

Abstract
Nipah virus (NiV) is a nonsegmented, single-stranded, negative-sense RNA virus belonging to the genus Henipavirus, family Paramyxoviridae. NiV causes acute encephalitis and respiratory disease in humans, is associated with high mortality, and poses a threat in southern Asia. The genomes of henipaviruses are about 18,246 nucleotides (nt) long, which is longer than those of other paramyxoviruses (around 15,384 nt). This difference is caused by the noncoding RNA region, particularly the 3′ untranslated region (UTR), which occupies more than half of the noncoding RNA region. To determine the function(s) of the NiV noncoding RNA region, we investigated the effects of NiV 3′ UTRs on reporter gene expression. The NiV N 3′ UTR (nt 1 to 100) demonstrated strong repressor activity associated with hnRNP D protein binding to that region. Mutation of the hnRNP D binding site or knockdown of hnRNP D resulted in increased expression of the NiV N 3′ UTR reporter. Our findings suggest that NiV N expression is repressed by hnRNP D through the NiV N 3′ UTR and demonstrate the involvement of posttranscriptional regulation in the NiV life cycle. To the best of our knowledge, this provides the first report of the functions of the NiV noncoding RNA region.
INTRODUCTION
Nipah virus (NiV) belongs to the genus Henipavirus, family Paramyxoviridae (1). NiV causes acute encephalitis and respiratory disease, characterized by fever, headaches, vomiting, dizziness, and sore throat. An outbreak of NiV in Bangladesh in 2004 was associated with mortality of around 75% (2). Human NiV infections may be asymptomatic or cause fatal encephalitis. NiV was first isolated in Malaysia in 1999 (3), and since then, it has been responsible for outbreaks in Bangladesh and India and poses a major threat to populations in southern and Southeast Asia.
The NiV genome was found to comprise a single-stranded, nonsegmented, negative-sense RNA molecule of 18,246 nucleotides (nt) in a Malaysian isolate and 18,252 nt in a Bangladeshi isolate (4). It includes six genes corresponding to the nucleocapsid (N), phosphoprotein (P), matrix protein (M), fusion protein (F), glycoprotein (G), and large protein (L). The P gene also encodes accessory proteins C, V, and W, which are synthesized by translation from another initiation site or by insertion of a G nucleotide (5). In other paramyxoviruses, the genomic RNA and N proteins are thought to form the nucleocapsid, which is the template for transcription and replication, while the P and L proteins form viral RNA polymerase. The promoter is located at the 3′ end of the RNA genome, which is where RNA synthesis occurs.
Henipavirus genomes are longer than those of most other paramyxoviruses, most of which have genomes of around 15,384 nt (5–7). These differences in genome size are mainly attributed to the noncoding RNA region, particularly the 3′ UTR, which occupies more than half of the noncoding RNA region. In mammalian cells, the 3′ UTR is known to be the site recognized by many factors involved in posttranscriptional regulation of gene expression.
In this study, we investigated the function of the noncoding RNA region in the NiV genome by examining the ability of the NiV UTR to regulate gene expression.
MATERIALS AND METHODS
Plasmid construction.
NiV 3′ UTR reporter vectors were constructed by amplification of 3′ UTRs using specific primers: Nipah N 3′ UTR NheI, F, 5′-tat GCT AGC cgt cta ttt cca ata ttc tac (restriction enzyme sites are in uppercase); Nipah N 3′ UTR XbaI, R, 5′-tat TCT AGA ttt ttc tta atg agt tag tag; Nipah P3′ UTR NheI, F, 5′-tat GCT AGC tca ctg aat tgt cag cag aaa; Nipah P 3′ UTR XbaI, R, 5′-tat TCT AGA ttt ttc tta att gat atg caa; Nipah M 3′ UTR NheI, F, 5′-tat GCT AGC aca gaa ttc ttc taa aat tta; Nipah M 3′ UTR XbaI, R, 5′-tat TCT AGA ttt ttt gta ata gtt atc taa; Nipah F 3′ UTR NheI, F, 5′-tat GCT AGC tgt att cag att gat gaa att; Nipah F 3′ UTR XbaI, R, 5′-tat TCT AGA ttt ttt att aaa ttt cta aag; Nipah G 3′ UTR NheI, F, 5′-tat GCT AGC aaa tca acc tca taa ttt aat; Nipah G 3′ UTR XbaI, R, 5′-tat TCT AGA ttt ttc tta att tac act aat; Nipah L 3′ UTR NheI, F, 5′-tat GCT AGC ttt aac ctt cca aat cca aga; and Nipah L 3′ UTR XbaI, R, 5′-tat TCT AGA ttt ttc tta ata act gaa cct. PCR products were digested with NheI and XbaI and cloned into the XbaI site of pGL3control (Promega). NiV N 3′ UTR deletion variants were constructed by inverted PCR using Phusion DNA polymerase (Finzymes) with various combinations of primers. After gel extraction, the purified PCR products were phosphorylated and self-ligated using T4 PNK (Toyobo) and T4 DNA ligase (Promega). NiV N supporting plasmids were constructed by amplification of the NiV N coding sequence or the NiV N coding sequence with the 3′ UTR using the following primers: Nipah N 5′ UTR MluI, F, 5′-ata ACG CGT agg aac caa gac aaa cac ttt tgg; Nipah N ORF XbaI, R, 5′-tat TCT AGA tca cac atc agc tct gac gaa a; and Nipah N 3UTR XbaI, R, 5′-tat TCT AGA ttt ttc tta atg agt tag tag. The PCR products were digested and cloned between the MluI and XbaI sites of the pTNT vector (Promega). Chemically competent Escherichia coli DH5α was used routinely for transformation of recombinant plasmids (Toyobo). All plasmids were purified using a Genopure Plasmid Midi Kit (Roche Applied Science).
Reporter assay.
Reporter plasmids and Renilla luciferase plasmid (pRLTK; Promega) were mixed and transfected into HeLa cells using Lipofectamine LTX (Invitrogen). Luciferase activities were measured after the appropriate transfection times using a Dual-Luciferase Reporter Assay System (Promega). For small interfering RNA (siRNA) experiments, siRNA transfection was carried out for 48 h, and reporter vectors were transfected into knockdown (KD) HeLa cells using Lipofectamine 2000 (Invitrogen). Luciferase activity was measured at 24 h posttransfection.
Northern blotting.
Transfection was conducted using Lipofectamine LTX (Invitrogen). At 24 h posttransfection with the reporter plasmid, cells were exposed to 5 μg/ml actinomycin D (Wako) for the prescribed times. Total RNA was separated by formaldehyde-denaturing 1.5% (wt/vol) agarose gel electrophoresis. The RNA was then transferred to Hybond N+ membranes (GE Healthcare) by capillary transfer. After UV cross-linking, the membranes were stained with methylene blue to visualize RNA, prehybridized, and then hybridized using DIG Easy Hyb (Roche). The membranes were then washed, and detection of digoxigenin (DIG)-labeled RNA probes was carried out using a DIG Northern Starter kit (Roche) according to the manufacturer's instructions. The DIG-labeled RNA probe was synthesized using a DIG RNA-labeling kit (Roche).
RNA pulldown assay.
A NiV N 3′ UTR fragment was cloned into pcDNA3 (Invitrogen) and then used as a PCR template. Templates for RNA synthesis were prepared by PCR (KOD-plus DNA polymerase; Toyobo). RNAs were synthesized using a T7 RiboMax Express Large Scale RNA Production System (Promega) and purified with Isogen (Nippongene). 3′-end biotin-labeled RNA probes were prepared using an RNA 3′-End Biotinylation Kit (Pierce). After chloroform extraction, biotin-labeled RNA probes were purified using MicroSpin G-25 Columns (GE Healthcare).
Cytoplasmic extracts were prepared by trypsinization of HeLa cells and resuspension in hypotonic buffer (10 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol [DTT]). After 10 min on ice, the HeLa cells were collected and resuspended in NP-40 lysis buffer (10 mM HEPES-KOH, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40) supplemented with a protease inhibitor (Complete Mini, EDTA-free; Roche Applied Science). After incubation for 10 min followed by centrifugation, the supernatant containing the HeLa cytoplasmic extract was collected. HeLa cytoplasmic extracts were precleared with Streptavidin Sepharose High Performance (GE Healthcare) prior to performing the RNA pulldown assays.
Biotin-labeled RNA probes, precleared HeLa cytoplasmic extracts, streptavidin Sepharose, protease inhibitor, RNase inhibitor, and 10 μg/ml polyinosine-polycytidine (pIpC) (Sigma-Aldrich) were mixed and incubated at 4°C for 2 h. Samples were prepared for mass spectrometry analysis using NP-40 lysis buffer as a binding buffer. hnRNP protein binding to the NiV N 3′ UTR was examined using more stringent conditions involving an in vitro binding buffer (50 mM HEPES-KOH, pH 7.4, 100 mM NaCl, 3 mM MgCl2, 0.1% Triton X-100). Binding complexes were washed three times with NP-40 lysis buffer or in vitro binding buffer and then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), silver staining, and Western blotting.
Cell fractionation.
Cell fractionation was conducted as described previously (8; http://www.lamondlab.com/).
SDS-PAGE, silver staining, and Western blotting.
Silver staining was conducted using a Silver Stain Plus kit (Bio-Rad). SDS-PAGE and Western blotting were conducted using general protocols. The wash buffer consisted of 10 mM Tris-HCl, pH 7.4, 100 mM NaCl, and 0.1% Tween 20 (TBST), and the blocking buffer was 5% (wt/vol) skim milk in TBST. To detect NiV N proteins, HeLa cells were infected with vTF7-3 for 1 h. Following a medium change, the cells were transfected with the NiV N supporting plasmid for 24 h. Cellular proteins were extracted and analyzed by Western blotting.
The primary antibodies used were hnRNP D/AUF1 (Abcam; ab50692), hnRNP A/B (Santa Cruz; sc-133664), α-tubulin (MBL; M175-3), and lamin A/C (Santa Cruz; sc-6215). Anti-NiV N serum was produced in our laboratory (9). The secondary antibodies were polyclonal horseradish peroxidase-conjugated anti-mouse IgG (Dako), polyclonal anti-rabbit IgG (Dako), and polyclonal anti-goat IgG. The ECL Plus or ECL Western Blotting Detection System (GE Healthcare) was used to detect proteins on membranes.
Protein expression and RNA gel shift assays.
Purified hnRNP proteins were obtained using an E. coli protein expression system. Expression vectors for hnRNP were constructed by inserting hnRNP open reading frames (ORFs) into pGEX-4T-1 (GE Healthcare), followed by transformation into Rosetta2 cells (TaKaRa). The cells were induced with 1 mM isopropyl thio-β-galactoside and glutathione S-transferase fusion proteins. They were then extracted by sonication, purified with glutathione Sepharose 4B (GE Healthcare), and digested with thrombin (GE Healthcare). Thrombin proteases were removed using benzamidine Sepharose 6B (GE Healthcare). Radio isotope (RI)-labeled RNA probes were prepared with [32P]pCp and T4 RNA ligase (TaKaRa) and purified using MicroSpin G-25 columns. The RNA probes, purified proteins, and other additives were mixed and incubated in in vitro binding buffer for 10 min at room temperature. pIpC (Sigma-Aldrich; P1530) was used as a nonspecific RNA for standardization of the amounts of total RNAs in the reaction complex. After the addition of 4% (wt/vol) sucrose, RNA-protein complexes were separated on 5% (wt/vol) polyacrylamide gels with 0.5× Tris-borate EDTA buffer.
siRNA experiments.
The siRNAs used in our study were purchased from RNAi Inc. or Sigma-Aldrich. The siRNA sequences were 5′-GCC ACA ACG UCU AUA UCA UUU-3′ (siRNA for enhanced green fluorescent protein [siEGFP], sense), 5′-AUG AUA UAG ACG UUG UGG CUU-3′ (siEGFP, antisense), 5′-GAU UUA AAG GAU UAU UUU ACU-3′ (siRNA for hnRNP A/B [sihnRNP A/B], sense), 5′-UAA AAU AAU CCU UUA AAU CUU-3′ (sihnRNP A/B, antisense), 5′-GAU CCU AUC ACA GGG CGA UUU-3′ (siRNA for hnRNP D [sihnRNP D], sense), and 5′-AUC GCC CUG UGA UAG GAU CUU-3′ (sihnRNP D, antisense). Transfection of siRNAs was performed using Lipofectamine 2000, with all siRNAs used at a final concentration of 50 nM. Repression of target genes was confirmed by extraction and analysis of cellular proteins by Western blotting at 72 h posttransfection.
RESULTS
The NiV N 3′ UTR downregulates reporter gene expression.
Firefly luciferase reporters were attached to NiV 3′ UTRs, and the plasmids were transfected into HeLa cells. Some of the NiV 3′ UTRs regulated reporter gene expression (Fig. 1A). The NiV N 3′ UTR showed the strongest suppressor activity, reducing luciferase expression to about 40% compared with the control. We performed Western blotting to determine if the suppressor activity of the NiV N 3′ UTR reduced NiV N protein expression. Because of restrictions on the use of recombinant living NiV, we used a technique similar to that for NiV rescue, which involves transfection with supporting plasmids after infection of vaccinia T7 virus (10). Transfection with a supporting plasmid containing the NiV N 3′ UTR reduced the amount of NiV N protein synthesized (Fig. 1B). The effects of the NiV N 3′ UTRs on RNA stability were examined by Northern blotting. Reporter mRNAs with NiV N 3′ UTRs degraded more quickly than mock RNAs, but the amounts of 18S rRNAs were similar (Fig. 1C).
Fig 1.
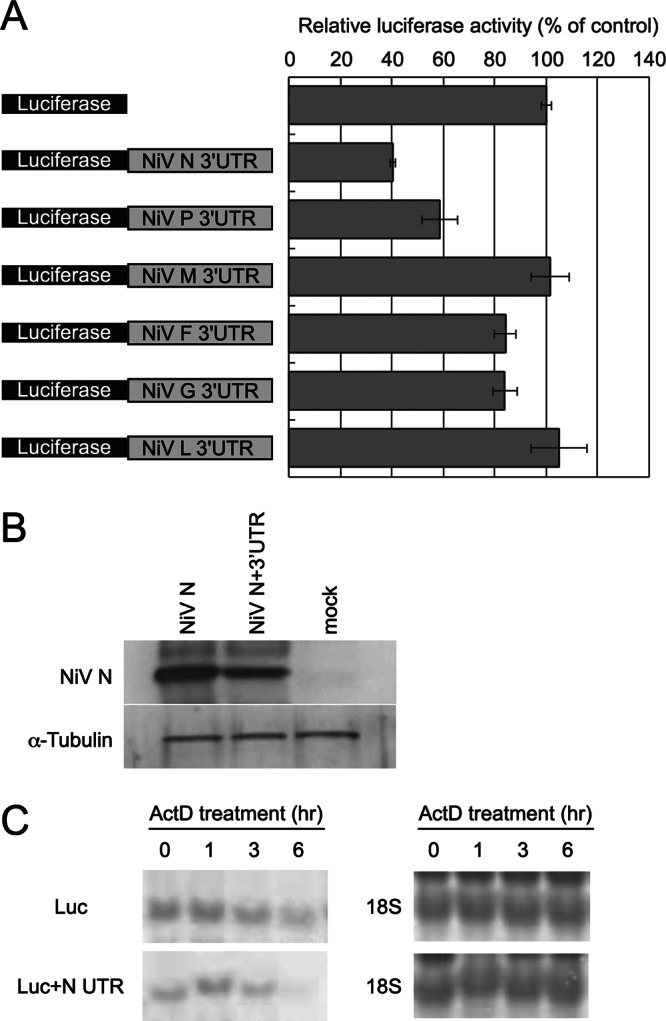
Effects of NiV 3′ UTRs on reporter gene expression. (A) UTR reporters were constructed by insertion of NiV 3′ UTRs downstream of the luciferase gene in pGL3control. Luciferase activities were measured at 24 h posttransfection and normalized to Renilla luciferase activities. The value of pGL3control was set to 100. The error bars indicate standard deviations. (B) Downregulation by the NiV N UTR resulted in a decrease in NiV N protein levels. Proteins were detected by Western blotting. (C) Reporter RNAs were detected by Northern blotting. For endogenous controls, 18S rRNA was stained with methylene blue.
Participation of cis-elements in NiV N 3′ UTR downregulation.
To identify cis-regulatory elements within the NiV N 3′ UTR, we constructed reporter plasmids containing different fragments of the NiV N 3′ UTR. These NiV N 3′ UTR fragments were associated with different levels of regulation (Fig. 2). The region encoded by nt 1 to 100 of the NiV N 3′ UTR showed the strongest repression activity. The ability of this 100-nt region to form tertiary structures was predicted using two RNA-folding prediction websites, centroidfold (11) and mfold (12), but no rigid or significant RNA structures were detected in the 100-nt region (data not shown).
Fig 2.

Deletion analysis of the NiV N 3′ UTR. (Left) cis-Acting elements in the NiV N 3′ UTR were identified using luciferase reporters attached to NiV N 3′ UTR fragments. (Right) Luciferase activities were measured at 24 h posttransfection and normalized to Renilla luciferase activities. The value of the pGL3control was set to 100. The 7th and 14th graph bars from the top are identical. Also, the 1st and 7th graph bars from the bottom are identical. The error bars indicate standard deviations.
Binding proteins participate in NiV N 3′ UTR downregulation.
We identified three specific proteins that bound to the 100-nt region within the NiV N 3′ UTR (Fig. 3A). These proteins were identified by liquid chromatography-mass spectrometry as hnRNP U (GeneID 3192), hnRNP D (GeneID 3184), and hnRNP A/B (GeneID 3812). myc-tagged hnRNP A/B and hnRNP D were detected mainly in the nucleus and to a lesser extent in the cytoplasm. No cytoplasmic myc-tagged hnRNP U protein was detected (data not shown).
Fig 3.

Identification of NiV N 3′ UTR binding proteins. (A) Binding proteins were identified using RNA pulldown assays with 3′-end biotin-labeled RNAs corresponding to the full-length NiV N 3′ UTR (586 nt), followed by silver staining. Candidate bands are indicated by arrowheads. (B) Both hnRNP A/B and D localized in the nucleus and cytoplasm. (C) Endogenous hnRNP A/B and D bound to the NiV N 3′ UTR. The bands indicated by arrowheads are hnRNP A/B or D.
We confirmed the cytoplasmic localization of endogenous hnRNP A/B and D using nuclear/cytoplasmic fractionation and Western blotting (Fig. 3B). hnRNP A/B was detected in both nuclear and cytoplasmic fractions, with the smaller isoform apparently more permeable than the larger isoform. The smaller isoform was more abundant, as determined by reverse transcription (RT)-PCR (data not shown). siRNA experiments indicated that the larger isoform might overlap a nonspecific band. hnRNP D was detected mainly in the nuclear fraction, and to a lesser extent in cytoplasmic samples. hnRNP D also showed isoform-dependent localization.
Endogenous hnRNP A/B and D proteins were detected in pulldown samples, confirming that they bound to the NiV N 3′ UTR (Fig. 3C). The intensities of specific bands were decreased when competitor RNAs corresponding to NiV N 3′ UTR nt 1 to 100 or 1 to 568 were added to the RNA-protein complexes. RNA pulldown assays in the presence of various competitors were used to identify the binding site (Fig. 4). RNA probes (nt 1 to 100, 1 to 80, and 1 to 60) functioned as competitors, though an nt 1-to-40 RNA probe showed weaker competitor activity than the longer competitors. The binding site for hnRNP A/B and hnRNP D thus appears to include nt 41 to 60 of the NiV N 3′ UTR but is also likely to require additional nucleotides.
Fig 4.
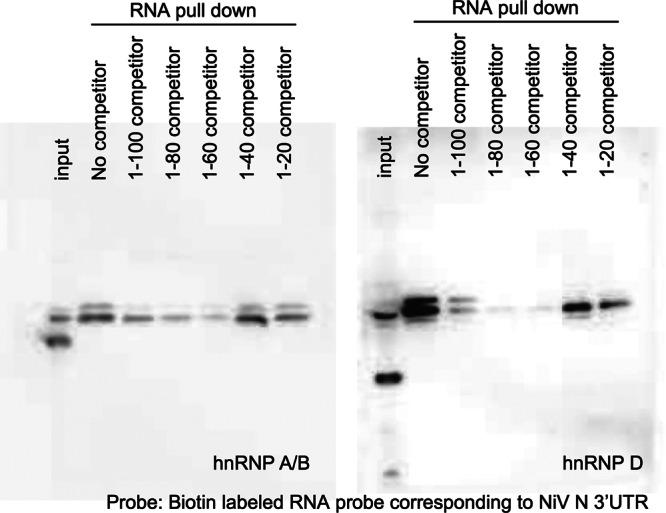
Identification of the hnRNP binding site in the NiV N 3′ UTR. The hnRNP binding site was identified using RNA pulldown assays in the presence of competitors of different lengths. RNAs corresponding to the full-length NiV N 3′ UTR (586 nt) were labeled at the 3′ end with biotin and used as RNA probes. hnRNP protein was detected by Western blotting.
Mutations in the hnRNP binding site restore expression of the NiV N 3′ UTR reporter.
As shown in Fig. 5A, nt 41 to 60 of the NiV N 3′ UTR is an AU-rich sequence. We replaced this binding region (nt 41 to 60) with a GC-rich sequence (Fig. 5A) and used RNA gel shift assays to determine if hnRNP binding to the RNA probe was impaired by mutation. hnRNP A/B isoform B and hnRNP D isoform C and isoform D were observed in the cytoplasmic fractions and were therefore used for these experiments (Fig. 3B). Proteins and 3′-end biotin-labeled RNA probes were mixed with or without competitors, and the RNA-protein complexes were loaded on polyacrylamide gels (Fig. 5B). RNA-protein complexes were observed in the absence of competitor, but a shifted band disappeared in the presence of the wild-type competitor. Competitor activity was impaired by mutation. Reporter activity was partially recovered by a NiV N 3′ UTR reporter vector with a mutation in the nt 41-to-60 sequence (Fig. 5C). RT-PCR was used to examine the effects of the mutations on RNA stability. Reporter mRNAs attached to the mutated 100-nt region of the NiV N 3′ UTR and were more stable than mRNAs that attached to the wild-type 100-nt NiV N 3′ UTR (Fig. 5D).
Fig 5.
Mutations in the hnRNP binding site impaired downregulation by the NiV N 3′ UTR. (A) Wild-type (wt) (above) and mutated (mut) (below) sequences around the hnRNP binding site. Underlining indicates the region in which nucleotides are changed. (B) Mutations in the hnRNP binding site impaired the binding of hnRNP proteins to the RNA probe. pIpC was used as a nonspecific RNA to standardize the amount of total RNAs in the reaction complex. EMSA, electrophoretic mobility shift assay. (C) The 100-nt NiV N 3′ UTR region with or without mutations at nt 41 to 60 was transfected into HeLa cells. Luciferase activities were measured at 24 h posttransfection and normalized to Renilla luciferase activities. The value of pGL3control was set to 100. (D) RT-PCR was used to detect mRNAs that attached to the 100-nt wild-type or mutated NiV N UTR. 18S rRNAs were detected as endogenous controls. The half-lives were calculated from the intensities of the bands.
Knockdown of hnRNP D increases expression of the NiV N 3′ UTR reporter.
The full-length (nt 1-to-568) UTR competitor was more effective than the nt 1-to-100 UTR competitor (Fig. 3C). Therefore, we could not exclude the possibility that hnRNP proteins also regulated gene expression, directly or indirectly, through other regions in the NiV N 3′ UTR. To investigate the relationship between hnRNP proteins and the full-length NiV N 3′ UTR, we examined the effects on the UTR reporter when hnRNP proteins were repressed. The efficiencies of the siRNAs were confirmed prior to use in the reporter assay. hnRNP A/B and D protein levels detected by Western blotting at 72 h posttransfection were repressed (Fig. 6A). Expression of the NiV N 3′ UTR reporter was markedly increased by knockdown of hnRNP D (Fig. 6B). However, knockdown of hnRNP A/B slightly increased expression of the UTR reporter. To determine if hnRNP D was solely involved in the regulation of NiV N, six NiV UTR reporters were transfected into hnRNP KD cells. The expression of reporters attached to the NiV N, P, or G UTR was also increased in response to hnRNP D knockdown (Fig. 6C), while the expression of reporters attached to the NiV F or L UTR was barely affected.
Fig 6.
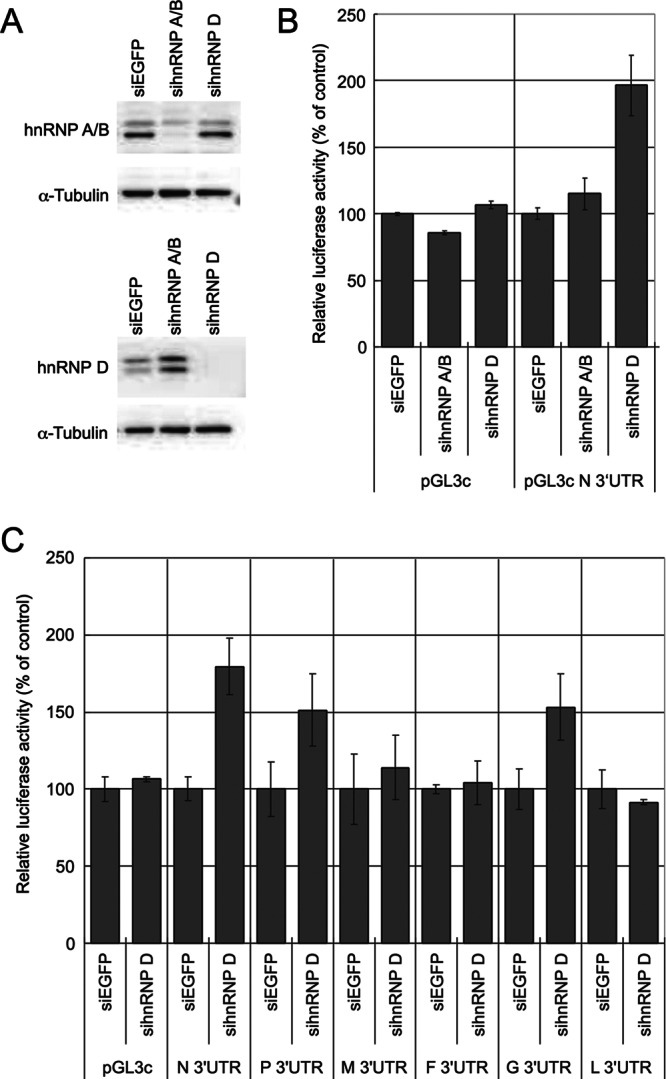
Expression of NiV N 3′ UTR reporters was increased by knockdown of hnRNP D. (A) siRNAs designed to knock down hnRNP A/B, hnRNP D, or EGFP (designated sihnRNP A/B, sihnRNP D, or siEGFP) were transfected into HeLa cells. The membranes were cut at the 50-kDa marker before addition of antibody. (B) Expression levels of NiV N 3′ UTR reporters when hnRNP A/B or D was repressed. Luciferase activities were measured and normalized to Renilla luciferase activities. The value of siEGFP-transfected cells was set to 100. The error bars indicate standard deviations. (C) Expression levels of six NiV UTR reporters when hnRNP D was repressed. The six NiV 3′ UTR reporters were transfected into hnRNP D KD cells. Luciferase activities were measured and normalized to Renilla luciferase activities. The value of siEGFP-transfected cells was set to 100.
DISCUSSION
The results of this study demonstrated that the NiV N 3′ UTR was downregulated through its interaction with hnRNP D. hnRNP proteins are localized within the cell, indicating that this interaction does not occur until transcription is initiated and is therefore not a part of the viral infection cycle.
The transcription start site in paramyxoviruses is located at the 3′ end of the genome, and transcription proceeds in a 3′-to-5′ direction, with transcriptional attenuation. The gene located furthest upstream is therefore thought to be transcribed most effectively. The N gene is the most upstream gene in the NiV genome, suggesting that N mRNA corresponding to the NiV N UTR is likely to be most abundant in infected cells. We hypothesized that gene expression regulation through the NiV N 3′ UTR would represent the most important mechanism for posttranscriptional regulation of the virus.
The genomes of other paramyxoviruses, particularly morbilliviruses, have exceptionally long UTRs (M 3′ UTR and F 5′ UTR). Little information is available regarding viral gene regulation of the 3′ UTR, though Takeda et al. reported that the measles virus M 3′ UTR was required for effective translation whereas the measles virus F 5′ UTR inhibited protein production (13). Filoviruses have long noncoding regions similar to those seen in henipaviruses. Neumann et al. found that the Ebola virus 3′ nontranslated region (NTR) (NTR is used in the original reference; it appears to be almost the same as UTR) attenuated protein expression from upstream genes, while the 5′ NTR affected efficient protein expression of downstream genes (14). Our results, together with these observations, strongly suggest that viral UTRs play a key role in regulating viral gene expression through posttranscriptional mechanisms.
As previously reported, NiV infects and replicates effectively in HeLa cells (10). We assumed that viruses operate a posttranscriptional regulation system to control expression of the required proteins. The ratio of N, P, and L proteins is an important factor for effective replication (15, 16). Because resources in a single cell are limited, posttranscriptional regulation would avoid the synthesis of unwanted proteins, optimize the ratios, and enable effective and resource-efficient replication.
In our experiments, the NiV N 3′ UTR was downregulated by hnRNP D through a viral AU-rich region, in accordance with previous results (17–19). AU-rich elements are often found in the 3′ UTRs of inflammatory cytokines, oncogenes, transcription factors, and growth factors. These mRNAs are recognized by AU-rich element-binding proteins, such as AUF1 (hnRNP D) and tristetraprolin, resulting in rapid degradation. Although HuR is also an AU-rich element-binding protein, it stabilizes mRNAs containing AU-rich elements (20). Additionally, AU-rich elements are recognized by small RNAs that regulate gene expression posttranscriptionally (21). We did not detect any other AU-rich element-binding factors in our experiments, but further studies are needed to investigate the possibility that they might regulate NiV N gene expression through the NiV N 3′ UTR under other conditions.
The AU-rich elements consist of an AUUUA motif within U-rich sequences. They are classified as either class I, II, or III, based on the number of AUUUA motifs (22). Classes I and II contain multiple copies of AUUUA, while class III motifs do not contain typical AUUUA sequences. We were unable to detect any AUUUA motifs around the NiV N 3′ UTR nt 41-to-60 sequence and therefore concluded that the NiV N 3′ UTR is a class III AU-rich element.
hnRNP A/B was localized in the cytoplasm. Although the function of hnRNP A/B in the cytoplasm is unknown, our results suggest that it might act as a novel posttranscriptional regulator. Both hnRNP A/B and hnRNP D bound strongly to the NiV N 3′ UTR. Moreover, we observed that knockdown of hnRNP A/B slightly increased the amount of hnRNP D (Fig. 6A). We hypothesized that hnRNP A/B bound to the hnRNP D binding site and possibly interacted with hnRNP D to control its function in the cytoplasm, although we cannot exclude the possibility that knockdown of hnRNP A/B affected the expression of hnRNP D.
We excluded hnRNP U from the candidate NiV N UTR binding proteins because it was not detected in the cytoplasm of uninfected HeLa cells. However, hnRNP U was reported to relocalize from the nucleus to the cytoplasm following vesicular stomatitis virus (VSV) infection (23). Similar relocalizations of hnRNP A1, C1/C2, and K were also reported (24). In addition to VSV, other RNA viruses, such as enterovirus (25, 26), rhinovirus (27), Japanese encephalitis virus (28), and Junin virus (29), have also been shown to relocalize hnRNP proteins. To the best of our knowledge, relocalization of hnRNP proteins by NiV infection has not been reported previously. However, if host RNA-binding proteins do relocalize to the cytoplasm upon NiV infection, these proteins, such as hnRNP U, might interact with NiV N mRNAs.
Many host RNA-binding proteins, such as hnRNP proteins, are known to interact with viral noncoding RNA regions. They play diverse roles in the gene expression and replication of RNA viruses (30). Our results contribute to an understanding of the interaction between host RNA-binding proteins and viral noncoding RNA regions.
To reveal the function of a noncoding RNA region in the NiV genome, we identified hnRNP A/B and D as proteins that bind to the NiV N 3′ UTR. Using mutagenesis and siRNAs, we revealed that hnRNP D downregulated NiV N expression by binding to the viral 3′ UTR. This is the first report to reveal the function of the NiV noncoding RNA region. Although this study focused on the NiV N 3′ UTR, our data and other reports indicate that various UTRs may play roles in posttranscriptional regulation.
ACKNOWLEDGMENTS
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (JSPS) and by the Global COE Program Center of Education and Research for Advanced Genome-Based Medicine: for Personalized Medicine and the Control of Worldwide Infectious Diseases (MEXT, Japan).
Liquid chromatography-mass spectrometry analysis was conducted by the Laboratory Center for Proteomics Research (Institute of Medical Science, University of Tokyo).
Footnotes
Published ahead of print 20 March 2013
REFERENCES
- 1. Eaton BT, Broder CC, Middleton D, Wang LF. 2006. Hendra and Nipah viruses: different and dangerous. Nat. Rev. Microbiol. 4:23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anonymous 2004. Nipah encephalitis outbreak over wide area of western Bangladesh, 2004. Health Sci. Bull. 2:7–11 [Google Scholar]
- 3. Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435 [DOI] [PubMed] [Google Scholar]
- 4. Harcourt BH, Lowe L, Tamin A, Liu X, Bankamp B, Bowden N, Rollin PE, Comer JA, Ksiazek TG, Hossain MJ, Gurley ES, Breiman RF, Bellini WJ, Rota PA. 2005. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis. 11:1594–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harcourt BH, Tamin A, Ksiazek TG, Rollin PE, Anderson LJ, Bellini WJ, Rota PA. 2000. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 271:334–349 [DOI] [PubMed] [Google Scholar]
- 6. Wang LF, Yu M, Hansson E, Pritchard LI, Shiell B, Michalski WP, Eaton BT. 2000. The exceptionally large genome of Hendra virus: support for creation of a new genus within the family Paramyxoviridae. J. Virol. 74:9972–9979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harcourt BH, Tamin A, Halpin K, Ksiazek TG, Rollin PE, Bellini WJ, Rota PA. 2001. Molecular characterization of the polymerase gene and genomic termini of Nipah virus. Virology 287:192–201 [DOI] [PubMed] [Google Scholar]
- 8. Celis JE, Carter N, Simons K, Small JV, Hunter T, Shotton D. 2005. Cell biology, four-volume set: a laboratory handbook. Elsevier Science, Burlington, MA [Google Scholar]
- 9. Huang M, Sato H, Hagiwara K, Watanabe A, Sugai A, Ikeda F, Kozuka-Hata H, Oyama M, Yoneda M, Kai C. 2011. Determination of a phosphorylation site in Nipah virus nucleoprotein and its involvement in virus transcription. J. Gen. Virol. 92:2133–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoneda M, Guillaume V, Ikeda F, Sakuma Y, Sato H, Wild TF, Kai C. 2006. Establishment of a Nipah virus rescue system. Proc. Natl. Acad. Sci. U. S. A. 103:16508–16513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hamada M, Sato K, Kiryu H, Mituyama T, Asai K. 2009. Predictions of RNA secondary structure by combining homologous sequence information. Bioinformatics 25:i330–i338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeda M, Ohno S, Seki F, Nakatsu Y, Tahara M, Yanagi Y. 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J. Virol. 79:14346–14354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neumann G, Watanabe S, Kawaoka Y. 2009. Characterization of Ebolavirus regulatory genomic regions. Virus Res. 144:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freiberg A, Dolores LK, Enterlein S, Flick R. 2008. Establishment and characterization of plasmid-driven minigenome rescue systems for Nipah virus: RNA polymerase I- and T7-catalyzed generation of functional paramyxoviral RNA. Virology 370:33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Halpin K, Bankamp B, Harcourt BH, Bellini WJ, Rota PA. 2004. Nipah virus conforms to the rule of six in a minigenome replication assay. J. Gen. Virol. 85:701–707 [DOI] [PubMed] [Google Scholar]
- 17. Barreau C, Paillard L, Osborne HB. 2005. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33:7138–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gingerich TJ, Feige JJ, LaMarre J. 2004. AU-rich elements and the control of gene expression through regulated mRNA stability. Anim. Health Res. Rev. 5:49–63 [DOI] [PubMed] [Google Scholar]
- 19. Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. 2003. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J. Cell Physiol. 195:356–372 [DOI] [PubMed] [Google Scholar]
- 20. Brennan CM, Steitz JA. 2001. HuR and mRNA stability. Cell Mol. Life Sci. 58:266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. 2005. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120:623–634 [DOI] [PubMed] [Google Scholar]
- 22. Chen CY, Shyu AB. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465–470 [DOI] [PubMed] [Google Scholar]
- 23. Gupta AK, Drazba JA, Banerjee AK. 1998. Specific interaction of heterogeneous nuclear ribonucleoprotein particle U with the leader RNA sequence of vesicular stomatitis virus. J. Virol. 72:8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pettit Kneller EL, Connor JH, Lyles DS. 2009. hnRNPs relocalize to the cytoplasm following infection with vesicular stomatitis virus. J. Virol. 83:770–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin JY, Li ML, Huang PN, Chien KY, Horng JT, Shih SR. 2008. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5′ untranslated region and participates in virus replication. J. Gen. Virol. 89:2540–2549 [DOI] [PubMed] [Google Scholar]
- 26. Lin JY, Shih SR, Pan M, Li C, Lue CF, Stollar V, Li ML. 2009. hnRNP A1 interacts with the 5′ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J. Virol. 83:6106–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cammas A, Pileur F, Bonnal S, Lewis SM, Leveque N, Holcik M, Vagner S. 2007. Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein A1 controls translation initiation of specific mRNAs. Mol. Biol. Cell 18:5048–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katoh H, Mori Y, Kambara H, Abe T, Fukuhara T, Morita E, Moriishi K, Kamitani W, Matsuura Y. 2011. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J. Virol. 85:10976–10988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maeto CA, Knott ME, Linero FN, Ellenberg PC, Scolaro LA, Castilla V. 2011. Differential effect of acute and persistent Junin virus infections on the nucleo-cytoplasmic trafficking and expression of heterogeneous nuclear ribonucleoproteins type A and B. J. Gen. Virol. 92:2181–2190 [DOI] [PubMed] [Google Scholar]
- 30. Li Z, Nagy PD. 2011. Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol. 8:305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]