

Abstract
The innate host defense against influenza virus is largely dependent on the type I interferon (IFN) system. However, surprisingly little is known about the cellular source of IFN in the infected lung. To clarify this question, we employed a reporter mouse that contains the firefly luciferase gene in place of the IFN-β-coding region. IFN-β-producing cells were identified either by simultaneous immunostaining of lungs for luciferase and cellular markers or by generating conditional reporter mice that express luciferase exclusively in defined cell types. Two different strains of influenza A virus were employed that either do or do not code for nonstructural protein 1 (NS1), which strongly suppresses innate immune responses of infected cells. We found that epithelial cells and lung macrophages, which represent the prime host cells for influenza viruses, showed vigorous IFN-β responses which, however, were severely reduced and delayed if the infecting virus was able to produce NS1. Interestingly, CD11c+ cell populations that were either expressing or lacking macrophage markers produced the bulk of IFN-β at 48 h after infection with wild-type influenza A virus. Our results demonstrate that the virus-encoded IFN-antagonistic factor NS1 disarms specifically epithelial cells and lung macrophages, which otherwise would serve as main mediators of the early response against infection by influenza virus.
INTRODUCTION
Type I interferons (IFN) represent essential components of the antiviral host defense which link innate immunity and adaptive immunity (1). The family of type I IFN in mammals typically includes more than 10 different IFN-α subtypes, a single IFN-β, and minor subtypes such as IFN-ω or IFN-ε. These various IFN genes are usually induced simultaneously in response to virus infection, although the expression profiles of different IFN genes might differ considerably depending on the producer cell type and nature of the stimulus (2–4). IFN-β is considered to be a master regulator of type I IFN synthesis, as it appears early after viral infection and thereby primes expression of IFN genes (5). For signaling, all type I IFN family members bind to the same heterodimeric IFN-α/β receptor complex, which is present on most if not all nucleated cells. Binding of IFN to the receptor complex will result in transcriptional activation of a large number of IFN-stimulated genes, which eventually confer antiviral activity by several different mechanisms (6).
Viral pathogens induce the synthesis of type I IFN and other cytokines by triggering pathogen recognition receptors (PRR) such as Toll-like receptors (TLR) and retinoic acid-inducible gene I (RIG-I)-like helicases. Interestingly, most viruses trigger type I IFN responses both in productively infected host cells and in immune cells which patrol infected organs. Early IFN synthesis is responsible for efficient host protection. However, successful viruses frequently encode factors that suppress production of IFN (1, 7). At present, our knowledge about the contributions of various cell types to the production of IFN during infection is incomplete. Detection of IFN-producing cells by standard immunological methods is complicated by the fact that type I IFN is rapidly secreted from virus-triggered cells and thus usually does not accumulate to high intracellular levels. Transgenic mice in which reporter genes are inserted into the coding regions of the IFN-α or -β genes are powerful tools to study virus-induced expression of IFN in vivo. Recently, a reporter mouse which expresses green fluorescent protein in place of IFN-α6 was used to demonstrate that alveolar macrophages contribute to IFN synthesis in the lungs of Newcastle disease virus (NDV)-infected animals (8). To visualize IFN synthesis in various cell types of the brain during infection with La Crosse bunyavirus (3), we had employed a different reporter mouse in which the IFN-β-coding region was replaced by firefly luciferase (9). These studies clearly showed that virus-encoded IFN-antagonistic factors may display their activities in particular cell types of infected organs but, at the same time, may fail to inhibit IFN synthesis by immune cells (3, 8).
Influenza virus defense heavily relies on the innate immune system and uses virus-triggered IFN as an effector. Accordingly, recombinant mice unable to respond to type I IFN are highly susceptible to influenza A viruses (10). Resistance of mice against influenza virus further strongly depends on IFN-regulated genes which encode antiviral factors such as Mx (11) and IFITM3 (12, 13). This is in line with the view that IFN plays a decisive role in defense against influenza virus. In spite of intensive investigations, surprisingly little is known about the cellular origin of type I IFN in influenza virus-infected lungs. Previous studies demonstrated that various cell types, including macrophages, pneumocytes, and dendritic cells, are able to produce IFN after influenza virus infection (14–18). However, the relative contributions of these cells to IFN production remained unclear.
In this study, we used our IFN-β reporter mice to analyze the composition of the IFN-β source in lungs of animals infected with either wild-type influenza A virus or a virus mutant that cannot produce the IFN-antagonistic factor NS1. We show that virus-infected epithelial cells and macrophages produced the bulk of IFN-β after infection with the highly attenuated NS1-deficient mutant virus, whereas IFN-β synthesized in response to infection with wild-type influenza A virus originated mostly from CD11c+ cells expressing or lacking macrophage markers.
MATERIALS AND METHODS
Mice.
Mice were bred in the animal facility of the Department of Virology at the University of Freiburg. All mice used in this study were on the C57BL/6 background. Global (IFN-β+/Δβ-luc) and conditional reporter (IFN-β+/floxβ-luc) mice have been described previously (4). IFN-βfloxβ-luc/floxβ-luc mice were crossed to LysM-Cre (19), Nkx2-1-Cre (Jackson Lab, stock number 008661) (20), or CD11c-Cre mice (21) to generate mice that express the reporter gene in macrophages/monocytes and granulocytes, lung epithelial cells, or CD11c+ cells, respectively. IFN-βfloxβ-luc/floxβ-luc-LysM-Cre+/+ mice were crossed to CD11c-Cre mice to generate double-transgenic conditional reporter mice expressing the reporter gene simultaneously in macrophages/monocytes and CD11c+ cells.
Viruses and infection of mice.
Wild-type SC35M and mutant SC35M-ΔNS1, which cannot express the IFN-antagonistic factor NS1, were previously described (10). Virus stocks were generated in Vero cells, MDCK cells, or MDCK cells expressing NS1 (22). Age-matched animals (6 to 11 weeks old) were infected intranasally with 5 × 104 PFU of wild-type or mutant SC35M in 40 μl of phosphate-buffered saline (PBS). Lungs either were collected without fixation for measuring luciferase activity or were perfusion fixed for immunohistochemical analyses as described below.
Ex vivo luciferase measurement.
Lungs were homogenized in 800 μl of PBS using FastPrep-24 equipment and lysing matrix A (MP Biomedicals). Samples (200 μl) were treated with 50 μl of 5× cell culture lysis buffer (Promega), and luciferase activity was measured in a Sirius tube luminometer (Berthold Technologies) using the single luciferase assay system (Promega) according to the manufacturer's protocol.
Immunohistochemistry.
Animals were sacrificed with a mixture of ketamine (3.7%), xylazine (0.2%), and acepromacine (0.02%) before transcardial perfusion with 0.9% NaCl followed by 4% buffered paraformaldehyde in PBS. Lungs were postfixed in the latter solution for 6 more hours. Fixed lungs were dehydrated with 15% sucrose in PBS followed by 30% sucrose in PBS for 16 h each. Dehydrated lungs were embedded in optimal cutting tissue cover (OCT), frozen on a liquid-nitrogen-cooled aluminum block, and stored at −80°C. Frozen lungs were sectioned into 5- to 10-μm-thick sections using a Leica cryostat. Tissue sections were dried for 30 min before rehydration with PBS. After endogenous peroxidase was blocked with H2O2 or Peroxo-Block (Invitrogen), samples were processed with PBS containing 5% normal donkey serum and 0.1% Triton X-100 for 20 min. Sections were then incubated with rabbit antiluciferase antibody (Fitzgerald, 70C-CR2020RAP) in PBS containing 3% normal donkey serum at 4°C overnight. For detection of luciferase, signal amplification with the TSA fluorescein system (Perkin-Elmer) was performed according to the manufacturer's instructions using a biotin-conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch). Anti-influenza virus (AbD Serotec OBT1551), anti-CD11c (Abcam, ab33483), anti-EpCAM (BD Pharmingen, 552370) and DyLight488-, DyLight549-, DyLight649-, Cy3-, or Cy2-conjugated secondary antibodies (Jackson ImmunoResearch) were used as recommended by the manufacturers. Macrophages were labeled with fluorescein isothiocyanate (FITC)-conjugated BS-1 lectin (Sigma-Aldrich, L9381). Slides were mounted in DAPI (4′,6′-diamidino-2-phenylindole)-containing IS mounting medium (Dianova). Digital images were taken with an ApoTome fluorescence microscope (Zeiss) using AxioVision software.
RESULTS
Epithelial cells and macrophages are the main producers of IFN-β in lungs of mice infected with influenza A virus mutant SC35M-ΔNS1.
We previously reported that a highly attenuated variant of the influenza A virus strain SC35M lacking the IFN-antagonist factor NS1 (SC35M-ΔNS1) induces a rapid IFN-β response in lungs of mice that peaks at about 24 h postinfection (9). To visualize IFN-β-producing cells in virus-infected lungs during such responses, we inoculated luciferase reporter mice with 5 × 104 PFU of SC35M-ΔNS1. Lungs were harvested at 24 h postinfection, and lung sections were stained with antibodies against luciferase and viral antigen. Numerous cells which expressed the luciferase reporter gene were located either in or on top of the epithelium that lines the bronchus and broncheolus (Fig. 1A, left and middle panels). Rarely, luciferase-positive cells were also present in the alveoli (Fig. 1A, right panel). Luciferase-positive cells were usually also positive for viral antigen (Fig. 1A).
Fig 1.
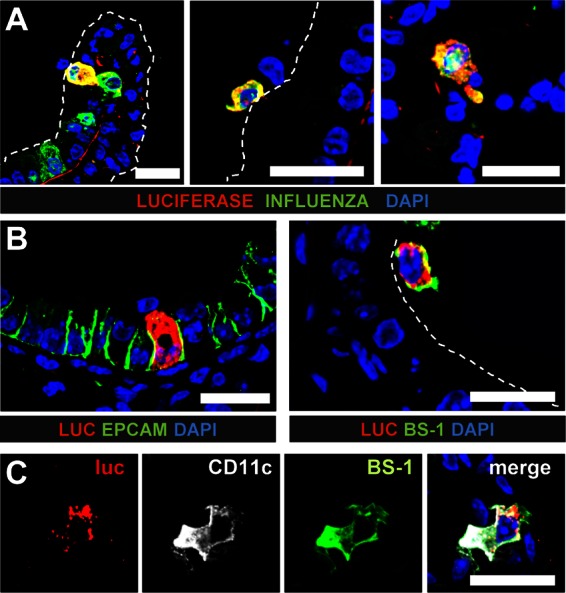
Epithelial cells and macrophages are main producers of IFN-β in lungs of mice infected with SC35M-ΔNS1. Infected global reporter mice were sacrificed at 24 h postinfection, and lung slices were simultaneously stained for luciferase and either viral antigen (A) or cellular markers (B and C). (A) Luciferase-producing cells typically expressed viral antigen and were found in (left panel) or on top of (middle panel) the epithelium of bronchi and bronchioles. A few luciferase-positive cells were also found in the alveoli (right panel). (B) Luciferase-positive cells were either epithelial cells expressing EpCAM (left panel) or lectin BS-1-positive lung macrophages (right panel). (C) Luciferase-positive cells which expressed CD11c usually also reacted with BS-1, a lectin that can specifically bind to lung macrophages. Dashed lines indicate the apical surface of the lung epithelium. Staining of nuclei (blue) was achieved with DAPI. Size bars, 20 μm.
To identify the cells producing IFN-β, we stained lung sections simultaneously for luciferase and cell type-specific markers. The most prominent IFN-β producers under these conditions were epithelial cells which line the airways and express the epithelial cell adhesion molecule (EpCAM) (Fig. 1B, left panel) (23). In addition, lung macrophages which could be stained by the lectin BS-1 expressed IFN-β (Fig. 1B, right panel) (24, 25).
To quantify IFN-β production by epithelial cells and macrophages, we used the Cre-LoxP system to establish reporter mice which express the transgene only in defined cell populations and measured luciferase activity in lung lysates (4). For the current study, we used Nkx-Cre mice to generate animals in which IFN-β promoter-controlled luciferase expression is restricted to lung epithelial cells (20). Further, we used LysM-Cre mice to generate reporter mice which express the luciferase gene in macrophages, granulocytes, and, at a low frequency, monocytes (19). In addition, we used CD11c-Cre mice, which can express the luciferase gene in dendritic cells and certain macrophages (4), as well as double-transgenic LysM/CD11c-Cre mice, in which both LysM+ and CD11c+ cells can express the IFN-β-regulated luciferase gene. “Global” reporter mice, which are able to express the luciferase gene in all cell types (9), were included as a reference. Age-matched groups of the different reporter mice were infected with SC35M-ΔNS1, lungs were extracted at various time points, and luciferase activity in lung lysates was measured (Fig. 2A). In line with our findings from the histological analysis discussed above, we detected a strong IFN-β response in Nkx-Cre and LysM-Cre reporter mice, whereas luciferase activity measured in CD11c-Cre reporter mice was comparably low (Fig. 2A). In accordance, luciferase activity in lungs of infected LysM/CD11c-Cre reporter mice was not significantly higher than that in LysM-Cre reporter mice (Fig. 2A). The LysM gene in mice is highly active in alveolar macrophages but shows only low activity in dendritic cells (26). On the other hand, macrophages and dendritic cells both express high levels of CD11c (26). Our data thus suggest that alveolar as well as CD11c+ macrophages contribute to the IFN-β response under these conditions. To confirm this interpretation of the data, we performed additional staining experiments in which the lectin BS-1 was used in combination with antibodies for CD11c. We observed that most luciferase-positive cells which expressed CD11c could also be stained with BS-1 (Fig. 1C), indicating that they indeed represent lung macrophages.
Fig 2.

Conditional reporter mice confirm that epithelial cells and macrophages are the dominant IFN-β producers in mouse lungs during infection with SC35M-ΔNS1. Conditional reporter mice expressing Cre recombinase in epithelial cells (Nkx), macrophages/monocytes (LysM), CD11c+ cells (CD11c), or macrophages/monocytes and CD11c+ cells (LysM/CD11c) were infected with SC35M-ΔNS1. (A) Lungs were removed and tested for luciferase activity at the indicated time points. Values represent means ± standard errors of the means (SEM) for groups of animals (n ≥ 6) from each time point. (B) Luciferase values from infected conditional reporter mice expressing the luciferase gene exclusively in the indicated cell types were compared to luciferase values from infected “global” reporter mice. Values from “global” reporter mice were set to 100% and depicted as full-sized pies at each time point. The sizes of the pies are proportional to total luciferase activities at a particular time point postinfection.
Careful quantitative analysis of the data revealed that the combined luciferase signals of Nkx-Cre, LysM-Cre, and CD11c-Cre reporter mice at any given time after infection with SC35M-ΔNS1 reached about 55% to 70% of the values measured in lungs of infected “global” reporter mice, in which all cell types can produce luciferase (Fig. 2B). The difference (white areas in Fig. 2B) might be due to either IFN-β production by an additional, uncharacterized cell type or incomplete rearrangement of the modified IFN-β locus in the conditional reporter mice.
CD11c+ cells located in infectious foci are the main producers of IFN-β after challenge with wild-type influenza virus.
To visualize IFN-β production during infection with wild-type influenza A virus, we stained lung sections of global reporter mice infected with wild-type SC35M, a virus strain that can efficiently suppress early type I IFN synthesis (9). Lungs were removed at various time points postinfection and stained with antibodies that specifically recognize luciferase or viral antigen. Numerous virus-infected cells were present in lungs analyzed at 24 h postinfection, but no luciferase-positive cells were detected (Fig. 3A). In lungs analyzed at 48 h postinfection, single scattered luciferase-expressing cells were present in heavily infected (Fig. 3B) but not in uninfected (Fig. 3C) lung areas. The luciferase-positive cells appeared to contain either no or only small amounts of viral antigen. It should be noted that our staining approach cannot distinguish between actively infected cells and cells that had simply engulfed viral antigen. Nevertheless, overall, these results are in good agreement with earlier findings which showed that during lethal SC35M infection, IFN-β production is low during the early stages of infection but reaches high levels at later stages of infection (9). It also nicely agrees with findings by others (27) that wild-type influenza viruses remain undetected by the immune system during the first 2 days after infection of the lung.
Fig 3.
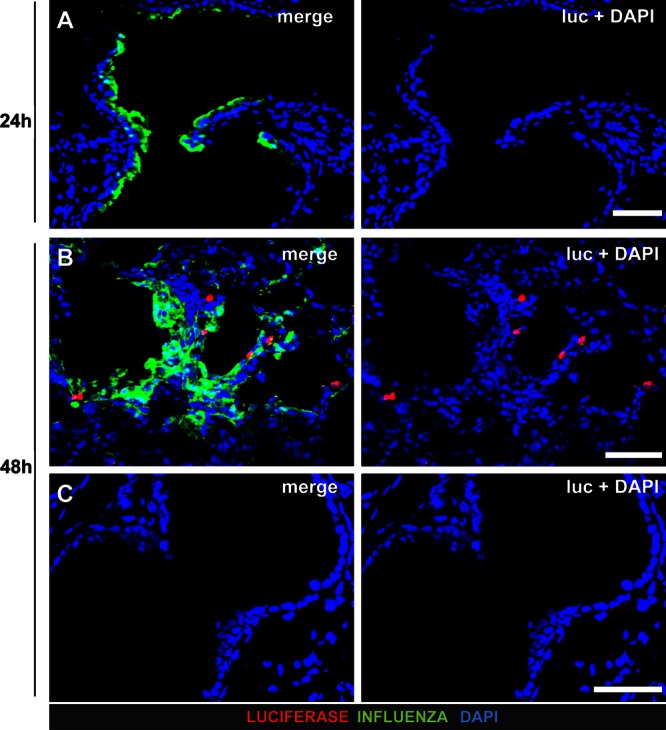
IFN-β production in lungs of mice infected with wild-type SC35M. Lungs of infected “global” reporter mice were stained for luciferase and virus antigen. Counterstaining (blue) was done with DAPI. (A) No luciferase-positive cells were detectable at 24 h postinfection. (B and C) At 48 h postinfection, single dispersed luciferase-positive cells were detected in heavily infected (B) but not in virus antigen-free (C) lung areas. Note that luciferase-positive cells did not contain detectable levels of viral antigen. Size bars, 50 μm.
When lung sections of SC35M-infected reporter mice were stained with antibodies for luciferase and different cell markers, we observed that the majority of luciferase-positive cells in infected lungs at 48 h postinfection expressed CD11c (Fig. 4). Interestingly, a fraction of the CD11c+ cells was also positive for the lung macrophage marker BS-1 (Fig. 4A and B).
Fig 4.
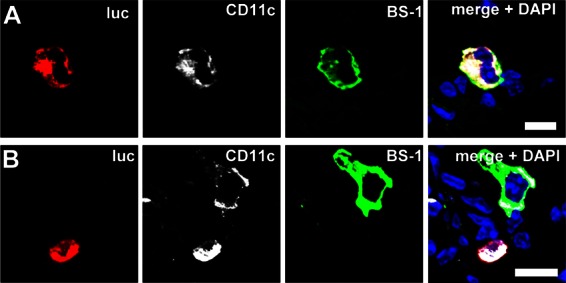
A fraction of CD11c+ cells producing IFN-β in lungs of mice infected with wild-type SC35M are macrophages. Lungs of virus-infected “global” reporter mice were harvested at 48 h postinfection and stained simultaneously with antibodies against luciferase (luc) and CD11c and with lectin BS-1. Luciferase-positive cells were positive for both CD11c and the lung macrophage marker BS-1 (A) or for CD11c but not BS-1 (B). Counterstaining (blue) was done with DAPI. Size bars, 10 μm.
To quantify IFN-β production by different cell types during lethal infection with SC35M, we determined luciferase activity in lungs by using the same set of conditional reporter mice as described above for the SC35M-ΔNS1 mutant virus. In agreement with the histological findings, we found that wild-type SC35M virus induced the IFN-β promoter-driven luciferase gene in infected lungs with delayed kinetics. In contrast to the situation in mice infected with SC35M-ΔNS1, substantial reporter gene expression was not observed before 48 h postinfection (Fig. 5). At all time points analyzed, expression of the luciferase reporter gene was rather weak in epithelial cells (Nkx-Cre mice), although it increased continuously over time (Fig. 5A). Cells expressing LysM produced similar amounts of luciferase at 48, 72, and 96 h postinfection, whereas luciferase activity in CD11c-Cre reporter mice clearly peaked at 48 h postinfection and returned to substantially reduced levels at 72 and 96 h postinfection (Fig. 5A). In contrast to the situation in lungs of mice infected with SC35M-ΔNS1, after infection with wild-type SC35M, luciferase gene expression in double-transgenic LysM/CD11c-Cre mice was only slightly different from that in CD11c-Cre single-transgenic mice (Fig. 5A). These observations suggest that transiently appearing CD11c+ dendritic cells lacking macrophage markers are important IFN-β producers at intermediate stages of lethal infections with wild-type influenza A virus.
Fig 5.

Lung CD11c+ cells are the main producers of IFN-β at late stages of infection with wild-type SC35M. Conditional reporter mice selectively expressing luciferase in epithelial cells (Nkx), macrophages/monocytes (LysM), CD11c+ cells (CD11c), or macrophages/monocytes and CD11c+ cells (LysM/CD11c) were infected with wild-type SC35M, and luciferase activity in lungs was determined at the indicated time points. Values represent means ± SEM for groups of animals (n ≥ 6) from each time point. (B) Luciferase values from infected conditional mice expressing the reporter gene in the indicated cell types were compared to luciferase values from infected global reporter mice. Values from global reporter mice were set to 100% and depicted as full-sized pies at each time point. The sizes of the pies are proportional to total luciferase activity at a particular time point postinfection.
Again, as described above for infections with SC35M-ΔNS1, the combined signals of Nkx-Cre, LysM-Cre, and CD11c-Cre reporter mice at any given time of the analysis reached only about 70% of the corresponding signals of “global” reporter mice at 48 h postinfection, and these values dropped to 30 to 40% at 72 and 96 h postinfection (Fig. 5B). The difference observed at 48 h postinfection may mostly be due to incomplete rearrangement of the modified IFN-β locus. The larger differences observed at the later time points are probably due to IFN-β production by additional, unidentified immune cells that infiltrate the lungs of moribund animals.
DISCUSSION
This study showed that various cell types contribute to type I IFN synthesis in the lungs of influenza virus-infected mice. If an attenuated mutant virus unable to produce the IFN-antagonistic factor NS1 was used, infected epithelial cells and CD11c− lung macrophages responded vigorously and synthesized large amounts of IFN-β at very early stages of the infection, whereas CD11c+ cells played a minor role. In contrast, in lungs of mice infected with virulent wild-type virus, IFN-β was produced mainly by CD11c+ cells expressing or lacking macrophage markers, whereas the contribution of the huge number of infected epithelial cells was comparatively moderate. Thus, the IFN-antagonistic factor NS1 of virulent influenza viruses efficiently disarms epithelial cells and CD11c− lung macrophages, which appear to constitute the first line of antiviral defense against these pathogens.
Considering the fact that comparatively high levels of luciferase activity were measured in lungs of mice infected with the NS1-deficent influenza A virus for 24 h, we were surprised to find that luciferase synthesis was restricted to relatively few infected epithelial cells and virus-positive macrophages. This finding supports earlier observations that vigorous IFN synthesis by a small number of cells in an infected organ may confer an astonishingly high degree of antiviral protection (28, 29). Our results clearly demonstrate that virus-infected epithelial cells and macrophages are able to mediate antiviral protection via type I IFN and presumably other cytokines, unless this process is actively blocked by virus-encoded IFN-antagonistic factors.
As expected, in the situation where the virus-encoded NS1 protein suppressed IFN induction very efficiently in infected epithelial cells and pulmonary macrophages, these cells remained luciferase negative during the initial period of infection with wild-type virus. Interestingly, at 48 h postinfection, few luciferase-positive cells had appeared in lung areas that contained many infected cells and showed severe tissue damage. Most of these luciferase-positive cells expressed CD11c, and a substantial fraction of these cells further bound the lectin BS-1. This suggests that IFN detected at 48 h postinfection is produced mainly by dendritic cells and CD11c+ macrophages. Thus, the source of IFN in lungs of mice infected with wild-type influenza A virus is fundamentally different from that in lungs of mice infected with NS1-deficient virus mutants.
Another remarkable finding of our study is that the source of IFN changed during the course of infection. The IFN-producing CD11c+ cells found in wild-type influenza virus-infected lungs at 48 h postinfection fell into two distinct classes, namely, cells able and cells unable to bind BS-1. IFN-producing cells which exclusively expressed CD11c were only transiently present and had almost completely disappeared from infected lungs at 72 h postinfection (Fig. 5). It is likely that the disappearing CD11c+ cells are dendritic cells which migrate to draining lymph nodes soon after contact with viral antigen, as recently demonstrated by others (30, 31). Those authors further demonstrated that the migratory dendritic cells were not productively infected with influenza virus, which explains why IFN production by these cells was not inhibited by virus-encoded NS1. Our study showed that IFN-producing CD11c+ cells which express LysM and bind to BS-1 remained in the infected tissue, suggesting that these cells are bona fide lung macrophages. However, it should be noted that plasmacytoid dendritic cells (pDCs) are also addressed by both LysM-Cre and CD11c-Cre, although at lower frequency than lung macrophages (32). A recent study suggested that pDCs which appeared to sense virus using TLR7 contribute to IFN production at late stages of infections with influenza virus (17). Thus, it seems likely that at least a fraction of the luciferase activity observed in our LysM/CD11c-Cre reporter mice originated from pDCs.
Conditional reporter mice expressing Cre recombinase exclusively in specific cell populations such as lung epithelial cells, macrophages/monocytes, or CD11c+ cells were used here. We noted that the sum of the luciferase signals measured in infected Nkx-Cre, LysM-Cre, and CD11c-Cre mice was lower than the value measured in global reporter mice in which the modified IFN-β locus was rearranged in all cell types. There are at least two different explanations for these findings. The first possibility is that Cre-mediated recombination in the conditional reporter mice was incomplete, as previously suggested by others (32). The second possibility is that additional cell types which are not addressed by LysM-, CD11c-, or Nkx-Cre contributed to IFN synthesis in virus-infected lungs. We believe that the second possibility most likely accounts for at least a fraction of the unknown activity at very late times after infection with wild-type influenza virus. It is well known that lethal influenza virus infection induces massive recruitment of monocytes, natural killer cells, T cells, and B cells to the lung (33–35). It is conceivable that these cells contributed substantially to the production of IFN observed at late stages of lethal infection with wild-type influenza virus in our experimental setting.
Previously, other researchers used a reporter mouse that synthesizes green fluorescent protein under the control of the IFN-α6 gene promoter to study the innate immune response in the virus-infected lung (8). Those authors reported that alveolar macrophages and conventional dendritic cells were the main producers of type I IFN in the lung after intranasal infection of mice with Newcastle disease virus (NDV), whereas epithelial cells apparently did not synthesize substantial amounts of IFN. In the light of our current findings, this result is surprising, as NDV is well known for its ability to trigger robust synthesis of type I IFN in a wide range of cells from many species (36, 37). Since no information was provided on whether NDV, which is a pathogen of chickens, is able to infect lung epithelial cells in mice, it remains unclear why NDV and NS1-deficient influenza viruses appear to differ in this respect.
In summary, our work demonstrates that wild-type influenza virus uses NS1 to selectively inhibit production of type I IFN in cells that trigger very early antiviral responses, namely, epithelial cells and lung macrophages. Moreover, our work demonstrates that the high levels of IFN seen at late times after infection with wild-type influenza virus originate mainly from different cells which are less affected by the virus-encoded IFN-antagonistic factor NS1.
ACKNOWLEDGMENTS
We thank Alexandra Sontheimer for technical support and Bernd Heimrich and Thomas Michiels for helpful discussions.
This work was supported by grants from the Deutsche Forschungsgemeinschaft to P.S. (SFB 620) and to S.W. (SFB 566) as well as from the Bundesministerium für Bildung und Forschung (BMBF) to S.W.
Footnotes
Published ahead of print 10 April 2013
REFERENCES
- 1. Haller O, Kochs G, Weber F. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344:119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Genin P, Vaccaro A, Civas A. 2009. The role of differential expression of human interferon-α genes in antiviral immunity. Cytokine Growth Factor Rev. 20:283–295 [DOI] [PubMed] [Google Scholar]
- 3. Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P. 2012. Visualizing production of Beta interferon by astrocytes and microglia in brain of la crosse virus-infected mice. J. Virol. 86:11223–11230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Solodova E, Jablonska J, Weiss S, Lienenklaus S. 2011. Production of IFN-beta during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS One 6:e18543 doi: 10.1371/journal.pone.0018543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, Leanderson T. 1998. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr. Biol. 8:223–226 [DOI] [PubMed] [Google Scholar]
- 6. Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Versteeg GA, Garcia-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 13:508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. 2007. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 27:240–252 [DOI] [PubMed] [Google Scholar]
- 9. Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, Staeheli P, Weiss S. 2009. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol. 183:3229–3236 [DOI] [PubMed] [Google Scholar]
- 10. Kochs G, Koerner I, Thiel L, Kothlow S, Kaspers B, Ruggli N, Summerfield A, Pavlovic J, Stech J, Staeheli P. 2007. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J. Gen. Virol. 88:1403–1409 [DOI] [PubMed] [Google Scholar]
- 11. Haller O, Staeheli P, Kochs G. 2007. Interferon-induced Mx proteins in antiviral host defense. Biochimie 89:812–818 [DOI] [PubMed] [Google Scholar]
- 12. Bailey CC, Huang IC, Kam C, Farzan M. 2012. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog. 8:e1002909 doi: 10.1371/journal.ppat.1002909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JS. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837 [DOI] [PubMed] [Google Scholar]
- 15. Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe-Schlosser P, Matrosovich M, Seeger W, Lohmeyer J, Herold S. 2013. Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 9:e1003188 doi: 10.1371/journal.ppat.1003188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS, Jr, Bakaletz LO, Durbin RK, Flano E, Durbin JE. 2007. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J. Virol. 81:9790–9800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaminski MM, Ohnemus A, Cornitescu M, Staeheli P. 2012. Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J. Gen. Virol. 93:555–559 [DOI] [PubMed] [Google Scholar]
- 18. Phipps-Yonas H, Seto J, Sealfon SC, Moran TM, Fernandez-Sesma A. 2008. Interferon-beta pretreatment of conventional and plasmacytoid human dendritic cells enhances their activation by influenza virus. PLoS Pathog. 4:e1000193 doi: 10.1371/journal.ppat.1000193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8:265–277 [DOI] [PubMed] [Google Scholar]
- 20. Tiozzo C, De Langhe S, Yu M, Londhe VA, Carraro G, Li M, Li C, Xing Y, Anderson S, Borok Z, Bellusci S, Minoo P. 2009. Deletion of Pten expands lung epithelial progenitor pools and confers resistance to airway injury. Am. J. Respir. Crit. Care Med. 180:701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J. signaling controls the homeostasis of CD8− dendritic cells in the spleen. J. Exp. Med. 204:1653–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kochs G, Martinez-Sobrido L, Lienenklaus S, Weiss S, Garcia-Sastre A, Staeheli P. 2009. Strong interferon-inducing capacity of a highly virulent variant of influenza A virus strain PR8 with deletions in the NS1 gene. J. Gen. Virol. 90:2990–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kasper M, Behrens J, Schuh D, Muller M. 1995. Distribution of E-cadherin and Ep-CAM in the human lung during development and after injury. Histochem. Cell Biol. 103:281–286 [DOI] [PubMed] [Google Scholar]
- 24. Paine R, III, Morris SB, Jin H, Wilcoxen SE, Phare SM, Moore BB, Coffey MJ, Toews GB. 2001. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L1210–L1218 [DOI] [PubMed] [Google Scholar]
- 25. Simon RH, McCoy JP, Jr, Chu AE, Dehart PD, Goldstein IJ. 1986. Binding of Griffonia simplicifolia I lectin to rat pulmonary alveolar macrophages and its use in purifying type II alveolar epithelial cells. Biochim. Biophys. Acta 885:34–42 [DOI] [PubMed] [Google Scholar]
- 26. Siracusa MC, Reece JJ, Urban JF, Jr, Scott AL. 2008. Dynamics of lung macrophage activation in response to helminth infection. J. Leukoc. Biol. 84:1422–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moltedo B, Lopez CB, Pazos M, Becker MI, Hermesh T, Moran TM. 2009. Stealth influenza virus replication precedes the initiation of adaptive immunity. J. Immunol. 183:3569–3573 [DOI] [PubMed] [Google Scholar]
- 28. Rand U, Rinas M, Schwerk J, Nohren G, Linnes M, Kroger A, Flossdorf M, Kaly-Kullai K, Hauser H, Hofer T, Koster M. 2012. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol. Syst. Biol. 8:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T. 2012. Stochastic expression of the interferon-beta gene. PLoS Biol. 10:e1001249 doi: 10.1371/journal.pbio.1001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, Murphy KM, Garcia-Sastre A, Merad M. 2012. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Invest. 122:4037–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ho AW, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, Lew FC, Wong KL, Hanson BJ, Macary PA, Kemeny DM. 2011. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J. Immunol. 187:6011–6021 [DOI] [PubMed] [Google Scholar]
- 32. Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ. 2008. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J. Exp. Med. 205:2839–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kato A, Schleimer RP. 2007. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr. Opin. Immunol. 19:711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kohlmeier JE, Woodland DL. 2009. Immunity to respiratory viruses. Annu. Rev. Immunol. 27:61–82 [DOI] [PubMed] [Google Scholar]
- 35. McGill J, Heusel JW, Legge KL. 2009. Innate immune control and regulation of influenza virus infections. J. Leukoc. Biol. 86:803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang Z, Krishnamurthy S, Panda A, Samal SK. 2003. Newcastle disease virus V protein is associated with viral pathogenesis and functions as an alpha interferon antagonist. J. Virol. 77:8676–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 77:1501–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]