

Abstract
This study identified specific and avid RNA aptamers consisting of 2′-hydroxyl- or 2′-fluoropyrimidines against hepatitis C virus (HCV) NS5B replicase, an enzyme that is essential for HCV replication. These aptamers acted as potent decoys to competitively impede replicase-catalyzed RNA synthesis activity. Cytoplasmic expression of the 2′-hydroxyl aptamer efficiently inhibited HCV replicon replication in human liver cells through specific interaction with, and sequestration of, the target protein without either off-target effects or escape mutant generation. A selected 2′-fluoro aptamer could be truncated to a chemically manufacturable length of 29 nucleotides (nt), with increase in the affinity to HCV NS5B. Noticeably, transfection of the truncated aptamer efficiently suppressed HCV replication in cells without escape mutant appearance. The aptamer was further modified through conjugation of a cholesterol or galactose-polyethylene glycol ligand for in vivo availability and liver-specific delivery. The conjugated aptamer efficiently entered cells and inhibited genotype 1b subgenomic and genotype 2a full-length HCV JFH-1 RNA replication without toxicity and innate immunity induction. Importantly, a therapeutically feasible amount of the conjugated aptamer was delivered in vivo to liver tissue in mice. Therefore, cytoplasmic expression of 2′-hydroxyl aptamer or direct administration of chemically synthesized and ligand-conjugated 2′-fluoro aptamer against HCV NS5B could be a potent anti-HCV approach.
INTRODUCTION
Hepatitis C virus (HCV) is the main causative agent of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (1, 2). Although HCV infection causes worldwide health problems, efficient and specific antiviral therapy has not yet been developed.
HCV belongs to the genus Hepacivirus in the family Flaviviridae. The enveloped virus contains a single, positive-stranded RNA genome about 9.6 kb in length encoding a single polyprotein of about 3,010 amino acids (3). This polyprotein precursor is co- or posttranslationally processed by cellular and viral proteases to yield functional structural and nonstructural proteins (4). The structural proteins include the core protein, C, and the envelope glycoproteins E1 and E2. The nonstructural (NS) proteins comprise the protease NS2, the multifunctional protein NS3 consisting of serine protease and helicase, the serine protease cofactor NS4A, the proteins NS4B and NS5A, and the RNA-dependent RNA polymerase NS5B, which are components of a complex responsible for HCV replication (5). NS5B is the central catalytic enzyme in HCV RNA replication. A number of drugs targeting HCV NS5B replicase are under development. However, the rapid appearance of drug-resistant escape mutant viruses has been reported (6).
RNA aptamers are small structured single-stranded RNAs which have emerged as attractive and feasible alternatives to small-molecule and antibody-based therapy. Aptamers can be evolved by systematic evolution of ligands by exponential enrichment (SELEX), an iterative selection method, and bind target proteins with high affinity and specificity through formation of well-defined complementary three-dimensional structures (7, 8). RNA aptamers have clinical value because of features including great specificity, high affinity, easy and large-scale synthesis by chemical methods, pharmaceutical flexibility, and poor immunogenicity (9, 10).
In this study, we isolated two kinds of RNA aptamers with 2′-hydroxyl nucleotides or 2′-fluoro pyrimidines specific to the HCV NS5B replicase. Biochemical characteristics of the aptamers, including their affinity and specificity, were analyzed, and the efficacy of the aptamers at inhibiting HCV replication was assessed. We observed whether aptamer-resistant escape mutants of HCV could be generated. Cellular internalization and toxicity of the aptamer were analyzed, and the in vivo efficacy of liver tissue uptake of the aptamer was assessed.
MATERIALS AND METHODS
Cells and HCV constructs.
The human hepatoma cell line Huh-7 and its derivative Huh-7.5 were maintained in Dulbecco's modified Eagle medium (DMEM) with high glucose (HyClone, Thermo Fisher Scientific Inc., South Logan, UT) with 10% fetal bovine serum. Huh-7.5 cells have mutational inactivation of retinoic acid-inducible gene I (RIG-I) and hence are highly permissive for the initiation of HCV replication (11–13). For HCV RNA synthesis, genotype 1b (con 1 strain) plasmid pFKI389neo/NS3–3′/5.1, containing two cell-culture adaptive mutations in NS3 and one in NS5A (provided by R. Bartenschlager), was restricted with AseI and ScaI, or genotype 2a plasmid pJFH-1 containing the full-length JFH-1 cDNA downstream of the T7 RNA promoter construct (provided by T. Wakita) was digested with XbaI and treated with mung bean nuclease (New England BioLabs, Ipswich, MA) for the correct 3′ end of the HCV cDNA. HCV RNA was synthesized in vitro with the digested plasmids using T7 RNA polymerase (TaKaRa, Otsu, Japan). The HCV 1b subgenomic replicon cell line and JFH-1 genomic replicon cell line were generated by previously described methods (14, 15).
Oligonucleotides and primers.
The sequences of random pool library RNA and primers for reverse transcription and PCR are available upon request.
Protein purification.
pCP11, a genotype 1b (BK strain) NS5B recombinant protein expression construct, was kindly provided by H. Myung at Hankuk University of Foreign Studies (Yongin, South Korea). We constructed a genotype 2a JFH-1 NS5B recombinant protein expression vector by PCR with pJFH-1 as a template and cloned it into the pET28-a(+) expression vector (Invitrogen, Carlsbad, CA). Each recombinant protein was tagged with a hexahistidine at the N terminus. Proteins were overexpressed in Escherichia coli BL21(DE3), induced with isopropyl-β-d-1-thiogalactopyranoside, and purified with nickel-chelate resin (nickel-nitrilotriacetic acid [Ni-NTA] agarose; Qiagen, Hilden, Germany).
Selection procedure.
A random pool of RNA oligonucleotides was produced by in vitro transcription of synthetic DNA templates using T7 RNA polymerase (TaKaRa) for 2′-hydroxyl RNA aptamer selection or a DuraScribe T7 transcription kit (Epicentre Technologies, Madison, WI) for 2′-deoxy-2′-fluoropyrimidine-modified-aptamer selection. SELEX was performed to isolate RNA aptamers specific to HCV 1b NS5B, essentially as described previously (7, 8)
In vitro replicase activity assay.
The NS5B replicase assay was performed with a chemically synthesized 36-nucleotide (nt) RNA (5′-GGAAAAAAAAAAAAAAAAAAAAAAAAAUAUAUAUAU-3′) as a template. This template has a weak loop structure at the 3′ end because of AU repeat sequences. HCV NS5B was capable of using RNA templates that can fold back intramolecularly at the 3′ terminus to produce a near-dimer-size hairpin product. Reaction mixtures contained 20 mM HEPES (pH 7.5), 7.5 mM dithiothreitol, 50 mM NaCl, 5 mM MgCl2, 0.025% glycerol, 0.2 μM RNA template, 0.01 to 0.2 μM aptamer, 100 μM UTP and CTP, 10 μCi of [α-32P]UTP, and 300 ng of HCV NS5B in a volume of 40 μl.
Gel shift assay.
Internally radiolabeled RNA aptamer (6 fmol) or library RNA was incubated with 10 fmol to 6.4 pmol of NS5B protein at room temperature for 20 min. The protein-RNA complexes were then analyzed on a 6% nondenaturing polyacrylamide gel containing 2% glycerol. For the competition assay, internally radiolabeled RNA aptamer (6 pmol) was mixed with 100- to 10,000-fold excesses of unlabeled library RNA or selected RNA aptamers.
Surface plasmon resonance (SPR) assay.
SPR assay was performed using a Biacore 2000 system. To immobilize His-tagged HCV NS5B protein on the carboxymethylated sensor chip (CM5 chip; GE Healthcare, Piscataway, NJ) surface, 0.1 M N-hydroxysuccinimide and 0.4 M N-ethyl-N′-(dimethylaminopropyl)carbodiimide were used. After NS5B protein immobilization, the chip surface was deactivated using 1 M ethanolamine hydrochloride (pH 8.5). To measure the binding constants and other kinetic parameters, at least 5 different concentrations (5 nM ∼ 100 nM) of RNAs were injected into the flow cells. The protein surface was regenerated using 50 mM NaOH after injection of each sample. The BIAevaluation program was used to analyze collected sensograms.
Aptamer expression constructs.
To create an R-OH (2′-hydroxyl aptamer) expression vector, we inserted a PCR-amplified R-OH aptamer sequence into the U6+1 plasmid or the pAV 7SL vector, whose expression is under the control of the PolIII promoter. To produce stable aptamer expression constructs, we digested a U6+1-R-OH or 7SL-R-OH construct with SpeI and BamHI and inserted it into the pcDNA3.1 GS vector (Invitrogen). The cloned constructs were designated pcU6+1-R-OH and pc7SL–R-OH, respectively.
Aptamer minimization.
To obtain the minimal sequence required for specific binding to HCV NS5B protein, we made three forms of truncated R-F, the 2′-fluoro aptamer, by in vitro transcription of PCR-amplified templates.
Chemical synthesis of aptamers.
Chemically synthesized cholesterol- and galactose-polyethylene glycol (Gal-PEG)-conjugated R-F t2 aptamers (chol–R-F t2 and Gal-PEG–R-F t2), a 2′-deoxy-2′-fluoropyrimidine-modified NS5B aptamer, was purchased from ST Pharm Co., LTD (Seoul, South Korea).
Colony-forming assay.
To observe long term effects of aptamers on HCV replication, HCV replicon cells were transfected with pc7SL-R-OH plasmid or control plasmid using the DMRIE-C reagent (Invitrogen) according to the manufacturer's instruction. The medium was then replaced with fresh medium containing 600 μg/ml zeocin (Invitrogen) every three days. After zeocin-resistant pools had been obtained, cells were treated with 500 μg/ml G418 (Invitrogen) every two or three days for 1 month to select cells that supported the replication of HCV subgenomic RNA. In the case of the aptamer R-F t2, 2 × 105 of HCV replicon cells on a 35-mm dish were transfected with 8 nmol of chemically synthesized R-F t2 aptamer or control RNA using Lipofectamine 2000 (Invitrogen) every two days according to the manufacturer's instructions. Cells were treated with 500 μg/ml G418 for 2 weeks. One-tenth of the cells were then replated in 35-mm dishes and treated as mentioned above until viable colonies were obtained. Colony formation was confirmed by methylene blue staining.
Immunoblot analysis.
Immunoblotting was performed as previously described (16) using the following primary antibodies: rabbit polyclonal anti-NS5B (266-A; Virogen, Watertown, MA), mouse monoclonal anti-NS5A (MAB8694; Millipore, Billerica, MA), mouse monoclonal core antibody (MA1-080; Thermo Scientific, Rockford, IL), and mouse anti-actin (sc-8432; Santa Cruz Biotechnology, Santa Cruz, CA). The protein bands were visualized by an enhanced chemiluminescence protocol (Amersham Pharmacia, Cardiff, United Kingdom).
Real-time PCR.
Real-time PCR was performed as previously described (16). For analyzing HCV replicon RNA in the aptamer-administered HCV 1b replicon cell, isolated total RNA was reverse transcribed with a 3′ primer specific for the negative strand of HCV cDNA or a random primer for 18S cDNA. The resultant cDNAs were subsequently amplified with 2× real-time PCR premix (Solgent Co., Daejeon, South Korea) with primers specific for the neomycin resistance marker gene of the HCV replicon construct. In the case of HCV 2a JFH-1 RNA detection, RNA was reverse transcribed with 3′ primer specific for the NS2 coding region of JFH-1. PCR was performed using the Rotor-Gene (Corbett, Valencia, CA) and the SYBR green PCR method. The threshold levels obtained from the HCV assay were adjusted to the threshold levels found in the 18S PCR to correct for minor variations in cDNA loading.
RNA immunoprecipitation.
HCV-replicon cells were seeded in 100-mm plates at a density of 2 × 106 cells/plate. The next day, Gal-PEG or Gal-PEG–R-F t2 was treated at 4 μM concentration. For R-OH aptamer, plasmids were transfected using DMRIE-C transfection reagent. After 24 h, cells were washed with ice-cold 1× PBS and incubated with 1% formaldehyde for 10 min (17). We treated glycine (pH 7.0) at final concentration of 0.25 M for 5 min. Cells were lysed with RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.2% sodium azide, 0.1% SDS, 0.1% NP-40, and 0.5% sodium deoxycholate) and sonicated. Sonicated lysates were immunoprecipitated with either normal IgG or rabbit polyclonal anti-NS5B (Virogen). Pellets and supernatant were incubated at 70°C for 1 h to reverse the cross-links, and the RNA was purified and subjected to RT-PCR for aptamer detection.
Cell toxicity assay.
Huh-7 cells were seeded in 96-well plates at a density of 2 × 104 cells/well and incubated overnight at 37°C. Triplicate wells were treated with the chol–R-F t2 or Gal-PEG–R-F t2 aptamer. The concentrations used for treatment ranged from 51.2 pM to 500 μM. Cell toxicity assays were performed 48 h after aptamer treatment using the Celltiter 96 Aqueous One solution reagent (Promega, Madison, WI) according to manufacturer's instructions. Cell viability was estimated by assessing the absorbance at 490 nm.
Assay of the innate immune gene expression.
Huh-7 cells were seeded in 12-well plates at a density of 2 × 105 cell/well. The next day, cells were treated with cholesterol, R-F t2, chol–R-F t2, Gal-PEG, or Gal-PEG–R-F t2 at a 4 μM concentration. As a positive control, poly(I·C) (Sigma-Aldrich, St. Louis, MO) was transfected with TransIT-TKO transfection reagent (Mirus Bio, Madison, WI). Total RNA was isolated from each RNA-treated cell and reverse transcribed using random primers (Promega). qRT-PCR was performed using the Rotor-Gene (Corbett) and SYBR green PCR method to analyze the innate immune gene expression with primers as previously described (18).
Statistics.
The significance of differences between the mean values within groups was tested by using a paired one-tailed t test (Student's t test). All data were expressed as averages ± standard deviations (SD). Differences were considered statistically significant at a P value of <0.05.
Animal studies.
This study was reviewed and approved (document 2010-14-033) by the Institutional Animal Care and Use Committee (IACUC) of the Asan Institute for Life Sciences, Asan Medical Center (Seoul, South Korea). The committee abides by the dictates of the Institute of Laboratory Animal Resources (ILAR) guide. Male BALB/c mice (weight, 18 to 20 g), aged 8 weeks, were obtained from Orient Bio, Inc. (Sungnam, South Korea), and were allowed to acclimate to their new environment for at least 1 week before study initiation.
Aptamer administration into mice and sampling.
Mice were given aptamer intravenously in the tail vein. Five mice were used for each destructive sample per time point and used in real-time PCR for determining aptamer levels in plasma and liver. At preset times (0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h postdose), using a destructive sampling technique, mice were deeply anesthetized with 8% sevoflurane in oxygen (100%), and then the blood sample was obtained by cardiac puncture and liver tissue was collected using the cardiac perfusion method (19). Briefly, a perfusion catheter was inserted in the aorta after the exposure of the thoracic cavity, and then the animal was exsanguinated with heparinized saline. For real-time PCR analysis, perfusions were performed using RNase-free water and the perfused livers were collected and stored in liquid N2 until analysis.
Aptamer detection in mouse plasma and liver tissue.
For detecting aptamers in mouse blood, 1 μl of plasma was directly reverse transcribed using MultiScribe reverse transcriptase (Applied Biosystems, Carlsbad, CA) according to the manufacturer's protocols. The aptamer concentrations in blood samples were then analyzed with quantitative PCR analysis by real-time PCR using Rotor-Gene (Corbett) and Kapa SYBR fast universal 2× qPCR master mix (KK4602; Kapa Biosystems, Woburn, MA). For detecting aptamer in mouse liver tissue, 1 μg of mouse liver tissue RNA was reverse transcribed and analyzed with quantitative PCR as described above.
RESULTS
Selection of 2′-hydroxyl RNA aptamers specific to HCV NS5B replicase.
Four groups of RNAs with 2′-hydroxyl nucleotides that specifically and avidly bound HCV 1b NS5B replicase were selected (Fig. 1A). Aptamer 2, designated R-OH, was chosen because of its intense binding to HCV NS5B and low Kd value (Fig. 1A). Nonlabeled R-OH aptamer competitively inhibited the interaction between radiolabeled R-OH aptamer and HCV NS5B, but library RNA did not, even when present at a 10,000-fold excess (Fig. 1B). Moreover, the R-OH aptamer selectively bound HCV NS5B replicase in HCV-replicating whole-cell extracts (data not shown).
Fig 1.

Specific binding to and competitive inhibition against RNA replicase activity of HCV 1b NS5B by the 2′-hydroxyl aptamer. (A) Selected RNA sequences were determined and grouped according to their sequence similarity. Underlined letters indicate the same or similar sequences between groups. Kd values and binding percentages of the aptamer and NS5B are shown. (B) Internally radiolabeled R-OH aptamers were incubated with increasing amounts of unlabeled library or R-OH aptamers. (C) Copy-back RNA synthesis using a 36-nt RNA template, in which sequences at the 3′ end folded back intramolecularly and hybridized, with increasing amounts of tRNA or R-OH aptamer. (D) De novo initiation of RNA synthesis using HCV 3′ UTR RNA as a template with increasing amounts of tRNA or R-OH aptamer. (E) De novo RNA replicase assay with R-OH aptamer as the template. Asterisks indicate products of R-OH aptamer replication by HCV NS5B replicase.
R-OH aptamer competitively inhibits HCV NS5B replicase activity as a replicase substrate.
We determined whether the R-OH aptamer affected the biochemical activities of the HCV NS5B replicase. HCV NS5B prefers a primer-dependent initiation of RNA synthesis in vitro, either by elongation of a primer hybridized to an RNA homopolymer or via a copy-back mechanism (20). Moreover, HCV NS5B can initiate RNA synthesis de novo, as can the replicase of the closely related pestivirus bovine viral diarrhea virus (21). The R-OH aptamer was effective at inhibiting both copy-back RNA synthesis and de novo RNA replication by HCV NS5B in a concentration-dependent manner (Fig. 1C and D). Interestingly, unexpected bands were observed in the de novo replication assay containing the R-OH aptamer (Fig. 1D, asterisk). These bands could be NS5B-catalyzed, newly synthesized RNAs from the R-OH aptamer. To test this speculation, a de novo RNA replication assay was performed with R-OH aptamer as a template. Bands were evident on a concentration-dependent basis (Fig. 1E).
Cytoplasmic expression of R-OH aptamer inhibits replication of HCV replicon RNA without inducing escaping mutant viruses.
To test R-OH aptamer activities in cells, R-OH aptamer expression plasmids were constructed. Since HCV replicates in the cytoplasm, the R-OH aptamer was expressed in cytoplasm using 7SL promoter-based cassettes. As controls, nucleus-localized, RNA-producing U6+1 promoter-based cassettes were used. The 7SL and U6+1 RNA sequences were included in the cassettes to help drive folding of the RNA transcripts into discrete subdomains that should not interfere with the structure of the insert (22). NFATc- and HCV NS3 helicase-specific aptamers (23, 24) were also cloned. SPR analysis showed that 7SL and U6+1 sequences did not affect R-OH aptamer affinity for HCV NS5B (data not shown).
To evaluate anti-HCV activity of the R-OH aptamer, each construct was transfected into HCV replicon cells stably supporting HCV 1b subgenomic replication (14, 25), and cells were selected with zeocin. Expression of R-OH under the control of 7SL element almost completely inhibited HCV replication (Fig. 2A, top) and decreased NS5A protein level (Fig. 2A, bottom). Transient expression of NS3 aptamer inhibited HCV replication in our previous study (24). However, stable expression of NS3 aptamer did not affect HCV replication (Fig. 2A).
Fig 2.

Inhibition of HCV replicon replication by cytoplasmic expression of R-OH aptamer. (A) HCV replication was assessed at the HCV 1b subgenomic RNA level using RT-PCR (top) or at the HCV 1b NS5A protein level using immunoblotting (bottom) in Huh-7 cells. (B) HCV-replicon cells were transfected with 7SL (left) or U6+1-driven expression vectors (right), selected with zeocin and G418, and colony formation is shown. (C) Localization of 7SL and U6+1 R-OH aptamer. After subcellular fractionation, the subcellular distribution of the aptamer was determined by RT-PCR. Lamin B and tubulin were used as nuclear and cytoplasmic markers, respectively. (D) RNA immunoprecipitation was performed and amplified by RT-PCR in the aptamer-transfected HCV replicon cells. (E) Biotin-labeled R-OH (3 nM) was incubated with increasing amounts of recombinant JFH-1 NS5B protein. (F) HCV replication was assessed at the HCV 2a JFH-1 genomic and negative-strand RNA levels using qRT-PCR in Huh-7.5 cells transfected with each construct. Values are means ± SD from 3 independent experiments. *, P < 0.05; **, P < 0.01.
Next, we carried out a colony-forming assay to determine whether R-OH aptamer can induce drug-resistant escape viruses. HCV 1b subgenomic replicon contained the neomycin resistance gene, and 7SL and U6+1 constructs contained the zeocin resistance gene. Cells were transfected with the aptamer-coding vector along with HCV replicon RNA and were selected using G418 and zeocin. Of note, the G418-and-zeocin double selection almost completely inhibited colony formation in cells transfected with the 7SL-R-OH aptamer construct (Fig. 2B). In contrast, the U6+1-R-OH construct generated numerous colonies. The colony formation efficiencies of 7SL-NFATc, 7SL-NS3, and 7SL-R-OH were about 61%, 83%, and 1.7%, respectively, compared to the 7SL control. The efficiencies of U6-NFATc, U6-NS3, and U6-R-OH were 69%, 91%, and 79%, respectively, compared to the U6 control. These indicate that cytoplasmic expression of R-OH could inhibit HCV replicon replication with the least generation of escape mutant clones in cells. We confirmed the cellular localization of R-OH aptamer by fractionation assay of aptamer-transfected cells (Fig. 2C). 7SL-R-OH aptamer was expressed dominantly in the cytoplasm; in contrast, U6+1-R-OH aptamer was localized to the nuclear fraction, as reported previously (22).
RNA immunoprecipitation analysis showed that R-OH aptamer was efficiently precipitated with NS5B protein in HCV 1b replicon cells (Fig. 2D). The HCV NS3 aptamer was also precipitated, although to a lesser degree than R-OH. As the HCV replication complex contains all HCV nonstructural proteins, the NS3 aptamer could be precipitated indirectly with HCV NS5B replicase.
The R-OH aptamer could also bind genotype 2a JFH-1 NS5B protein (Fig. 2E), and 7SL-R-OH reduced the genotype 2a JFH-1 genomic RNA level in Huh-7.5 cells by up to ∼45% compared to that in 7SL-transfected cells (Fig. 2F). The JFH-1 negative strand was also decreased by up to ∼60%. Even though R-OH aptamer might be a more potent inhibitor of HCV type 1b due to a higher binding affinity for NS5B of type 1b, the results indicate that the R-OH aptamer is not genotype specific but is a genotype-independent general decoy for HCV NS5B.
The 2′-fluoro RNA aptamer specifically binds to HCV NS5B.
We selected an RNase-resistant 2′-fluoro RNA aptamer, named R-F, against HCV 1b NS5B replicase (Fig. 3A). The specificity of R-F was assessed by a competition assay between R-F and R-OH aptamers (Fig. 3B and C). Increasing amounts of R-OH aptamer efficiently inhibited biotin-labeled R-F-NS5B complex formation (Fig. 3B) as well as nonlabeled R-F did (Fig. 3C). Nonlabeled R-OH and R-F aptamers also competitively inhibited the biotin-labeled R-OH aptamer-NS5B complex formation (Fig. 3D and E). These results indicate that R-F could bind to the same sites as R-OH, although we could not completely exclude the possibility that R-F and R-OH allosterically regulate each other's binding to NS5B.
Fig 3.

HCV NS5B specific 2′-F RNA aptamer. (A) Sequence of R-F aptamer. (B and C) Competition assays for specific binding of R-F with increasing amounts of R-OH (B) or R-F (C). R-F-NS5B complexes are indicated by asterisks. (D and E) Competition assays for specific binding of R-OH with increasing amounts of R-OH (D) or R-F (E). R-F-NS5B complexes are indicated by arrows.
Minimization of R-F aptamer.
The R-F aptamer had a length of about 90 nt. For efficient chemical synthesis, the minimal aptamer sequences needed to be established. To this end, three truncated forms of R-F aptamer were made: R-F t1, R-F t2, and R-F t3 (Fig. 4A). Binding affinity of the truncated aptamer series was measured by SPR assay (Table 1). R-F t2, with a shortened length of 29 nt, displayed the highest affinity to HCV NS5B, with a 3-fold-lower KD(∼2.62 nM) than the full-length R-F aptamer. All truncated aptamers inhibited HCV 1b subgenomic replicon replication, and inhibition rates were consistent with their in vitro binding affinities (Fig. 4B and Table 1). R-F t2 suppressed HCV replication the most, by up to 65%. The reason for the higher efficiency of R-F t2 than full-length R-F aptamer could be a more stable and rigid stem-loop structural configuration suitable for HCV NS5B binding in the shortened aptamer, R-F t2.
Fig 4.

Minimization of 2′-F RNA aptamer and its inhibitory effect against the HCV 1b replicon. (A) Computer-predicted secondary structure of the R-F aptamer. Boxes indicate truncated forms of the aptamer. (B) Inhibition of HCV 1b replicon replication by transient transfection of truncated forms of R-F. HCV RNA level was assessed using qRT-PCR. Data are presented as means ± SD. *, P < 0.05; ***, P < 0.002. (C) Inhibition of HCV 1b replicon replication by the R-F t2 aptamer without the appearance of escape mutant viruses. HCV replicon cells were transfected with tRNA, 23-mer 2′-OMe RNA, or the R-F t2 aptamer and selected with G418, and colony formation is shown.
Table 1.
Affinity of the series of truncated R-F aptamer RNAs for HCV NS5B replicasea
| Aptamer | ka (1/ms) | kd (1/s) | KA (1/M) | KD (M) | χ2 |
|---|---|---|---|---|---|
| Library RNA | (3.80 ± 0.54) × 103 | (3.54 ± 0.40) × 10−3 | (1.08 ± 0.04) × 106 | (9.33 ± 0.31) × 10−7 | 3.52 |
| R-F full | (1.63 ± 0.76) × 105 | (1.08 ± 0.44) × 10−3 | (1.49 ± 0.11) × 108 | (6.73 ± 0.50) × 10−9 | 5.81 |
| R-F t1 | (1.60 ± 0.45) × 105 | (1.32 ± 0.21) × 10−3 | (1.20 ± 0.16) × 108 | (8.41 ± 1.10) × 10−9 | 1.71 |
| R-F t2 | (4.51 ± 1.05) × 105 | (1.13 ± 0.13) × 10−3 | (4.06 ± 1.38) × 108 | (2.62 ± 0.90) × 10−9 | 7.27 |
| R-F t3 | (8.58 ± 3.73) × 104 | (3.26 ± 0.11) × 10−3 | (2.63 ± 0.88) × 107 | (4.04 ± 1.37) × 10−8 | 1.52 |
Values were measured using the BIAevaluation program. ka, concentration of analyte binding to the target per millisecond; kd, concentration of analyte separating from the target per second; KA, equilibrium association constant; KD, equilibrium dissociation constant showing binding strength; χ2, a value showing the difference between the value calculated by the BIAevaluation program and data obtained from actual experiments, which should be 10 or less. At least 5 different concentrations (5 nM ∼ 100 nM) were used in each analysis.
Next, to determine whether the R-F t2 aptamer could induce drug-resistant escape viruses, we conducted a colony-forming assay using HCV 1b subgenomic replicon cells. The replicon cells were transfected with aptamer RNA and treated with G418 to select cells which can support HCV replicon replication. In contrast to the 88% efficiency of colony generation seen with 23-mer 2′-O-methyl (2′-OMe) RNA, compared to tRNA, R-F t2 completely inhibited HCV replicon colony formation (Fig. 4C). Consequently, we reduced the R-F aptamer to a chemically manufacturable length of 29 nt to improve binding activity to NS5B replicase and effectiveness of anti-HCV activity without inducing viral escape mutant clones.
Cholesterol- or galactose-PEG conjugated R-F t2 efficiently enters HCV-replicating cells and inhibits HCV 1b replication.
We further modified R-F t2 into two versions for direct delivery of the aptamer into cells and liver tissue. First, we conjugated cholesterol to the 5′ end of R-F t2. The conjugation has been shown to increase the circulation half-life of oligonucleotides, through association with plasma lipoproteins, and promote hepatic cell uptake (26–28). Inverted dT was attached to the 3′ end of R-F t2 to prevent RNase attack. The modified aptamer was designated chol–R-F t2. In addition, galactose was conjugated to R-F t2 through polyethylene glycol. The modified aptamer was designated Gal-PEG–R-F t2. Hepatocytes exclusively express large numbers of high-affinity cell surface receptors, specifically, the asialoglycoprotein receptor, which can recognize the galactose or N-acetylgalactosamine residues of desialylated glycoproteins (29).
Chol–R-F t2 and Gal-PEG–R-F t2 aptamers were internalized into HCV replicating human hepatocytes (data not shown). In HCV 1b subgenomic replicon cells, ∼12 × 105 copies of Gal-PEG–R-F t2 aptamers were detected per cell, which was about 5-fold more than those of internalized chol–R-F t2. Approximately 105 copies of Gal-PEG–R-F t2 aptamers were internalized per cell into JFH-1 stable cells, which was about 1.5-fold more than those of internalized chol–R-F t2. Both aptamers inhibited the replication of the HCV type 1b subgenomic replicon (Fig. 5A) by up to ∼80%, which was comparable to the previously reported inhibition achieved with cholesterol-conjugated antisense miR-122 (antagomiR-122) (28). In addition, Gal-PEG–R-F t2 and chol–R-F t2 efficiently reduced HCV NS5B and NS5A protein levels (Fig. 5B). Of note, Gal-PEG–R-F t2 aptamers were immunoprecipitated with HCV NS5B in HCV 1b replicon cells (Fig. 5C), indicating that anti-HCV effects could result from R-F-t2 aptamer and HCV NS5B interaction. Taken together, the data support the view that chemically modified R-F aptamer can efficiently internalize into HCV replicon cells, bind to HCV NS5B, and inhibit HCV replicon replication.
Fig 5.

Inhibition of HCV 1b replicon replication by Gal-PEG–R-F t2 and chol–R-F t2 without cytotoxic effect. (A) HCV subgenomic RNA levels were analyzed using qRT-PCR and expressed relative to the level in nontreated cells. Averages of measurements performed 3 independent times are shown with SD. **, P < 0.01. (B) HCV NS5A and NS5B protein levels were analyzed by immunoblotting after treating chol–R-F t2 (left) and Gal-PEG–R-F t2 (right). (C) RNA immunoprecipitation was performed in the aptamer-treated cells, RNA was amplified by RT-PCR (top), and NS5B proteins were assessed using immunoblotting (bottom). (D) Cell viability assessed with increasing amounts of the aptamer. (E and F) Innate immune gene induction levels in each treated cell were analyzed using qRT-PCR and are presented relative to the level in mock-treated cells.
Gal-PEG–R-F t2 and chol–R-F t2 do not induce cell toxicity and innate immune responses.
To determine if inhibition of HCV replication by both aptamers could be partially due to cell toxicity caused by a nonspecific antiviral response, such as an innate immune gene response, we performed a cell toxicity assay (Fig. 5D) and measured the induction level of TLR-3, MDA5, RIG-I, PKR, IFN-α2, IFN-β, OAS-1, and MX-1 mRNA from Gal-PEG–R-F t2- or chol–R-F t2-treated cells (Fig. 5E and F). Neither aptamers affected cell viability at concentrations up to 500 μM. In contrast to poly(I·C), no increase in the mRNA level of any innate immune response genes was observed in cells treated with either cholesterol-conjugated or Gal-PEG-conjugated aptamers. In contrast, R-F t2 induced TLR-3, MDA5, and IFN-α2 mRNA levels, and cholesterol induced TLR-3 mRNA levels. This could be due to following reasons. (i) Cholesterol- and Gal-PEG-conjugated R-F t2 bind to their cognate receptors and internalize through receptor-mediated endocytosis (29, 30). In contrast, native and nonconjugated R-F t2 might interact with cell surface or endosomal TLRs. Conjugation of cholesterol or Gal-PEG might disturb the interaction between aptamer and TLRs by its steric hindrance (31). (ii) Free cholesterol accumulation on plasma membrane is sufficient to activate TLRs in macrophages, perhaps due to associated changes in lipid raft microdomains (32, 33). The cholesterol may interact with proteins on plasma or endosomal membrane, such as TLRs, through hydrophobic interaction. (iii) According to recent work by Wada et al., cholesterol-conjugated oligonucleotides induce innate immune response depending on oligonucleotide composition (34). We speculate that R-F t2 may have a composition which does not induce an innate immune response when conjugated with cholesterol.
Gal-PEG–R-F t2 also inhibits genotype 2a JFH-1 virus replication.
We tested whether chol–R-F t2 and Gal-PEG–R-F t2 could suppress HCV replication of genotype 2a JFH-1 full-length genome, which produces infectious virus in tissue culture (15, 35). R-F aptamer specifically bound purified recombinant JFH-1 NS5B replicase (Fig. 6A). As shown in Fig. 6B, chol–R-F t2 decreased JFH-1 genomic RNA level in cells stably infected with virus by up to 33%, but Gal-PEG–R-F t2 more efficiently decreased JFH-1 genomic RNA (∼65%), similar to the effect by antagomiR-122. Consistently, level of the generated negative strand of JFH-1 was suppressed in Gal-PEG–R-F t2-treated cells (data not shown). Moreover, the JFH-1 core protein level was efficiently decreased by Gal-PEG–R-F t2 (Fig. 6C).
Fig 6.

Suppression of genotype 2a JFH-1 replication by the R-F aptamer. (A) Biotin labeled R-F (3 nM) was incubated with increasing amounts of recombinant JFH-1 NS5B protein. (B) JFH-1 RNA level was detected using qRT-PCR in the aptamer-treated Huh-7.5 cells stably supporting the virus replicon. JFH-1 RNA level was expressed relative to the level in nontreated cells. Data are presented as averages ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.002. (C) Core protein levels were analyzed with immunoblotting in the aptamer-treated cells. (D) Supernatants from the aptamer-treated cells were used to infect naive cells, and HCV JFH-1 genomic RNA levels were analyzed using qRT-PCR and expressed relative to the level in nontreated cells. *, P < 0.05; ***, P < 0.002.
To test if suppression of JFH-1 replication resulted in decreasing infectious virus particle formation, we collected supernatant from aptamer-treated JFH-1 stable cells and incubated the supernatant with naive Huh-7.5 cells. Gal-PEG–R-F t2 and antagomiR-122 efficiently decreased JFH-1 genomic RNA level by up to ∼86% and ∼95%, respectively, but chol–R-F t2 was less efficient (Fig. 6D). Negative-strand RNA levels were consistent with genomic RNA levels (data not shown). Overall, Gal-PEG–R-F t2 more efficiently entered the human hepatic cells and inhibited HCV replication of both genotype 1b and 2a viruses, and also hampered JFH-1 infectious-virus-particle formation.
Gal-PEG–R-F t2 is distributed to liver tissue in mice more efficiently than chol–R-F t2.
We tested whether both modified aptamers are efficiently distributed to liver tissue in vivo. To this end, we intravenously administered 25 mg/kg of Gal-PEG–R-F t2 or 100 mg/kg of chol–R-F-t2 aptamer to male BALB/c mice. In plasma, chol–R-F t2 was detected at much higher levels than Gal-PEG–RM9 t2 (Fig. 7A). The in vivo half-life of each aptamer in circulation was 11.03 ± 11.94 h for chol–R-F t2 and 3.79 ± 1.93 h for Gal-PEG–R-F t2. However, Gal-PEG–R-F t2 was distributed to mouse liver tissues more abundantly than chol–R-F t2 (Fig. 7B). The administered dose of Gal-PEG–R-F t2 was one-quarter that of chol–R-F t2; thus, 4- to 8-fold more Gal-PEG–R-F t2 was distributed to liver tissue than chol–RM9 t2. Therefore, Gal-PEG–R-F t2 could be more efficiently distributed to liver tissue than chol–R-F t2 in vivo, as shown in cells.
Fig 7.
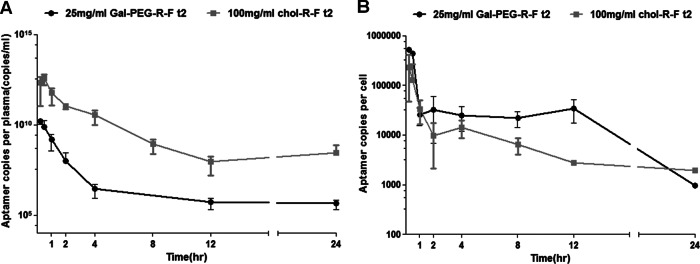
Gal-PEG–R-F t2 was efficiently distributed in mouse liver. Aptamer concentrations in plasma (A) and in liver tissue (B) of normal mice at various times after intravenous administration of chol–R-F t2 or Gal-PEG–R-F t2. Data are means ± SD (n = 5 for each administration).
DISCUSSION
Current anti-HCV treatments are associated with many side effects; they also have many contraindications, and on average, only roughly 50% of patients are able to support a sustained viral response (36). This has prompted international efforts to develop new antiviral drugs and vaccines that are active against all HCV genotypes.
A number of direct-acting antivirals (DAA) targeting HCV-encoding regulatory proteins show the rapid appearance of drug-resistant viruses (6). Since the HCV genome is present exclusively in RNA form during replication, various RNA-based therapeutics targeting the HCV genome have been suggested as tools against HCV (37, 38). However, the rapid emergence of HCV escape variants also could limit such HCV genome-targeting RNA approaches. Targeting host factors critical for HCV replication has been suggested as an alternative strategy to bypass the occurrence of drug-resistant escape mutants. A representative targetable host factor is miR-122. Recently, locked nucleic acid SPC3649-induced miR-122 antagonism was demonstrated to suppress HCV genotype 1a and 1b infection in vivo (39). However, escape variants resistant to SPC3649 could potentially occur (40). Additionally, the miR-122 level is significantly reduced in human primary hepatocellular carcinoma and negatively regulates tumorigenesis (41–43). Therefore, the possibility of nonspecific toxicity or pathogenesis due to host factor silencing in normal livers should be carefully considered (16).
Here, we isolated two kinds of specific RNA aptamers against HCV NS5B replicase composed of 2′-hydroxyl ribonucleotides and 2′-fluoro pyrimidine ribonucleotides. The 2′-hydroxyl aptamer, R-OH, competitively impeded RNA synthesis activity of the HCV replicase as a replicase substrate. In addition, the aptamer competitively inhibited the binding of authentic HCV substrate RNA, the HCV 3′ untranslated region (UTR), to the HCV replicase in vitro (data not shown). The 2′-fluoro aptamer, R-F, bound to HCV NS5B competitively with R-OH, indicating that R-F and R-OH bind to similar epitopes of the target protein. Aptamers in complex with target proteins that naturally bind nucleic acid were reported to generally interact with the nucleic acid binding site and mimic the natural interactions (44, 45). Therefore, the isolated aptamers could interact with the NS5B RNA substrate binding domain with or without the nearby motif and acted as a potent decoy to sequester NS5B from natural substrates in HCV-infected cells. Indeed, both the 7SL-driven cytoplasmic expression of R-OH and direct administration of R-F efficiently inhibited genotype 1b HCV subgenomic RNA replication and genotype 2a full-length JFH-1 replication in human liver cells.
The findings of intracellular interaction with NS5B without either off-target effects or induction of cellular toxicity and innate immune reaction by the aptamers strongly suggest that the aptamer-mediated HCV inhibition is mostly due to competitive sequestration of the target protein from binding to its viral RNA substrate in cells. Importantly, both R-OH and R-F aptamers failed to produce drug-resistant viruses. In contrast, many drug-resistant virus clones were produced by use of the NS3 aptamer. The NS3 region of the HCV genome may be a hot spot for mutation not deleterious to HCV replication, as evidenced by the fact that adaptive mutations in HCV replicon are frequently detected in the HCV NS3 helicase domain (25, 46, 47). For that reason, stable expression of NS3 aptamer might induce drug-resistant virus clones that adapted for efficient HCV 1b replicon replication. We speculate that isolated NS5B aptamers specifically bind to the essential HCV replicase substrate binding domain. Hence, drug-resistant clones may be deleterious to HCV replication. These observations imply that the isolated aptamers may be superior to any other DAA, in terms of both efficacy and safety.
For in vivo application of R-F, cholesterol or Gal-PEG was chemically conjugated. Of note, Gal-PEG–R-F t2 was more efficiently distributed in liver tissue than chol–R-F t2. HCV 1b replicon cells and stable JFH-1 cells are reported to produce thousands of copies of the HCV genome per cell (14, 15). In stable JFH-1 cells, about 105 copies of Gal-PEG–R-F t2 inhibited HCV replication by up to ∼65% and infectious virus particles by up to ∼86%. This indicates that a 10- to 100-fold excess of R-F t2 could successfully inhibit HCV replication. Using the hepatocellularity number that has been reported for mice (1.38 × 108 cells/g liver tissue) (48), we calculated that about 4.5 × 105 copies of aptamer were distributed per cell over 30 min and that a level of 3 × 104 copies per cell was maintained for 12 h after systemic injection of Gal-PEG–R-F t2 in vivo. In chronically infected patients, about 56% of hepatocytes contain HCV genomic RNA at any time and HCV replication occurs in only 14% of them at a low level (33 genomic RNA molecules/infected hepatocyte) (49). Based on these estimations, Gal-PEG–R-F t2 could subsist at a 100-fold excess compared with HCV genomic RNA in a chronically HCV-infected hepatocyte in vivo and, hence, achieve therapeutically effective quantities in patients.
In summary, we demonstrate that two kinds of RNA aptamers specifically and avidly bind to HCV NS5B replicase of both genotypes 1b and 2a and efficiently inhibit HCV replication of both genotypes in cells. Previously, other groups reported the use of DNA or RNA aptamers against HCV NS5B protein. Those aptamers were tested only for their binding ability and inhibitory effect on NS5B replicase using in vitro biochemical methods (50–52). Bellecave et al. reported DNA aptamers that inhibit HCV JFH-1 replication and viral particle formation in a cell culture system (53). However, those DNA aptamers were not examined with regard to cell toxicity profiles, distribution in animals, or side effects during long-term treatment. Therefore, concerns about safety and the possibility of escape mutant virus appearance during repeated treatment cannot be excluded with the DNA aptamers. To our knowledge, our aptamers are the first anti-HCV aptamers capable of inhibiting HCV replication of both genotypes 1b and 2a and suppressing HCV infectious virus particle formation without generation of escape mutant viruses and cellular toxicity. How the aptamers bind to both genotypes is not clear. Aptamers could present an extended molecular surface recognizing the native surface of target protein (54), indicating the possibility of broad genotype specificity for our HCV NS5B aptamers. The cytoplasmic expression of the R-OH aptamer or Gal-PEG–R-F t2 represents a new therapeutic tool and a potentially feasible alternative or additive to current HCV therapeutics. Further evaluations will be needed in animal models of HCV, such as human hepatocyte xenograft mouse models (55) or HCV-infected chimpanzees.
ACKNOWLEDGMENTS
We thank Ralf Bartenschlager (Heidelberg University, Germany) and Takaji Wakita (National Institute of Infectious Diseases, Japan) for supplying the HCV replicon construct, Charles Rice (Rockefeller University) for Huh-7.5 cells, and David Engelke (University of Michigan) for the U6+1 snRNA- and 7SL RNA-based expression vectors.
This study was supported by grants from the Korea Healthcare Technology R&D Project by the Korean Ministry for Health, Welfare & Family Affairs (A100399) and National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2012M3A9B6055200).
Footnotes
Published ahead of print 17 April 2013
REFERENCES
- 1. Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41–52 [DOI] [PubMed] [Google Scholar]
- 2. Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13:2436–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Egger D, Wölk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartenschlager R, Lohmann V. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631–1648 [DOI] [PubMed] [Google Scholar]
- 5. Moradpour D, Brass V, Gosert R, Wolk B, Blum H. 2002. Hepatitis C: molecular virology and antiviral targets. Trends Mol. Med. 8:476–482 [DOI] [PubMed] [Google Scholar]
- 6. Pawlotsky JM. 2011. Treatment failure and resistance with direct-acting antiviral drug against hepatitis C virus. Hepatology 53:1742–1751 [DOI] [PubMed] [Google Scholar]
- 7. Tuerk C, Gold L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510 [DOI] [PubMed] [Google Scholar]
- 8. Ellington AD, Szostak JW. 1990. In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–822 [DOI] [PubMed] [Google Scholar]
- 9. Que-Gewirth NS, Sullenger BA. 2007. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 14:283–291 [DOI] [PubMed] [Google Scholar]
- 10. Kaur G, Roy I. 2008. Therapeutic applications of aptamers. Expert Opin. Invest. Drugs 17:43–60 [DOI] [PubMed] [Google Scholar]
- 11. Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lohmann V, Korner F, Koch JO, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 15. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee CH, Kim JH, Kim HW, Myung HJ, Lee SW. 2012. Hepatitis C virus replication-specific inhibition of microRNA activity with self-cleavable allosteric ribozyme. Nucleic Acid Ther. 22:17–29 [DOI] [PubMed] [Google Scholar]
- 17. Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26:182–190 [DOI] [PubMed] [Google Scholar]
- 18. Chang BY, Lee CH, Lee JH, Lee SW. 2010. Comparative analysis of intracellular inhibition of hepatitis C virus replication by small interfering RNAs. Biotechnol. Lett. 32:1231–1237 [DOI] [PubMed] [Google Scholar]
- 19. Shibutani M. 2000. Anesthesia, artificial ventilation and perfusion fixation, p 511–521 In Krinke GJ. (ed), The laboratory rat. Academic Press, New York, NY [Google Scholar]
- 20. Behrens SE, Tomei L, Francesco RD. 1996. Identification and properties of the RA-dependent RNA polymerase of hepatitis C virus. EMBO J. 15:12–22 [PMC free article] [PubMed] [Google Scholar]
- 21. Luo G, Hamatake RK, Mathis DM, Racela J, Rigat KL, Lemm J, Colonno RJ. 2000. De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus. J. Virol. 74:851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paul CP, Good PD, Li SX, Kleihauer A, Rossi JJ, Engelke DR. 2003. Localized expression of small RNA inhibitors in human cells. Mol. Ther. 7:237–247 [DOI] [PubMed] [Google Scholar]
- 23. Bae SJ, Oum JH, Sharma S, Park J, Lee SW. 2002. In vitro selection of specific RNA inhibitors of NFATc. Biochem. Biophys. Res. Commun. 298:486–492 [DOI] [PubMed] [Google Scholar]
- 24. Hwang BH, Cho JS, Yeo HJ, Kim JH, Chung KM, Han KS, Jang SK, Lee SW. 2004. Isolation of specific and high-affinity RNA aptamers against NS3 helicase domain of hepatitis C virus. RNA 10:1277–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krieger N, Lohmann V, Bartenschlager R. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Healy JM, Lewis SD, Kurz M, Boomer RM, Thompson KM, Wilson C, McCauley TG. 2004. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm. Res. 21:2234–2246 [DOI] [PubMed] [Google Scholar]
- 27. de Smidt PC, Le Doan T, de Falco S, van Berkel TJ. 1991. Association of antisense oligonucleotides with lipoproteins prolongs the plasma half-life and modifies the tissue distribution. Nucleic Acids Res. 19:4695–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. 2005. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438:685–6899 [DOI] [PubMed] [Google Scholar]
- 29. Plank C, Zatloukal K, Cotton M, Mechtler K, Wagner E. 1992. Gene transfer into hepatocytes using asialoglycoprotein receptor mediated endocytosis of DNA complexed with an artificial tetra-antennary galactose ligand. Bioconjug. Chem. 3:533–539 [DOI] [PubMed] [Google Scholar]
- 30. Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M. 2007. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 25:1149–1157 [DOI] [PubMed] [Google Scholar]
- 31. Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. 2008. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320:379–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, Samokhin AO, Wang Y, Sayers S, Aikawa M, Jerome WG, Ostrowski MC, Bromme D, Libby P, Tabas IA, Welch CL, Tall AR. 2009. Free cholesterol accumulation in macrophage membranes activates toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 104:455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Azzam KM, Fessler MB. 2012. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol. Metab. 23:169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wada S, Obika S, Shibata MA, Yamamoto T, Nekatani M, Yamaoka T, Torigoe H, Harada-Shiba M. 2012. Development of a 2′,4′-BNA/LNA-based siRNA for dyslipidemia and assessment of the effects of its chemical modifications in vivo. Mol. Ther. Nucleic Acids 1:e45 doi: 10.1038/mtna.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Häussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975–982 [DOI] [PubMed] [Google Scholar]
- 37. Ryu KJ, Kim JH, Lee SW. 2003. Ribozyme-mediated selective induction of new gene activity in hepatitis C virus internal ribosome entry site-expressing cells by targeted trans-splicing. Mol. Ther. 7:386–395 [DOI] [PubMed] [Google Scholar]
- 38. Khaliq S, Khaliq SA, Zahur M, Ijaz B, Jahan S, Ansar M, Riazuddin S, Hassan S. 2010. RNAi as a new therapeutic strategy against HCV. Biotechnol. Adv. 28:27–34 [DOI] [PubMed] [Google Scholar]
- 39. Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. 2011. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc. Natl. Acad. Sci. U. S. A. 108:4991–4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, Croce CM, Bolondi L, Negrini M. 2007. Cyclin G1 Is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 67:6092–6099 [DOI] [PubMed] [Google Scholar]
- 42. Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, Hsu MT, Hsiao M, Huang HD, Tsou AP. 2009. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology 49:1571–1582 [DOI] [PubMed] [Google Scholar]
- 43. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, Chen HC, Lee CH, Tsai TF, Hsu MT, Wu JC, Huang HD, Shiao MS, Hsiao M, Tsou AP. 2012. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 122:2884–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang DB, Vu D, Cassiday LA, Zimmerman JM, Maher, Ghosh LJG. 2003. Crystal structure of NF-kB (p50)2 complexed to a high-affinity RNA aptamer. Proc. Natl. Acad. Sci. U. S. A. 100:9268–9273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosh G, Huang DB, Huxford T. 2004. Molecular mimicry of the NF-kB DNA target site by a selected RNA aptamer. Curr. Opin. Struct. Biol. 14:21–27 [DOI] [PubMed] [Google Scholar]
- 46. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1975 [DOI] [PubMed] [Google Scholar]
- 47. Grobler JA, Markel EJ, Fay JF, Graham DJ, Simcoe AL, Ludmerer SW, Murray EM, Migliaccio G, Flores OA. 2003. Identification of a key determinant of hepatitis C virus cell culture adaptation in domain II of NS3 helicase. J. Biol. Chem. 278:16741–16746 [DOI] [PubMed] [Google Scholar]
- 48. Sohlenius-Sternbeck AK. 2006. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol. In Vitro 20:1582–1586 [DOI] [PubMed] [Google Scholar]
- 49. Chang M, Williams O, Mittler J, Quintanilla A, Carithers RL, Jr, Perkins J, Corey L, Gretch DR. 2003. Dynamics of hepatitis C virus replication in human liver. Am. J. Pathol. 163:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bellecave P, Andreola ML, Ventura M, Tarrago-Litvak L, Litvak S, Astier-Gin T. 2003. Selection of DNA aptamers that bind the RNA-dependent RNA polymerase of hepatitis C virus and inhibit viral RNA synthesis in vitro. Oligonucleotides 13:455–463 [DOI] [PubMed] [Google Scholar]
- 51. Biroccio A, Hamm J, Incitti I, Francesco RD, Tomei L. 2002. Selection of RNA aptamers that are specific and high-affinity ligands of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 76:3688–3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jones LA, Clancy LE, Rawlinson WD, White PA. 2006. High-affinity aptamers to subtype 3a hepatitis C virus polymerase display genotypic specificity. Antimicrob. Agents Chemother. 50:3019–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bellecave P, Cazenave C, Rumi J, Staedel C, Cosnefroy O, Andreola ML, Ventura M, Tarrago-Litvak L, Astier-Gin T. 2008. Inhibition of hepatitis C virus (HCV) RNA polymerase by DNA aptamers: mechanism of inhibition of in vitro RNA synthesis and effect on HCV-infected cells. Antimicrob. Agents Chemother. 52:2097–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Long SB, Long MB, White RR, Sullenger BA. 2008. Crystal structure of an RNA aptamer bound to thrombin. RNA 14:2504–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bukh J. 2012. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 142:1279–1287.e3 [DOI] [PubMed] [Google Scholar]