CD14 and negatively sorted for CD4 T cells. An aliquot of the sorted cells was stained with anti-CD4 to confirm purity. SSC, side scatter. (b) IL-17 mRNA expression in purified peripheral blood CD4 T cells after short-term stimulation for 15 min with recombinant human IL-6. The fold change at days 7 and 35 p.i. relative to the levels for unstimulated control cells is shown. (c and d) IL-17 expression negatively correlates with LBP (c) and sCD14 (d) levels in plasma.
Next, we correlated IL-17 expression with the levels of plasma sCD14 (Fig. 2c) and LBP (Fig. 2d) to examine if suppressed IL-17 expression was associated with elevated levels of these markers of microbial translocation. Soluble CC14 levels were found to negatively correlate (r = −0.4336; P = 0.0796) with IL-17 expression, though this correlation was not significant. In contrast, LBP levels were found to have a significantly high negative correlation (r = −0.6853; P = 0.0070) with IL-17 expression levels in CD4 T cells.
Upregulated PIAS3, SHP2, and SOCS3 mRNA levels correlate with suppression of IL-17 expression.
IL-17 production is negatively regulated by a number of intracellular factors, such as PIAS3, SHP2, and SOCS3 (29, 30). To determine if the suppression of IL-17 expression during acute SIV infection was due to negative regulation, we evaluated the expression of PIAS3, SHP2, and SOCS3 mRNA (Fig. 3a) in purified populations of negatively sorted CD4 T cells at day 35 p.i. after short-term stimulation with IL-6 and compared it to that at day 7 p.i. We observed a significant upregulation in the expression of PIAS3, SHP2, and SOCS3 at day 35 p.i. compared to that at day 7 p.i.
Fig 3.

Expression of PIAS3, SHP2, and SOCS3 is significantly upregulated in CD4 T cells during acute SIV infection. (a) STAT3, PIAS3, SHP2, and SOCS3 mRNA expression in purified peripheral blood CD4 T cells at days 7 and 35 p.i. after short-term stimulation for 15 min with recombinant human IL-6. The fold change at days 7 and 35 p.i. relative to the levels for unstimulated control cells is shown. (b to d) IL-17 expression negatively correlates with PIAS3 (b), SHP2 (c), and SOCS3 (d) levels in peripheral blood CD4 T cells.
Next, we correlated the expression of PIAS3 (Fig. 3b), SHP2 (Fig. 3c), and SOCS3 (Fig. 3d) with the expression of IL-17. PIAS3 (r = −0.6434; P = 0.0120), SHP2 (r = −0.5804; P = 0.0239), and SOCS3 (r = −0.5315; P = 0.0377) levels had a significantly high negative correlation with IL-17 expression, suggesting that these negative regulators may contribute to the suppression of IL-17 expression during acute SIV infection.
Plasma sCD14 and LBP levels significantly correlate with IL-23 expression in CD14+ monocytes.
A number of cytokines, such as IL-6, IL-21, IL-23, and TGF-β, have been shown to be critical for the induction of IL-17 expression in CD4 T cells (31–33). To determine if suppressed IL-17 expression in CD4 T cells at day 35 p.i. was due to the paucity of IL-17-promoting cytokines, we evaluated the expression of IL-6, IL-21, IL-23, and TGF-β mRNA ex vivo in total PBMCs at day 7 p.i. and compared it to that at day 35 p.i. (Fig. 4a). There were no major changes in the expression levels of IL-6, IL-21, and TGF-β at day 35 p.i. relative to day 7 p.i. However, the expression of IL-23 was significantly elevated at day 35 p.i. compared to that at day 7 p.i.
Fig 4.

CD14+ monocytes upregulate the density of CD14 expression and IL-23 mRNA levels during acute SIV infection. (a) Ex vivo expression of IL-6, IL-21, IL-23, and TGF-β in total PBMCs at day 7 and day 35 p.i. The fold change relative to the level at day 7 p.i. is shown. The line indicates day 7 p.i. as the baseline. (b) Gating strategy used to sort purified populations of CD14+ monocytes by fluorescence-activated cell sorting. (c) Ex vivo expression of TLR4 and IL-23 expression in purified populations of CD14+ monocytes at days 7 and 35 p.i. The fold change relative to the level at day 7 p.i. is shown. The line indicates day 7 p.i. as the baseline. (d) Histogram showing the density of CD14 expression from a representative animal at days 7 and 35 p.i. and data showing the MFI of CD14 expression on CD14+ monocytes at days 7 and 35 p.i. (e to g) The correlations between the plasma levels of sCD14 and IL-23 expression on CD14+ monocytes (e), plasma levels of LBP and IL-23 expression on CD14+ monocytes (f), and plasma levels of LBP and MFI of CD14 expression on CD14+ monocytes (g) at days 7 and 35 p.i. are shown.
As IL-23 is primarily secreted by myeloid cells, we examined the ex vivo expression of IL-23 in purified populations of CD14+ monocytes from PBMCs at days 7 and 35 p.i. (Fig. 4b and c). Our results showed that IL-23 levels were significantly elevated in CD14+ monocytes at day 35 p.i. compared to day 7 p.i.
Previous studies have shown that translocated microbial products could activate blood monocytes (34). To determine if translocated microbial products could potentially activate monocytes, we evaluated the expression of TLR4 mRNA by quantitative reverse transcription-PCR (Fig. 4c) in purified populations of CD14+ monocytes and determined the density of CD14 expression on monocytes by flow cytometry (Fig. 4d) at day 7 p.i. and compared it to that at day 35 p.i. Both TLR4 and CD14 are primary receptors for LPS. There was no major change in the levels of TLR4 expression in monocytes. However, the density of CD14 expression was significantly upregulated on monocytes at day 35 p.i. compared to day 7 p.i.
Next, we correlated IL-23 expression with plasma sCD14 (Fig. 4e) and LBP (Fig. 4f) levels at days 7 and 35 p.i. IL-23 expression significantly correlated with both sCD14 (r = 0.5822; P = 0.0235) and LBP (r = 0.5860; P = 0.0226) levels, suggesting that translocated bacterial products could play a role in activating peripheral blood monocytes during acute stages of infection. Interestingly, we observed a significantly high positive correlation (r = 0.6783; P = 0.0077) between plasma LBP levels and the mean fluorescence intensity (MFI) of CD14 expression on monocytes (Fig. 4g).
CD8+ Ki-67+ T cells are significantly increased during acute SIV infection and correlate with plasma viral loads but not sCD14 or LBP levels.
Numerous studies have shown that translocation of microbial products is associated with systemic immune activation during HIV and SIV infections (6, 9). To determine if translocated microbial products play a role in acute immune activation, we evaluated the expression of Ki-67 on memory CD8 T cells at days 0, 7, 14, and 35 p.i. (Fig. 5a) and correlated that expression with sCD14 (Fig. 5b), LBP (Fig. 5c), and IL-17 (Fig. 5d) expression and plasma viral loads (Fig. 5e).
Fig 5.

CD3+ CD8+ Ki-67+ memory T cells significantly correlate with plasma viral loads but not sCD14 or LBP levels in plasma. (a) Percentage of CD3+ CD8+ Ki-67+ memory T cells in peripheral blood. (b to e) The correlations between the percentage of CD3+ CD8+ Ki-67+ memory T cells in peripheral blood and plasma sCD14 levels (b), plasma LBP levels (c), IL-17 expression in CD4 T cells (d), and plasma viral loads (e) are shown.
As previously reported (10–13), we observed a significant increase in CD8+ Ki-67+ T cells in peripheral blood during acute SIV infection. Surprisingly, we observed little or no correlation between CD8+ Ki-67+ T cells and sCD14 (r = −0.1652; P = 0.4744), LBP (r = 0.1704; P = 0.2496), or IL-17 (r = −0.3117; P = 0.1620) expression. In contrast, there was a significantly high positive correlation between CD8+ Ki-67+ T cells and plasma viral loads (r = 0.6798; P = 0.0010), suggesting that acute viral replication rather than translocated microbial products likely plays a primary role in driving acute immune activation.
DISCUSSION
Acute HIV and SIV infections are associated with significant perturbations in mucosal homeostasis that are characterized by a massive loss of CD4 T cells, viral replication, and damaged mucosal barrier (6, 25, 35–42). These changes occur within the first few weeks of infection and lead to a chronic state of inflammation that persists throughout the course of infection. Our findings suggest that systemic translocation of microbial products likely occurs very early during the course of infection.
Elevated sCD14 and LBP levels significantly correlated with the suppression of IL-17 production in CD4 T cells, supporting earlier observations that the loss of Th17 cells is associated with the translocation of microbial products (5, 43). Brenchley et al. (8) showed that peripheral blood CD4 T cells are skewed toward a Th1 phenotype, as opposed to a Th17 phenotype, during chronic HIV infection, whereas others have reported a loss of IL-17-producing CD4 T cells in peripheral blood during SIV infection (4, 16, 19, 44).
The suppression of IL-17 expression was not likely due to the paucity in IL-17-promoting cytokines, such as IL-6, IL-21, IL-23, and TGF-β, as we did not observe a suppression of these responses in PBMCs. Other studies have shown that plasma levels of IL-6, IL-21, and TGF-β were elevated during HIV and SIV infections (45–49), though studies have reported a decrease in IL-21 levels in the plasma (50) likely due to the loss of IL-21-producing Th17 cells (44). On the other hand, Zhao et al. showed that circulating IL-21 levels were increased during simian-human immunodeficiency virus infection (51). The relatively normal expression of IL-21 that we observed in total PBMCs may be due to the presence of CD8 T cells that were capable of producing IL-21, as some studies have reported (52).
Though altered cytokine levels may play a role in suppressing the induction of Th17 cells, the significantly low level of IL-17 expression at day 35 p.i. relative to that at day 7 p.i. after short-term stimulation with rIL-6 pointed to a role for other mechanisms in suppressing Th17 responses. In fact, we observed a significantly high negative correlation between IL-17 expression and the negative regulators of IL-17 production in CD4 T cells, namely, PIAS3, SHP2, and SOCS3.
PIAS3 is a potent inhibitor of activated STAT3 signaling (53). PIAS3 transcripts are absent in Th17 cells but are present Th1 and Th2 cells, and a downregulation of PIAS3 was found to induce Th17 cells and increase the severity of experimental autoimmune encephalomyelitis (EAE) in mice (54). On the other hand, SHP2 is a tyrosine phosphatase that has been shown to interfere with the STAT3 signaling pathway, and the transduction of SHP2 was accompanied by a failure to induce Th17 cells (55). Likewise, SOCS3 has been shown to inhibit cytokine-induced phosphorylation of STAT3 and IL-17 production (29). Miller et al. (56) showed that SOCS3 mRNA levels in CD4 T cells were higher in HIV-infected patients than healthy subjects, and the overexpression of SOCS3 was found to inhibit the JAK/STAT pathway. Others have shown that IL-6 stimulation induces SOCS3 expression via the activation of the STAT3 signaling pathway (57). Taken together these observations suggest that the elevated levels of PIAS3, SHP2, and SOCS3 could potentially play a role in suppressing IL-17 expression during acute SIV infection.
Given that IL-6-mediated signaling requires the activation of STAT3, it was surprising that STAT3 mRNA was not upregulated following short-term stimulation with rIL-6 for 15 min. However, IL-6 primarily acts at the level of STAT3 activation and phosphorylation, and it is highly likely that longer periods (1 to 3 h) of stimulation are required for IL-6-mediated expression of STAT3 mRNA, as some studies have suggested (58, 59).
It is not clear from our studies if protein levels for PIAS3, SHP2, and SOCS3 are increased in CD4 T cells, as we were unable to perform these studies for the lack of samples. The upregulated expression of the PIAS3, SHP2, and SOCS3 genes, however, along with the suppression of IL-17 expression, suggests that these negative regulatory factors may contribute to the suppression of Th17 cells very early during the course of infection.
It is highly likely that the suppression of IL-17 expression is due to the SIV-mediated loss of Th17 cells, as has been reported previously (8, 19, 60), or due to other mechanisms, such as those mediated by the induction of indoleamine 2,3-dioxygenase-1 (61, 62) or the paucity of IL-21-producing CD4 T cells (44). Our studies do not rule out a role for these mechanisms but suggest that additional mechanisms, such as those that negatively regulate the IL-17 signaling pathway, may play a role in suppressing the expression of Th17 cells during SIV infection and that these suppressive mechanisms were apparent very early during the course of infection.
Increased translocation of microbial products correlated with IL-23 expression in monocytes and was associated with a significant upregulation in the density of CD14 expression, suggesting that translocated products could contribute to the activation of monocytes during acute infection. Ancuta et al. (34) demonstrated that microbial translocation was associated with increased monocyte activation in HIV-infected patients, whereas Manuzak et al. (63) showed that bacterial antigens increase IL-23 production by peripheral blood monocytes. Louis et al. (64) showed that monocytes from patients with primary HIV infection significantly enhance IL-23 production in response to LPS stimulation. Kader et al. (16) showed that IL-23 mRNA levels were upregulated in the mucosa during acute stages of SIV infection and stayed high even after the loss of CD4 T cells.
Interestingly, neither the suppression of IL-17 expression nor increased levels of sCD14 or LBP directly correlated with acute immune activation, suggesting that mechanisms other than translocated microbial products likely drive the immune activation observed very early during infection. Acute infection is characterized by a massive amplification of viral infection that is accompanied by immune activation during the early stages of infection (10–13), likely due to the release of various proinflammatory mediators (65). In line with this argument, we observed a significantly high positive correlation between plasma viral loads and CD8+ Ki-67+ T cells in peripheral blood. Interestingly, immune activation appeared to precede elevated levels of plasma sCD14 and LBP; CD8+ Ki-67+ T cell levels were significantly higher at day 7 p.i. than at day 0, without an apparent change in sCD14 and LBP levels.
Though sCD14 and LBP levels did not correlate with acute immune activation, they significantly correlated with IL-23 expression in monocytes and the MFI of CD14 expression on monocytes, suggesting that translocated products could potentially interact with the higher levels of CD14 on monocytes and contribute to the generalized state of inflammation during acute SIV infection. The levels of sCD14 and LPB that we observed during acute stages of infection were lower than what has been reported in chronically infected animals (2) and in line with those described in previous reports showing low but detectable levels of LPS in the mucosa during early stages of SIV infection (6). Given the low levels of translocated products during early infection, it is highly likely that the direct effect of translocated microbial products on acute immune activation is probably masked by the release of inflammatory mediators associated with massive viral replication. Breed et al. (66) showed that rhesus macaques infected with the SIVmac239ΔGY mutant strain exhibited wild-type acute viremia and immune activation but maintained their mucosal CD4 T cells and displayed a lack of microbial translocation.
In conclusion, our studies show that acute SIV infection is characterized by a significant increase in markers of microbial translocation that correlate with suppressed Th17 responses. Elevated expression of various negative regulatory genes likely plays a role in suppressing IL-17 expression in CD4 T cells during acute stages of SIV infection. Active viral replication rather than translocated microbial products, however, correlated with Ki-67 expression on CD8 T cells, suggesting a primary role for viral replication in acute immune activation.
ACKNOWLEDGMENTS
We thank Jeffy George, Olusegun Onabajo, and Sean Maynard at the Uniformed Services University of the Health Sciences for assistance with the samples; Karen Wolcott and Kateryna Lund at the Biomedical Instrumentation Center and Jim Treece and Deborah Weiss at Advanced Bioscience Laboratories, Rockville, MD, for expert assistance with the animals; and Jeffrey D. Lifson and Michael Piatak at NCI Frederick, NIH, for their valuable assistance with plasma viral load analysis.
The described project was supported by DE019397, awarded to J.J.M. by the National Institute of Dental and Craniofacial Research (NIDCR).
The content is solely the responsibility of the authors and does not necessarily represent the official views of NIDCR or the National Institutes of Health.
S.L.B. and N.G.S. performed the experiments and analyzed the data; J.J.M. designed and supervised the study; S.L.B., N.G.S., D.C.D., and J.J.M. wrote the paper.
We declare no financial conflict of interest.
Footnotes
Published ahead of print 17 April 2013
REFERENCES
- 1. Douek DC, Picker LJ, Koup RA. 2003. T cell dynamics in HIV-1 infection. Annu. Rev. Immunol. 21:265–304 [DOI] [PubMed] [Google Scholar]
- 2. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–1371 [DOI] [PubMed] [Google Scholar]
- 3. Cecchinato V, Trindade CJ, Laurence A, Heraud JM, Brenchley JM, Ferrari MG, Zaffiri L, Tryniszewska E, Tsai WP, Vaccari M, Parks RW, Venzon D, Douek DC, O'Shea JJ, Franchini G. 2008. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 1:279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, Mold J, Swainson L, Barbour JD, Baskin CR, Palermo R, Pandrea I, Miller CJ, Katze MG, McCune JM. 2009. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 5:e1000295 doi: 10.1371/journal.ppat.1000295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, Godinez I, Sankaran S, Paixao TA, Gordon MA, Kolls JK, Dandekar S, Baumler AJ. 2008. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 14:421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, Hirsch VM, Silvestri G, Douek DC, Miller CJ, Haase AT, Lifson J, Brenchley JM. 2010. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6:e1001052 doi: 10.1371/journal.ppat.1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, Cervasi B, Yokomizo LK, Pan L, Vinton CL, Tabb B, Canary LA, Dang Q, Hirsch VM, Alter G, Belkaid Y, Lifson JD, Silvestri G, Milner JD, Paiardini M, Haddad EK, Brenchley JM. 2012. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 5:646–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, Scheinberg P, Price DA, Hage CA, Kholi LM, Khoruts A, Frank I, Else J, Schacker T, Silvestri G, Douek DC. 2008. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 112:2826–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciccone EJ, Greenwald JH, Lee PI, Biancotto A, Read SW, Yao MA, Hodge JN, Thompson WL, Kovacs SB, Chairez CL, Migueles SA, Kovacs JA, Margolis LB, Sereti I. 2011. CD4+ T cells, including Th17 and cycling subsets, are intact in the gut mucosa of HIV-1-infected long-term nonprogressors. J. Virol. 85:5880–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eberly MD, Kader M, Hassan W, Rogers KA, Zhou J, Mueller YM, Mattapallil MJ, Piatak M, Jr, Lifson JD, Katsikis PD, Roederer M, Villinger F, Mattapallil JJ. 2009. Increased IL-15 production is associated with higher susceptibility of memory CD4 T cells to simian immunodeficiency virus during acute infection. J. Immunol. 182:1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaur A, Hale CL, Ramanujan S, Jain RK, Johnson RP. 2000. Differential dynamics of CD4(+) and CD8(+) T-lymphocyte proliferation and activation in acute simian immunodeficiency virus infection. J. Virol. 74:8413–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Monceaux V, Viollet L, Petit F, Ho Tsong Fang R, Cumont MC, Zaunders J, Hurtrel B, Estaquier J. 2005. CD8+ T cell dynamics during primary simian immunodeficiency virus infection in macaques: relationship of effector cell differentiation with the extent of viral replication. J. Immunol. 174:6898–6908 [DOI] [PubMed] [Google Scholar]
- 13. Sopper S, Sauer U, Muller JG, Stahl-Hennig C, ter Meulen V. 2000. Early activation and proliferation of T cells in simian immunodeficiency virus-infected rhesus monkeys. AIDS Res. Hum. Retroviruses 16:689–697 [DOI] [PubMed] [Google Scholar]
- 14. Cline AN, Bess JW, Piatak M, Jr, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol 34:303–312 [DOI] [PubMed] [Google Scholar]
- 15. George J, Cofano EB, Lybarger E, Louder M, Lafont BA, Mascola JR, Robert-Guroff M, Mattapallil JJ. 2011. Early short-term antiretroviral therapy is associated with a reduced prevalence of CD8(+)FoxP3(+) T cells in simian immunodeficiency virus-infected controller rhesus macaques. AIDS Res. Hum. Retroviruses 27:763–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kader M, Bixler S, Piatak M, Jr, Lifson JD, Mattapallil JJ. 2009. Antiretroviral therapy fails to restore the severe Th-17:Tc-17 imbalance observed during simian immunodeficiency virus infection. J. Med. Primatol. 1:32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kader M, Bixler S, Roederer M, Veazey R, Mattapallil JJ. 2009. CD4 T cell subsets in the mucosa are CD28+Ki-67−HLA−DR−CD69+ but show differential infection based on alpha4beta7 receptor expression during acute SIV infection. J. Med. Primatol. 38(Suppl 1):24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kader M, Hassan WM, Eberly M, Piatak M, Lifson JD, Roederer M, Mattapallil JJ. 2008. Antiretroviral therapy prior to acute viral replication preserves CD4 T cells in the periphery but not in rectal mucosa during acute simian immunodeficiency virus infection. J. Virol. 82:11467–11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kader M, Wang X, Piatak M, Jr, Lifson J, Roederer M, Veazey R, Mattapallil JJ. 2009. Alpha4+beta7(hi)CD4+ memory T cells harbor most Th-17 cells and are preferentially infected during acute SIV infection. Mucosal Immunol. 2:439–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moore AC, Bixler SL, Lewis MG, Verthelyi D, Mattapallil JJ. 2012. Mucosal and peripheral Lin− HLA− DR+ CD11c/123− CD13+ CD14− mononuclear cells are preferentially infected during acute simian immunodeficiency virus infection. J. Virol. 86:1069–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Onabajo OO, George J, Lewis MG, Mattapallil JJ. 2013. Rhesus macaque lymph node PD-1(hi)CD4(+) T cells express high levels of CXCR5 and IL-21 and display a CCR7(lo)ICOS(+)Bcl6(+) T-follicular helper (Tfh) cell phenotype. PLoS One 8:e59758 doi: 10.1371/journal.pone.0059758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, Ambrozak DR, Sandler NG, Timmer KJ, Sun X, Pan L, Poholek A, Rao SS, Brenchley JM, Alam SM, Tomaras GD, Roederer M, Douek DC, Seder RA, Germain RN, Haddad EK, Koup RA. 2012. CD4 T follicular helper cell dynamics during SIV infection. J. Clin. Invest. 122:3281–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers, p 365–386 In Krawetz S, Misener S, (ed), Bioinformatics methods and protocols in the series methods in molecular biology. Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 24. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 25. Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093–1097 [DOI] [PubMed] [Google Scholar]
- 26. Heumann D, Lauener R, Ryffel B. 2003. The dual role of LBP and CD14 in response to Gram-negative bacteria or Gram-negative compounds. J. Endotoxin Res. 9:381–384 [DOI] [PubMed] [Google Scholar]
- 27. Jack RS, Fan X, Bernheiden M, Rune G, Ehlers M, Weber A, Kirsch G, Mentel R, Furll B, Freudenberg M, Schmitz G, Stelter F, Schutt C. 1997. Lipopolysaccharide-binding protein is required to combat a murine gram-negative bacterial infection. Nature 389:742–745 [DOI] [PubMed] [Google Scholar]
- 28. Kitchens RL, Thompson PA. 2005. Modulatory effects of sCD14 and LBP on LPS-host cell interactions. J. Endotoxin Res. 11:225–229 [DOI] [PubMed] [Google Scholar]
- 29. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. 2006. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. U. S. A. 103:8137–8142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yagil Z, Nechushtan H, Kay G, Yang CM, Kemeny DM, Razin E. 2010. The enigma of the role of protein inhibitor of activated STAT3 (PIAS3) in the immune response. Trends Immunol. 31:199–204 [DOI] [PubMed] [Google Scholar]
- 31. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. 2007. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 8:942–949 [DOI] [PubMed] [Google Scholar]
- 32. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. 2003. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 278:1910–1914 [DOI] [PubMed] [Google Scholar]
- 33. Manel N, Unutmaz D, Littman DR. 2008. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 9:641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D. 2008. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One 3:e2516 doi: 10.1371/journal.pone.0002516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 200:749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. George MD, Sankaran S, Reay E, Gelli AC, Dandekar S. 2003. High-throughput gene expression profiling indicates dysregulation of intestinal cell cycle mediators and growth factors during primary simian immunodeficiency virus infection. Virology 312:84–94 [DOI] [PubMed] [Google Scholar]
- 37. Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, Dandekar S. 2003. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 77:11708–11717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heise C, Vogel P, Miller CJ, Halsted CH, Dandekar S. 1993. Simian immunodeficiency virus infection of the gastrointestinal tract of rhesus macaques. Functional, pathological, and morphological changes. Am. J. Pathol. 142:1759–1771 [PMC free article] [PubMed] [Google Scholar]
- 39. Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434:1148–1152 [DOI] [PubMed] [Google Scholar]
- 40. Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reka S, Kotler DP. 1998. Correlation of intestinal structure and function in HIV infection. Gastrointest. Endosc. Clin. N. Am. 8:841–856 [PubMed] [Google Scholar]
- 42. Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. 1998. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280:427–431 [DOI] [PubMed] [Google Scholar]
- 43. Mucida D, Salek-Ardakani S. 2009. Regulation of TH17 cells in the mucosal surfaces. J. Allergy Clin. Immunol. 123:997–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Micci L, Cervasi B, Ende ZS, Iriele RI, Reyes-Aviles E, Vinton C, Else J, Silvestri G, Ansari AA, Villinger F, Pahwa S, Estes JD, Brenchley JM, Paiardini M. 2012. Paucity of IL-21-producing CD4(+) T cells is associated with Th17 cell depletion in SIV infection of rhesus macaques. Blood 120:3925–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Birx DL, Redfield RR, Tencer K, Fowler A, Burke DS, Tosato G. 1990. Induction of interleukin-6 during human immunodeficiency virus infection. Blood 76:2303–2310 [PubMed] [Google Scholar]
- 46. Emilie D, Peuchmaur M, Maillot MC, Crevon MC, Brousse N, Delfraissy JF, Dormont J, Galanaud P. 1990. Production of interleukins in human immunodeficiency virus-1-replicating lymph nodes. J. Clin. Invest. 86:148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pallikkuth S, Pilakka Kanthikeel S, Silva SY, Fischl M, Pahwa R, Pahwa S. 2011. Upregulation of IL-21 receptor on B cells and IL-21 secretion distinguishes novel 2009 H1N1 vaccine responders from nonresponders among HIV-infected persons on combination antiretroviral therapy. J. Immunol. 186:6173–6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strbo N, de Armas L, Liu H, Kolber MA, Lichtenheld M, Pahwa S. 2008. IL-21 augments natural killer effector functions in chronically HIV-infected individuals. AIDS 22:1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. White L, Krishnan S, Strbo N, Liu H, Kolber MA, Lichtenheld MG, Pahwa RN, Pahwa S. 2007. Differential effects of IL-21 and IL-15 on perforin expression, lysosomal degranulation, and proliferation in CD8 T cells of patients with human immunodeficiency virus-1 (HIV). Blood 109:3873–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Iannello A, Tremblay C, Routy JP, Boulassel MR, Toma E, Ahmad A. 2008. Decreased levels of circulating IL-21 in HIV-infected AIDS patients: correlation with CD4+ T-cell counts. Viral Immunol. 21:385–388 [DOI] [PubMed] [Google Scholar]
- 51. Zhao CC, Xue J, Cong Z, Gao XQ, Zhang WL, Chen T, Wu FX, Xiong J, Ju B, Su A, Wei Q, Qin C. 2013. Circulating IL-21 levels increase during early simian-human immunodeficiency virus infection in macaques. Arch. Virol. 158:853–858 [DOI] [PubMed] [Google Scholar]
- 52. Zhu X, Ma D, Zhang J, Peng J, Qu X, Ji C, Hou M. 2010. Elevated interleukin-21 correlated to Th17 and Th1 cells in patients with immune thrombocytopenia. J. Clin. Immunol. 30:253–259 [DOI] [PubMed] [Google Scholar]
- 53. Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. 1997. Specific inhibition of Stat3 signal transduction by PIAS3. Science 278:1803–1805 [DOI] [PubMed] [Google Scholar]
- 54. Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. 2012. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc. Natl. Acad. Sci. U. S. A. 109:E1248–E1257 doi: 10.1073/pnas.1114325109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, Aono H, Ishihara K, Huseby E, Betz UA, Murakami M, Hirano T. 2007. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 19:695–702 [DOI] [PubMed] [Google Scholar]
- 56. Miller RC, Schlaepfer E, Baenziger S, Crameri R, Zeller S, Byland R, Audige A, Nadal D, Speck RF. 2011. HIV interferes with SOCS-1 and -3 expression levels driving immune activation. Eur. J. Immunol. 41:1058–1069 [DOI] [PubMed] [Google Scholar]
- 57. Zhang L, Badgwell DB, Bevers JJ, III, Schlessinger K, Murray PJ, Levy DE, Watowich SS. 2006. IL-6 signaling via the STAT3/SOCS3 pathway: functional analysis of the conserved STAT3 N-domain. Mol. Cell. Biochem. 288:179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. 1994. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77:63–71 [DOI] [PubMed] [Google Scholar]
- 59. Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. 1998. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J. Biol. Chem. 273:6132–6138 [DOI] [PubMed] [Google Scholar]
- 60. Gosselin A, Monteiro P, Chomont N, Diaz-Griffero F, Said EA, Fonseca S, Wacleche V, El-Far M, Boulassel MR, Routy JP, Sekaly RP, Ancuta P. 2010. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J. Immunol. 184:1604–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, Barbour JD, Lowe MM, Jayawardene A, Aweeka F, Huang Y, Douek DC, Brenchley JM, Martin JN, Hecht FM, Deeks SG, McCune JM. 2010. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2:32ra36 doi: 10.1126/scitranslmed.3000632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, Kuzmichev YV, Carville A, Johnson RP. 2011. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood 118:3321–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Manuzak J, Dillon S, Wilson C. 2012. Differential interleukin-10 (IL-10) and IL-23 production by human blood monocytes and dendritic cells in response to commensal enteric bacteria. Clin. Vaccine Immunol. 19:1207–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Louis S, Dutertre CA, Vimeux L, Fery L, Henno L, Diocou S, Kahi S, Deveau C, Meyer L, Goujard C, Hosmalin A. 2010. IL-23 and IL-12p70 production by monocytes and dendritic cells in primary HIV-1 infection. J. Leukoc. Biol. 87:645–653 [DOI] [PubMed] [Google Scholar]
- 65. Valdez H, Lederman MM. 1997-1998. Cytokines and cytokine therapies in HIV infection. AIDS Clin. Rev. 1997-1998:187–228 [PubMed] [Google Scholar]
- 66. Breed MW, Jordan AP, Aye PP, Lichtveld CF, Midkiff CC, Schiro FR, Haggarty BS, Sugimoto C, Alvarez X, Sandler NG, Douek DC, Kuroda MJ, Pahar B, Piatak M, Jr, Lifson JD, Keele BF, Hoxie JA, Lackner AA. 2013. Loss of a tyrosine-dependent trafficking motif in the simian immunodeficiency virus envelope cytoplasmic tail spares mucosal CD4 cells but does not prevent disease progression. J. Virol. 87:1528–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]