

Abstract
It has been reported that HIV-1 Vpu mediates the degradation of interferon regulatory factor 3 (IRF-3) to avoid innate immune sensing. Here, we show that Vpu does not deplete IRF-3 from transfected cell lines or HIV-1-infected primary cells. Furthermore, the Vpu-dependent suppression of beta interferon expression described in previous studies could be ascribed to inhibition of NF-κB activation. Thus, Vpu suppresses innate immune activation through inhibition of NF-κB rather than degradation of IRF-3.
TEXT
The viral protein U (Vpu) of HIV-1 is a 16-kDa integral membrane protein produced together with Env during the late stage of the viral replication cycle. The Vpu proteins of pandemic HIV-1 group M (major) strains interact with the cytoplasmic tail of newly synthesized CD4 in the endoplasmatic reticulum to mediate its polyubiquitinylation and proteasomal degradation (1, 2). Degradation of the CD4 receptor may facilitate virion release, prevent superinfection, and enhance the incorporation of functional Env proteins into progeny viral particles. Second, Vpu promotes virion release (3, 4) by counteracting the restriction factor tetherin (BST-2), which tethers nascent virions to the cell surface (5, 6). Recent studies have suggested that Vpu also reduces cell surface expression of the natural killer (NK) cell ligands NTB-A and PVR (7, 8) and the lipid-antigen-presenting protein CD1d (9) to protect HIV-1-infected cells against NK cells and natural killer T (NKT) cells, respectively. Finally, it has been reported that Vpu mediates depletion of interferon regulatory factor 3 (IRF-3), a transcription factor that plays a central role in pathogen recognition receptor (PRR) signaling, to avoid innate immune sensing in virus-infected cells (10, 11).
Vpu is only encoded by HIV-1 and its simian immunodeficiency virus (SIV) precursors. We and others have shown that the Vpu proteins of group M, N, O, and P strains of HIV-1, which resulted from independent zoonotic transmissions, and their SIV counterparts exhibit fundamental functional differences (12–16). Perhaps most notably, only Vpus of pandemic group M strains have acquired the capability to antagonize tetherin while maintaining their CD4 function during adaptation to humans (12). In comparison, Vpu proteins of rare HIV-1 group N strains are usually weak tetherin antagonists and fail to degrade CD4, and those of nonpandemic HIV-1 group O and P strains lack significant anti-tetherin activity (12–16). Obviously, differences in the abilities of these viruses to avoid innate immune sensing of virally infected cells by the Vpu-mediated counteraction of IRF-3 may also play a role in their replication fitness and spread in the human population. Thus, the initial goal of the present study was to examine whether these primate lentiviral Vpus also differ in their abilities to degrade IRF-3 (10, 11).
First, we tried to confirm the published data that suggested that the HIV-1 NL4-3 Vpu induces effective IRF-3 degradation in established cell lines (10). To examine this, we transfected HeLa cells expressing endogenous IRF-3 with different doses of pCG vectors coexpressing AU1-tagged NL4-3 Vpu and enhanced green fluorescent protein (eGFP) (12) by using Lipofectamine (Invitrogen) according to the manufacturer's instructions. To monitor cellular and viral antigen expression, the cells were lysed in mammalian protein extraction reagent (Thermo Scientific) 2 days posttransfection, and cell lysates were separated in 4-to-12% bis-Tris gels (Invitrogen). Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes and probed with anti-IRF-3 antibody (Santa Cruz Biotechnology). Subsequently, blots were probed with anti-mouse or anti-rabbit IRDye Odyssey antibodies (Li-Cor), and proteins were detected using a Li-Cor Odyssey scanner. For controls, blots were incubated with antibodies specific for β-actin (Abcam) and AU1 (Covance). The results showed that NL4-3 Vpu was efficiently expressed but did not induce a reduction of IRF-3 expression levels (Fig. 1A). To further challenge this unanticipated finding, we analyzed the effect of Vpu on endogenous IRF-3 expression in 293T cells, which were also used in the previous studies (10, 11). In contrast to HeLa cells, only one IRF-3-specific band could be detected in unstimulated 293T cells, which is consistent with previous results (10, 24). Although Vpu was efficiently expressed in a dose-dependent manner, we did not observe an effect of Vpu on the levels of endogenously expressed IRF-3 (Fig. 1B). In agreement with published data, expression of NSP1-NCDV, a nonstructural protein of the Nebraska calf diarrhea rotavirus reduced IRF-3 expression levels, whereas NSP1 from a closely related porcine rotavirus (OSU) was inactive (13–15) (Fig. 1C). To examine the effect of Vpu on activated IRF-3, we treated the cells with poly(I·C), a synthetic analog of double-stranded RNA. Induction of innate immune signaling responses by poly(I·C) was verified by activation of the beta interferon (IFN-β) promoter (Fig. 1D). In agreement with our previous results, Vpu failed to reduce the expression levels of activated IRF-3 (Fig. 1D).
Fig 1.

Vpu does not reduce IRF-3 expression levels. (A) HeLa cells were transfected with increasing concentrations (0, 0.5, 1.5, and 4.5 μg in all experiments) of an expression plasmid for NL4-3 vpu. The values provided below panels A to D and F give the intensity of the IRF-3 signal, normalized to the β-actin control. For better comparison, the mock value was always set as 1.0 in each panel. (B) 293T cells were transfected with increasing concentrations of an expression plasmid for NL4-3 vpu. (C) 293T cells were transfected with 5 μg of control plasmid or expression plasmids for NCDV (active against IRF-3) or OSU (not active against IRF-3) NSP1 (15). Western blotting was performed 24 h later with an IRF-3 antibody kindly provided by Michael David. (D) 293T cells were transfected with increasing concentrations of Vpu expression plasmid. To stimulate IRF-3-dependent signaling, cells were cotransfected with 2 μg poly(I·C). Poly(I·C) stimulation was monitored in an IFN-β promoter reporter luciferase assay (right panel). At 2 days posttransfection, cells were lysed and Western blotting was performed. The membrane was incubated with antibodies against IRF-3 and AU1. β-Actin was used as a loading control and for normalization (left panel). (E) SupT1 cells were transduced with VSV-G-pseudotyped virus stocks of wild-type or vpu-defective HIV-1 NL4-3. To induce IRF-3-dependent signaling, cells were coinfected with SeV 24 h before lysis at 3 days postransduction. Western blot analysis was performed using three different anti-IRF-3 antibodies (from Santa Cruz Biotechnology [SC], a gift from Michael David [MD], and AR1). Staining for the viral p55 and p24 Gag antigens was performed to monitor HIV-1 infection. β-Actin was used as loading control. (F) SupT1 cells were transduced with the indicated VSV-G-pseudotyped HIV-1 constructs and analyzed by Western blotting 3 days later. IRES-env proviral constructs allowed the expression of vpu alleles independent of overlapping env sequences.
To examine a possible effect of Vpu on IRF-3 expression in HIV-1-infected T cells, we transduced SupT1 cells with vesicular stomatitis virus G protein (VSV-G)-pseudotyped HIV-1 NL4-3 constructs containing intact or defective vpu genes (20). Virus stocks were generated by transient transfection of 293T cells as described previously (21). To activate immune signaling, cells were infected with Sendai virus (SeV) 2 days postransduction, as reported in previous studies (10, 11, 16). SeV infection and immune activation were confirmed by cytopathic effects and increased NF-κB and IFN-β reporter gene activities (data not shown). Three days postransduction, cells were lysed and analyzed by Western blotting. To further validate our results, we utilized three different IRF-3 antibodies, including the one (designated MD) used in the previous studies of Doehle and colleagues (10, 11, 17). Irrespective of the antibody used, we detected similar levels of IRF-3 in cells infected with wild-type or vpu-defective HIV-1 constructs (Fig. 1E). To exclude that we might miss Vpu-mediated effects on IRF-3 expression levels due to allele-specific differences, we infected cells with NL4-3-based constructs expressing different vpu alleles without overlapping env sequences. To generate these constructs, we first eliminated the vpu-env overlap and subsequently inserted an internal ribosome entry site (IRES) element downstream of vpu to restore env expression. Just like wild-type HIV-1 NL4-3, derivatives expressing JR-CSF or repaired YU2 Vpu failed to induce significant degradation of IRF-3 in transduced SupT1 cells (Fig. 1F). The expression of functional Vpu from the IRES-env constructs has been shown before (12) and was confirmed by a reduction of mature virions (i.e., p24) in the cell lysates due to the counteraction of tetherin and/or degradation of CD4 (Fig. 1F).
To exclude the possibility that some subtle effects of Vpu on IRF-3 were missed because the cells were examined in bulk, we next performed fluorescence-activated cell sorting (FACS)-based assays. The pCG vectors used in these experiments coexpress the gene of interest and eGFP from the same RNA via an IRES and thus allow the direct comparison of IRF-3 expression levels in transfected and untransfected cells (18). Since previous studies suggested that Vpr and Vif may also affect IRF-3 expression (16, 17), we included constructs expressing these accessory genes in our studies. 293T cells were transfected with these expression constructs, and 24 h later the cells were fixed, permeabilized (Fix & Perm cell permeabilization kit; ADG), and examined for IRF-3 expression by flow cytometric analysis using unconjugated anti-IRF-3 (Santa Cruz Biotechnology) and Alexa Fluor 647-conjugated anti-rabbit antibodies (Invitrogen) (Fig. 2A). To determine the relative IRF-3 expression levels, the mean AF647 fluorescence intensity (MFI) of transfected (eGFP+) cells was normalized to the MFI of the untransfected (eGFP−) cell population after subtraction of isotype control values. We found that Vpu, Vpr, and Vif had no significant effect on IRF-3 expression levels, whereas NSP1 (NCDV) reduced it by about 40% (Fig. 2A and B).
Fig 2.
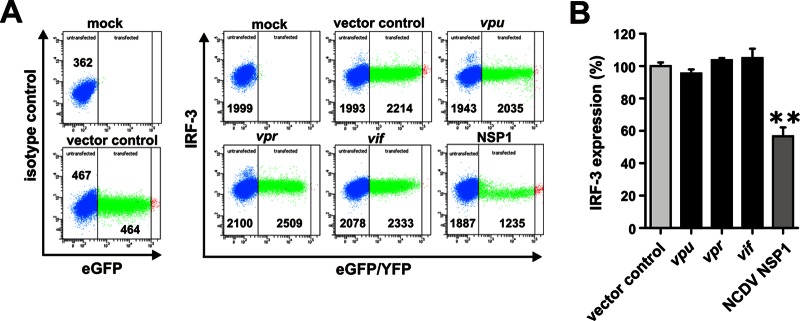
Vpu, Vpr, and Vif do not affect the level of IRF-3 expression. (A) 293T cells were transfected with 5 μg of pCG vectors coexpressing NL4-3 Vpu, Vpr, or Vif proteins and eGFP via an IRES. A vector coexpressing NCDV NSP1 and yellow fluorescent protein (YFP) was used as a positive control. The cells were permeabilized at 24 h posttransfection and stained for IRF-3 (Santa Cruz Biotechnology) for FACS analysis. (B) Levels of IRF-3 expression in the presence of the indicated expression constructs relative to the empty vector control (set as 100%). Data are mean values (± the standard errors of the means) derived from three independent experiments.
To examine the effects in HIV-1-infected SupT1 cells we utilized a variety of HIV-1 NL4-3-based proviral constructs coexpressing eGFP via an IRES element (12, 19, 20). These constructs have the advantage that all viral genes are expressed from the wild-type HIV-1 long terminal repeat promoter and via the regular splicing sites. Thus, they represent a highly sensitive system to examine the effects of accessory proteins, such as Vpu, on the expression levels of cellular proteins in HIV-1-infected cells (12, 19, 21). Our results showed that individual or combined deletions in the viral accessory vpu, vpr, and nef genes had no significant effect on IRF-3 expression levels in HIV-1-infected T cells (Fig. 3A and B). To examine the effects in primary target cells of HIV-1, we transduced peripheral blood mononuclear cells (PBMCs) with wild-type and vpu-deficient HIV-1 virions. Flow cytometric analyses revealed that an intact vpu gene had no effect on IRF-3 expression levels but reduced tetherin expression by about 40% (Fig. 3C and D). This magnitude of tetherin degradation is in agreement with published data (22–26) and verified functional expression of Vpu.
Fig 3.

Lack of vpu, nef, or vpr expression does not affect IRF-3 expression levels in HIV-1-infected T cells. (A) SupT1 cells were infected with virus stocks of different NL4-3 IRES eGFP mutants. At 3 days postinfection, cells were permeabilized and stained for IRF-3 (Santa Cruz Biotechnology) to perform FACS analysis. (B) Levels of IRF-3 expression in cells infected with the indicated mutant NL4-3 constructs relative to the wild-type virus (wt; set as 100%). Data are means (± standard errors of the means [SEM]) derived from three independent experiments. (C) PBMCs were transduced with VSV-G-pseudotyped virus stocks of wild-type or vpu-defective NL4-3 IRES eGFP mutants. PBMCs were stimulated with interleukin-2 and phytohemagglutinin for 3 days prior to transduction. At 3 days postransduction, the cells were permeabilized and stained for IRF-3 (Santa Cruz Biotechnology) or tetherin (eBioscience) for FACS analyses. (D) Mean levels (± SEM; n = 3) of IRF-3 and tetherin expression in cells transduced with the vpu-defective virus relative to the wild-type control (set as 100%) are indicated.
It has previously been shown that Vpu reduces IRF-3-dependent expression of IFN-β (10, 11). To examine this, we cotransfected 293T cells with a pCG-based plasmid expressing HIV-1 WITO Vpu (or an empty vector as control) and constructs containing the firefly and Gaussia luciferases (in a 5:1 ratio) under the control of the IFN-β and pTAL promoters, respectively. We used the WITO vpu for these experiments because it is derived from a transmitted/founder subtype B HIV-1 group M strain (27) and is thus more relevant for the in vivo situation than the T cell line-adapted molecular HIV-1 NL4-3 clone. Previous studies showed that the WITO Vpu is highly active in degrading CD4 and counteracting tetherin (12). The pTAL promoter construct contains a minimal TATA-like promoter (pTAL) region from the herpes simplex virus thymidine kinase (HSV-TK) promoter (Clontech) that is nonresponsive to IRF-3 and NF-κB, and this served as an internal control for transfection efficiencies. For immune activation, cells were infected with SeV at 24 h posttransfection, and the luciferase activities were determined 1 day later. The firefly luciferase signals were normalized to the corresponding Gaussia luciferase signals. In agreement with published data (10), SeV induced IFN-β promoter activity >20-fold and Vpu substantially reduced this activation (Fig. 4A). Control experiments confirmed, however, that the IRF-3 expression levels remained unchanged in the presence of WITO Vpu (Fig. 4B), suggesting that Vpu may inhibit IFN-β promoter activity by an IRF-3-independent mechanism. Recently, it has been reported that Vpu impairs viral immune sensing by suppressing tetherin-induced NF-κB activation (28). Thus, we examined whether NF-κB may play a role in the induction of IFN-β. In agreement with this possibility, we found that the IFN-β promoter contains binding sites for both IRF-3 and NF-κB (Fig. 4C). Thus, the reporter construct used in previous studies (11, 12) may not be specific for IRF-3 activation. To determine whether the IFN-β promoter is responsive to NF-κB, the activity of the IFN-β promoter-dependent firefly luciferase was examined after activation of NF-κB through cotransfection of a constitutively active mutant of IKKβ (IKKβ ca). Luciferase activities were determined 2 days posttransfection as described above. The results demonstrated that IFN-β promoter activity was about 40-fold enhanced by NF-κB activation and that this effect was greatly diminished by Vpu (Fig. 4D). Notably, expression of constitutively active IKKβ did not increase the levels of endogenous IRF-3 in transiently transfected 293T cells (Fig. 4E). Next, we examined the effect of SeV and Vpu on NF-κB activation with the help of a reporter vector that expressed firefly luciferase under the control of three NF-κB binding sites. We found that SeV induced the NF-κB-dependent firefly luciferase expression about 6-fold and that this induction was greatly diminished in the presence of Vpu (Fig. 4F). The analysis of 293T cells cotransfected with NF-κB-dependent firefly luciferase, the pTAL promoter Gaussia luciferase construct, and expression plasmids for constitutively active IKKβ and Vpu confirmed that Vpu inhibits NF-κB induction (Fig. 4G). This inhibition is not allele specific and has been confirmed for a variety of different primate lentiviral Vpu proteins, including that encoded by the NL4-3 molecular clone (D. Sauter and F. Kirchhoff, unpublished observations). These results confirmed that Vpu may suppress viral immune sensing and the secretion of inflammatory cytokines such as IFN-β but further suggest that these effects are due to inhibition of NF-κB activation rather than IRF-3 degradation.
Fig 4.
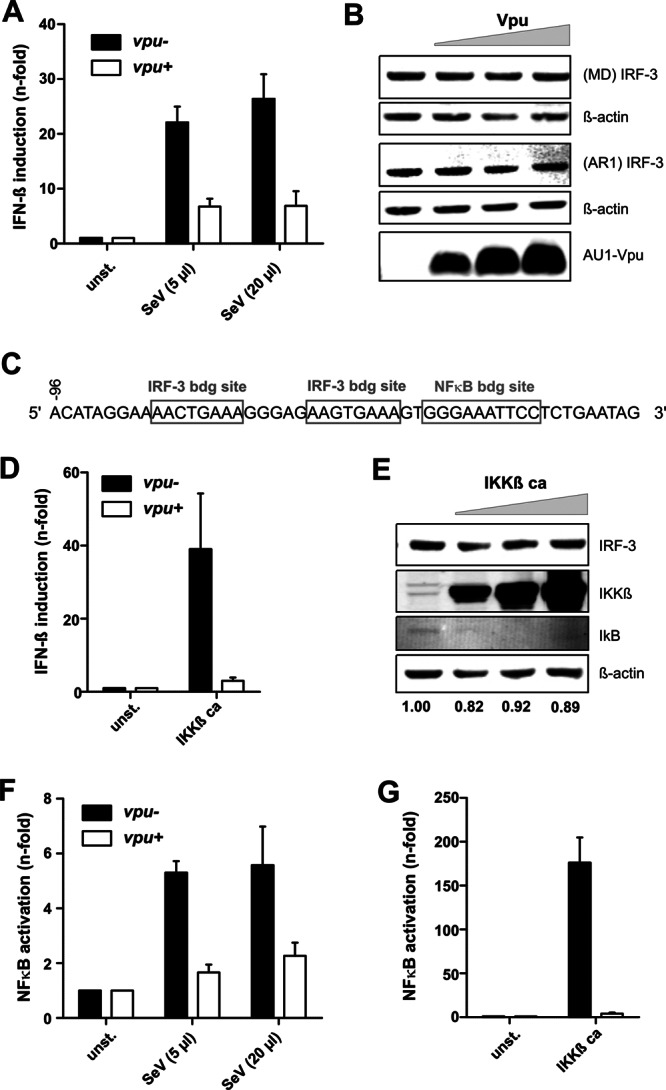
Vpu reduces innate immune activation by suppressing NF-κB activation. (A) 293T cells were cotransfected with vectors expressing HIV-1 WITO Vpu (or control plasmid) and constructs containing the firefly and Gaussia luciferases under the control of the IFN-β and pTAL promoters, respectively. For immune activation, cells were infected with SeV at 24 h posttransfection, and the luciferase activities were determined 1 day later. The results in panels A, D, F, and G are from three independent experiments (± the SEM). (B) 293T cells were transfected with increasing concentrations of WITO Vpu expression plasmid, and IRF-3 expression was monitored by using two different antibodies, as described for Fig. 1. (C) IRF-3 and NF-κB binding sites in the IFN-β promoter. (D) 293T cells were cotransfected with vectors expressing Vpu and IKKβ ca together with IFN-β firefly and pTAL Gaussia luciferase vectors. Induction of the IFN-β promoter was determined by luciferase assays as described above. (E) 293T cells were transfected with increasing concentrations (0, 0.5, 1.5, and 4.5 μg) of expression plasmids for constitutively active IKKβ to activate NF-κB and analyzed by Western blotting using antibodies against IRF-3 (M. David), IKKβ, IκB, and β-actin. The values provided below panels B and E show the intensity of the IRF-3 signal normalized to the β-actin control. For better comparisons, the mock value was always set to 1.0 in each panel. (F) 293T cells were cotransfected with NF-κB promoter firefly luciferase and pTAL promoter Gaussia luciferase constructs together with Vpu expression or control vectors and stimulated by infection with Sendai virus 24 h posttransfection, prior to luciferase assays at 48 h posttransfection. (G) 293T cells cotransfected with NF-κB-dependent firefly luciferase, the pTAL promoter Gaussia luciferase construct, and expression plasmids for constitutively active IKKβ were examined in luciferase assays as described above. Firefly luciferase signals were generally normalized to the corresponding Gaussia luciferase signals to account for differences in transfection efficiencies.
In summary, we showed here that Vpu does not significantly deplete IRF-3, even at very high expression levels. Notably, our studies involved the analysis of primary human cells infected with wild-type and vpu-defective HIV-1 strains, which allowed us to readily distinguish between infected and uninfected cells and to monitor other Vpu functions, such as degradation of tetherin (12, 21). The fact that even this highly sensitive experimental system failed to reveal a significant effect of Vpu on IRF-3 expression levels strongly argues against Vpu-mediated degradation of IRF-3. Our results confirm, however, that Vpu reduces viral immune sensing (10). However, this effect involved modulation of NF-κB rather than IRF-3-dependent signaling. Vpu-mediated inhibition of NF-κB signaling may explain most results from previous reports (10, 11). Notably, data supporting a direct role of Vpu on IRF-3 expression were sparse in the studies of Doehle and coworkers, and the effects of HIV-1 infection on IRF-3 expression levels have been variable for different data sets; for example, a complete lack of IRF-3 protein expression was observed in some Western blot assays, but IRF-3 was still readily detectable by microscopy and remained unchanged in one coimmunoprecipitation experiment in the presence of Vpu (10). Furthermore, unspecific effects of virus infection on cell viability may have contributed to the reduction of IRF-3 expression in some experiments. In either case, our observation that Vpu inhibits NF-κB activation may have important implications, since this transcription factor plays a major role in the induction of antiviral immune responses. Currently, we are investigating whether the effect of Vpu on NF-κB activity is conserved between different primate lentiviruses and the mechanism(s) underlying this Vpu function.
ACKNOWLEDGMENTS
We thank Susanne Engelhart, Daniela Krnavek, Kerstin Regensburger, and Martha Mayer for excellent technical assistance. SeV was kindly provided by Georg Kochs. The IRF-3 expression vector was kindly provided by John Hiscott. Michael Gale provided the IFN-β promoter reporter construct and AR1. The construct for NF-κB-dependent firefly luciferase and constitutively active IKKβ were kindly provided by Bernd Baumann. The NSP1 expression constructs were kindly provided by Michele Hardy.
This work was supported by the Deutsche Forschungsgemeinschaft (Leibniz Award to F.K.) and the Hector Foundation.
Footnotes
Published ahead of print 3 April 2013
REFERENCES
- 1. Bour S, Schubert U, Strebel K. 1995. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation. J. Virol. 69:1510–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 66:7193–7200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Strebel K, Klimkait T, Maldarelli F, Martin MA. 1989. Molecular and biochemical analyses of human immunodeficiency virus type 1 vpu protein. J. Virol. 63:3784–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Göttlinger HG, Dorfman T, Cohen EA, Haseltine WA. 1993. Vpu protein of human immunodeficiency virus type 1 enhances the release of capsids produced by gag gene constructs of widely divergent retroviruses. Proc. Natl. Acad. Sci. U. S. A. 90:7381–7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neil SJD, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430 [DOI] [PubMed] [Google Scholar]
- 6. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, Barker E. 2010. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 8:397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matusali G, Potestà M, Santoni A, Cerboni C, Doria M. 2012. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J. Virol. 86:4496–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moll M, Andersson SK, Smed-Sörensen A, Sandberg JK. 2010. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 116:1876–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M., Jr 2012. Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J. Virol. 86:8367–8374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doehle BP, Chang K, Fleming L, McNevin J, Hladik F, McElrath MJ, Gale M., Jr 2012. Vpu-deficient HIV strains stimulate innate immune signaling responses in target cells. J. Virol. 86:8499–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sauter D, Schindler M, Specht A, Landford WN, Münch J, Kim Votteler K-AJ, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U. S. A. 102:4114–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graff JW, Ewen J, Ettayebi K, Hardy ME. 2007. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J. Gen. Virol. 88:613–620 [DOI] [PubMed] [Google Scholar]
- 15. Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. 2009. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J. Virol. 83:10322–10335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. 2008. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr 2009. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83:10395–10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greenberg ME, Iafrate AJ, Skowronski J. 1998. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17:2777–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schindler M, Münch J, Kutsch O, Li H, Santiago ML, Bibollet-Ruche F, Müller-Trutwin MC, Novembre FJ, Peeters M, Courgnaud V, Bailes E, Roques P, Sodora DL, Silvestri G, Sharp PM, Hahn BH, Kirchhoff F. 2006. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 125:1055–1067 [DOI] [PubMed] [Google Scholar]
- 20. Wildum S, Schindler M, Münch J, Kirchhoff F. 2006. Contribution of Vpu, Env, and Nef to CD4 downmodulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 80:8047–8059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sauter D, Unterweger D, Vogl M, Usmani SM, Heigele A, Kluge SF, Hermkes E, Moll M, Barker E, Peeters M, Learn GH, Bibollet-Ruche F, Fritz JV, Fackler OT, Hahn BH, Kirchhoff F. 2012. Human tetherin exerts strong selection pressure on the HIV-1 group N Vpu protein. PLoS Pathog. 8:e1003093 doi: 10.1371/journal.ppat.1003093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Früh K, Moses AV. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J. Virol. 83:7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goffinet C, Allespach I, Homann S, Tervo Habermann H-MA, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, Krijnse-Locker J, Banting G, Kräusslich Fackler H-GOT, Keppler OT. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285–297 [DOI] [PubMed] [Google Scholar]
- 24. Gupta RK, Hué S, Schaller T, Verschoor E, Pillay D, Towers GJ. 2009. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 5:e1000443 doi: 10.1371/journal.ppat.1000443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. 2009. HIV-1 Vpu neutralizes the antiviral factor tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 5:e1000574 doi: 10.1371/journal.ppat.1000574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJD, Bieniasz PD. 2009. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5:e1000300 doi: 10.1371/journal.ppat.1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galão RP, Le Tortorec A, Pickering S, Kueck T, Neil SJD. 2012. Innate sensing of HIV-1 assembly by tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 12:633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]