

Abstract
Previous experiments identified a 12-amino-acid (aa) peptide that was sufficient to interact with the herpes simplex virus 1 (HSV-1) portal protein and was necessary to incorporate the portal into capsids. In the present study, cells were treated at various times postinfection with peptides consisting of a portion of the Drosophila antennapedia protein, previously shown to enter cells efficiently, fused to either wild-type HSV-1 scaffold peptide (YPYYPGEARGAP) or a control peptide that contained changes at positions 4 and 5. These 4-tyrosine and 5-proline residues are highly conserved in herpesvirus scaffold proteins and were previously shown to be critical for the portal interaction. Treatment early in infection with subtoxic levels of wild-type peptide reduced viral infectivity by over 1,000-fold, while the mutant peptide had little effect on viral yields. In cells infected for 3 h in the presence of wild-type peptide, capsids were observed to transit to the nuclear rim normally, as viewed by fluorescence microscopy. However, observation by electron microscopy in thin sections revealed an aberrant and significant increase of DNA-containing capsids compared to infected cells treated with the mutant peptide. Early treatment with peptide also prevented formation of viral DNA replication compartments. These data suggest that the antiviral peptide stabilizes capsids early in infection, causing retention of DNA within them, and that this activity correlates with peptide binding to the portal protein. The data are consistent with the hypothesis that the portal vertex is the conduit through which DNA is ejected to initiate infection.
INTRODUCTION
Herpesviruses cause a number of important diseases in animals and humans, including recurrent skin lesions, blindness, birth defects, transplant rejection, encephalitis, and lymphoid neoplasia. The viral thymidine kinase is by far the most common target of existing antiherpesvirus therapies. This kinase phosphorylates acyclovir and many of its derivatives, making them bioavailable to the replicating viral DNA polymerase (1). Depending on the compound, incorporation of the triphosphorylated drug into the elongating DNA either acts as a competitor for nucleotides, resulting in stalling of the DNA polymerase, or, like acyclovir, precludes formation of further phosphodiester bonds and terminates DNA replication because the drug lacks a 3′ hydroxyl group (2). Because acyclovir and its derivatives represent the most common treatment for herpesvirus infections, and thymidine kinase is ultimately dispensable for viral replication, viral resistance to this group of compounds is common and represents an important threat to human health (3, 4).
Several novel compounds have recently yielded promising results and may eventually expand the antiherpesvirus pharmacopeia (reviewed in reference 5). Different compounds target the viral helicase/primase (6), a conserved viral protein kinase (7–11), and the viral terminase, which is a 3-component enzyme that endonucleolytically cleaves viral DNA from concatemers within the infected cell nucleus and pumps the cleaved genomes into a preformed capsid through a portal vertex (10, 12–16). Others, such as cidofovir, likely affect the viral DNA polymerase (17, 18).
In this report, we explore and evaluate the efficacy of an inhibitor of another antiviral target, specifically the portal vertex. This structure has been targeted previously for antiviral purposes using thiourea compounds (19, 20). In those studies, drug treatment precluded efficient incorporation of the portal into the capsid.
Capsid assembly begins in the nucleus of infected cells when the portal protein encoded by UL6 interacts with a single peptide that is located within the scaffold protein or the identical region within the C terminus of pUL26 (21–24). This peptide (YPYYPGEARGAP; conserved residues italicized) is sufficient to interact with the portal protein in vitro, and the tyrosine and proline residues at positions 4 and 5, respectively, are necessary for coimmunoprecipitation with the portal protein (24, 25). Under normal conditions, the portal vertex, which comprises 12 copies of the human herpes virus 1 (HSV-1) UL6 gene product, acts as a nidus for formation of the rest of the procapsid (26–29). This strategy ensures that each capsid contains a single portal.
The completed herpes simplex virus procapsid is the progenitor of all other capsid types and consists of two proteinaceous layers, an outer shell and an inner scaffold (30, 31). The scaffold contains an abundant scaffold protein and a less abundant protease. The protease is located at the amino terminus of the product of the UL26 gene (pUL26), whereas the C terminus of pUL26 shares considerable sequence identity with the major scaffold protein (32–34). The protease cleaves pUL26 in cis after serine 247, thereby separating the N-terminal protease domain from the C-terminal scaffold domain (35–39). A second cleavage removes 25 amino acids (aa) from the C terminus of pUL26 (36, 37) and frees the scaffold protein from a linkage that otherwise binds it to the N terminus of the major external capsid protein VP5 (40–43). In productive reactions, DNA replaces the internal scaffold of the roughly spherical procapsid, and the external shell undergoes a morphological change that confers an icosahedral shape and increased stability. Two dead-end reactions can also occur in infected cells. In the first, B-type capsids are produced when the scaffold is cleaved by the protease but remains within the external shell and is not replaced by DNA. In the second, A capsids, which lack the internal scaffold and DNA, are formed. During the formation of A capsids, it is believed that DNA packaging is initiated but not completed, and the scaffold is expelled or degraded. Since the DNA is not sealed inside, it is believed to slip out through the portal, leaving an empty shell.
The opposite reaction of DNA packaging is DNA uncoating. After fusion of the virion envelope with the plasma or endosomal membranes, the capsid is delivered to the nuclear pore, where DNA uncoating takes place (44, 45). Subsequent to uncoating, empty external shells lacking an electron-dense internal core can be observed attached to the nuclear rim of the newly infected cell. The uncoated DNA is transported through the nuclear pore and into the nucleus to begin immediate early viral transcription. Although in vitro evidence indicates that DNA exits the capsid through a single capsid vertex (46–48), in vitro or in vivo data indicating that the portal is involved in DNA ejection are not yet available.
The current studies were undertaken to determine whether the scaffold peptide previously shown to interact with the portal protein could interfere with virus replication when delivered into infected cells. The resulting data provide a novel therapeutic avenue and support for the hypothesis that the portal constitutes the conduit through which DNA uncoats during the initiation of viral infection.
MATERIALS AND METHODS
Cells and viruses.
CV1 cells were purchased from ATCC and grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% newborn calf serum, 100 U/ml penicillin, and 100 μg/ml of streptomycin. CV6 cells were used for propagating UL6-null virus and were grown in the same medium supplemented with 200 μg/ml of hygromycin B as described previously (49). The herpes simplex virus 1 F strain [HSV-1(F)], used as the wild-type strain in this study, was from B. Roziman (50). The UL6 deletion mutant derived from the HSV-1 KOS strain was a gift from A. Patel (51), and the K26 green fluorescent protein (GFP) virus, in which GFP was fused in frame to the UL35 gene of the HSV-1 KOS strain, was from P. Desai (52).
Peptide synthesis.
Peptides with sequences of RQIKIWFQNRRMKWKKYPYYPGEARGAP (wild type, designated peptide 1) or RQIKIWFQNRRMKWKKYPYAAGEARGAP (mutant, designated peptide 2) or biotin-labeled versions of these peptides were commercially synthesized with 95% purity (Genemed Synthesis, Inc., San Antonio, TX). The sequence underlined in the wild-type peptide sequence is derived from residues 448 to 459 of pUL26 of HSV-1(F), which constitutes a domain that is sufficient to interact with the portal protein pUL6 and necessary for incorporation of the portal into capsids (24, 25). The tyrosine and proline residues at positions 4 and 5, respectively, were changed to alanines; these Y and P residues are shown as underlined and italicized above and are critical to the portal interaction (25). The sequences italicized and not underlined are derived from the antennapedia homeodomain, possessing the capability of facilitating uptake of covalently attached moieties into cells (53). The peptides were dissolved in double-distilled water at a concentration of 2 mM.
Toxicity testing.
Peptide cytotoxicity was determined by a cell proliferation assay according to the manufacturer's protocol (CellTiter 96 AQueous One Solution cell proliferation assay; Promega). Briefly, CV1 cells were grown in 12-well plates at 80% confluence. Medium only or medium containing either various concentrations of peptides or sodium azide (as a positive control for toxicity) was added. After incubation for 24 h at 37°C, 40 μl of the manufacturer's reagent was added to the cells. The plate was then incubated for 2 h at 37°C, and the absorbance at 490 nm was read with a plate reader (BLX800; Bio-TEX).
Virus replication assay and time course of peptide addition.
Subconfluent CV1 cells were infected with HSV-1(F) at a multiplicity of infection (MOI) of 0.1 PFU per cell and incubated for 1 h at 4°C for virus absorption and then for 60 min at 37°C. After incubation, the inoculum was aspirated, and the residual extracellular infectivity was inactivated in low-pH buffer (40 mM citric acid, 10 mM KCl, and 135 mM NaCl [pH 3.0]). Two milliliters of fresh growth medium containing 0.1% dimethyl sulfoxide (DMSO) and various concentrations of peptide was then added to the cells. At 24 h postinfection, the cells were frozen at −80°C, and after three freeze and thaw cycles, yields of infectious virus were determined by plaque assay on CV1 cells.
To determine the most effective time at which peptide treatment inhibited virus replication, the infected CV1 cells were treated with 100 μM wild-type or mutant peptide. The time of the addition of peptide is indicated in Fig. 1C. For the 0-min time point, peptide was present at the time of adsorption and infection and thereafter. At the other time points, the number of minutes indicated in Fig. 1C indicates the time incubated at 37°C between the low-pH wash and the addition of peptide. For these time points, the peptide then remained in the medium for the duration of the infection. For the 30-min time point, for example, virus was preadsorbed onto cells by incubation for 1 h at 4°C, shifted to 37°C for 1 h to allow viral entry, treated with a low-pH wash to reduce residual surface infectivity, and then incubated in growth medium for 30 min, at which time the peptide was added to the medium and remained in the overlay until lysis 24 h later.
Fig 1.
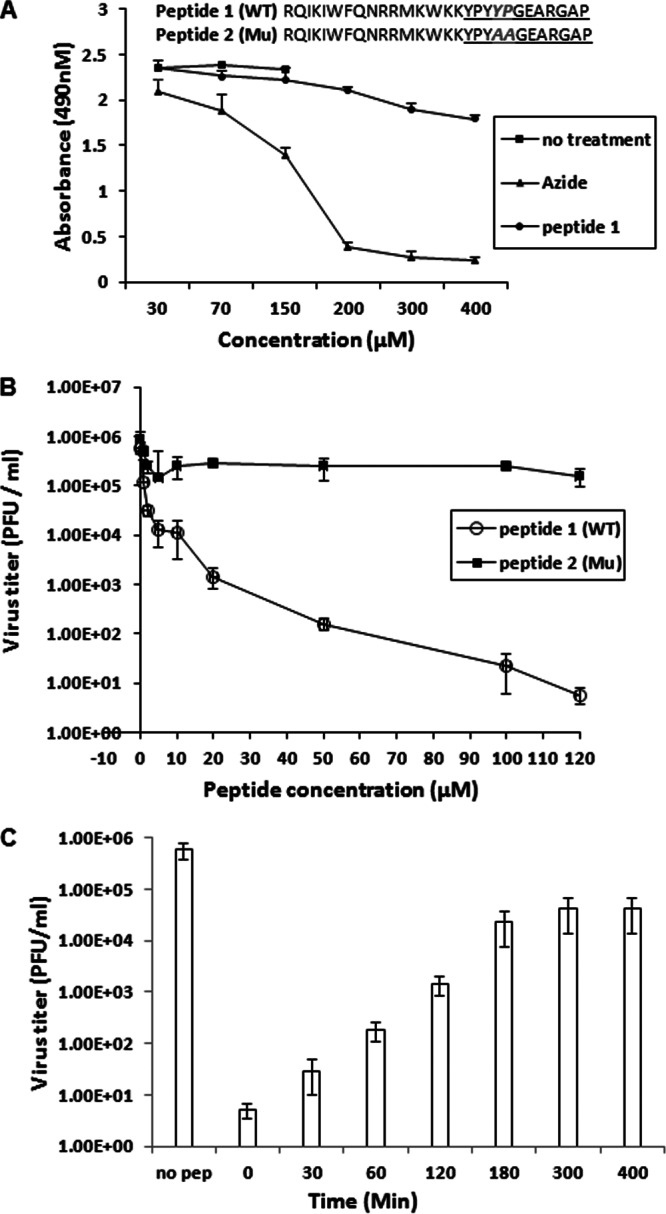
Antiviral activities of peptides. (A) Cytotoxic effects of peptides. CV1 cells in 12-well plates were treated with various concentrations of peptide overnight. The next day, cell proliferation reagent (Promega) was added to each well, and 2 h later, the absorbance at 490 nm was recorded. (B) Specific antiviral activities of peptides. Subconfluent CV1 cells in 6-well plates were infected with HSV-1(F) at an MOI of 0.1 PFU per cell. After a 1-h adsorption at 4°C and 60 min of incubation at 37°C, the inocula were removed, residual extracellular infectivity associated with the cells was reduced by treatment with a low-pH citrate buffer, and growth media containing the indicated concentrations of peptide were added. Twenty-four hours after virus infection, the amount of infectious virus was determined by plaque assay in CV1 cells. (C) Time dependence of inhibition of HSV replication. CV1 cells in 6-well plates were infected with HSV-1(F) at an MOI of 0.1 PFU per cell, allowed to adsorb for 1 h at 4°C, shifted to 37°C to allow viral entry, and then washed with citrate buffer (pH 3.0) to inactivate extracellular infectivity. For the 0-min time point, peptide was present at the time of adsorption and infection and thereafter. Otherwise, 100 μm of peptide 1 (wild type) was added at the indicated times after the citrate wash and was maintained in the medium until 24 h postinfection. The cells were then lysed by repeated freezing/thawing, and yields of infectivity were determined by plaque assay in CV1 cells. Each point and error bar for all three panels represents the mean ± standard deviation of results from three individual experiments. *, no treatment samples were done in parallel with the 30 μM, 70 μM, and 150 μM concentrations of azide or peptide.
All samples were lysed 24 h after infection, and viral infectivity was determined by plaque assay. All the experiments were repeated at least three times.
Immunoblotting.
CV1 cells in 6-well plates were infected with HSV-1(F) at an MOI of 10 PFU per cell and treated with various concentrations of peptides. At 6 or 24 h after infection, the cells were washed with phosphate-buffered saline (PBS) and solubilized in SDS sample buffer (62.5 mM Tris [pH 6.8], 2% SDS, 0.35 M β-mercaptoethanol, and 10% glycerol). The solubilized proteins were separated on 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes. The 6-h samples were probed with antibody to ICP4 (diluted 1:1,000 in PBS plus 1% bovine serum albumin [BSA]), whereas the 24-h samples were probed with antibody to pUL26 (MCA 406, diluted 1:2,000 in PBS with 1% BSA). The bound antibodies were detected by incubating with anti-mouse immunoglobulin conjugated with horseradish peroxidase (HRP) and visualized by enhanced chemiluminescence (ECL; Amersham). For a loading control, the blot was stripped and reprobed with anti-actin antibodies (Santa Cruz; diluted 1:200) and developed similarly.
Capsid purification.
Approximately 4 × 108 CV1 cells grown on two 850-cm2 roller bottles were infected with either HSV-1(F) or UL6-null mutant virus at an MOI of 10 PFU per cell. Twenty hours after infection, cells were scraped into PBS and pelleted by centrifugation at 4,000 rpm for 5 min at 4°C. Cell pellets were suspended in lysis buffer (20 mM Tris [pH 7.6], 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM dithiothreitol [DTT], and protease inhibitor) and sonicated, and the lysates were precleared by centrifugation at 10,000 rpm for 20 min. The viral capsids in the lysates were pelleted through a 35% (wt/vol) sucrose cushion prepared in TNE buffer (20 mM Tris [pH 7.6], 500 mM NaCl, and 1 mM EDTA) at 24,000 rpm in an SW28 rotor for 60 min. The capsid pellets were resuspended in TNE buffer and further purified by centrifugation through 20% to 50% sucrose gradients in an SW41 rotor at 24,500 rpm for 60 min. Capsids were visualized as light-scattering bands and collected with a Pasteur pipette.
Examination of viral capsid entry.
CV1 cells were grown on 18-mm by 18-mm glass coverslips in a 6-well plate, infected with HSV-1 K26GFP at an MOI of 20 PFU per cell, and treated with 100 μM peptides as described above. Three hours after infection, the cells were fixed with 3% paraformaldehyde (wt/vol), stained with Hoechst to locate the nucleus, and examined using a Leica TCS SP5 confocal microscope, equipped with a 63× oil immersion objective lens. Individual capsids associated with the nuclear rim were counted. Images for display were captured and exported as TIFF files, and the entire image was processed with Adobe Photoshop software.
PAA treatment.
CV1 cells grown on coverslips in 6-well plates were infected by adding medium containing 20 PFU HSV-1/cell followed by incubation for 1 h at 4°C. The plate was then transferred to a 37°C incubator for 30 min, and the medium was replaced with 1 ml of fresh DMEM containing either 100 μM peptide, 300 μg/ml of phosphonoacetic acid (PAA), or both. After incubation at 37°C for an additional 2.5 h, the cells were fixed and processed for fluorescence in situ hybridization of viral DNA as detailed below.
Fluorescence in situ hybridization of viral DNA.
CV1 cells grown on 18-mm by 18-mm glass coverslips in a 6-well plate were infected with HSV-1(F) at an MOI of 20 PFU per cell. After 1 h of absorption at 4°C and then shift to 37°C for 30 min, the inoculum was removed and fresh growth medium containing 0.1% DMSO and 100 μM peptides was added. Two and a half hours after the addition of peptide, the cells were washed with PBS and fixed by incubation at −20°C for 5 min with cold 95% ethanol-5% glacial acetic acid. Approximately 1 μg of plasmid pRB 162 containing the BamHI P fragment of the HSV-1 genome (a gift from B. Roizman) was labeled with Cy3-dCTP by nick translation according to the manufacturer's protocol (Amersham; N5500) using the protocol specific for production of probes to be used for in situ hybridization. The unincorporated Cy3-dCTP was removed by chromatography through a Micro Bio-Spin 6 column (Bio-Rad). After fixation, the cells were prehybridized by placing the coverslips facedown onto a drop of 30 μl of hybridization buffer (4× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 1× Dehart's (1% Ficoll, 1% polyvinylpyrrolidone, 1% BSA in water), 50% formamide, 10% dextran, 0.1 mg/ml of salmon sperm DNA) for 1 h at 37°C. The coverslips were then placed on 30 μl of hybridization buffer containing a 50-ng probe in a 6-well plate, incubated at 95°C for 2 min to denature the probe and sample, and then incubated at 37°C for 18 h for hybridization. The coverslips were washed sequentially with 2× SSC (0.3 M NaCl, 0.03 M sodium citrate [pH 7.0]) at 60°C for 5 min and 2× SSC at room temperature for 10 min, counterstained with Hoechst for 10 min, washed with PBS, and then mounted on glass slides. The samples were examined with a Leica TCS SP5 confocal microscope, using a 63× oil immersion objective lens. Images were captured and exported as TIFF files and then processed with Adobe Photoshop.
Electron microscopy.
CV1 cells grown in a 6-well plate (approximately 1.2 × 106) were infected with HSV-1(F) at an MOI of 20 PFU per cell and treated with 100 μM peptides as described above. Three hours after infection, the cells were fixed with 2.5% glutaraldehyde (Electron Microscopy Sciences) in 0.1 M sodium cacodylate buffer, pH 7.4, for 30 min at room temperature and then 90 min at 4°C. After three rinses for 5 min each with the same buffer, cells were treated with 2% osmium tetroxide overnight at 4°C, rinsed again with 0.1 M sodium cacodylate buffer, and then dehydrated with a graduated series of ethanol concentrations (20%, 50%, 70%, 90%, and 100%), followed by increasing concentrations of acetone (50%, and then two incubations with 100%). This was followed by stepwise infiltration with Epon-Araldite resin (Electron Microscopy Sciences) over the course of 48 h at room temperature. Samples were dispensed into Beem capsules, and the resin was polymerized at 65°C for 18 h. Thin sections (60 to 90 nm thick) were collected on 300-mesh nickel grids (EMS). Thin sections were counterstained with 2% aqueous uranyl acetate for 20 min and then Reynolds lead citrate for 7 min. Stained grids were viewed in a Philips 301 transmission electron microscope. All images were collected digitally and were processed using Adobe Photoshop software for illustrative purposes.
RESULTS
Initial experiments were performed to determine whether the scaffold peptide YPYYPGEARGAP was sufficient to inhibit viral replication. This peptide was linked to the antennapedia sequence RQIKIWFQNRRMKWKK that has been shown to enter cells efficiently and induce minimal toxicity (54, 55). As a control, a mutant peptide YPYAAGEARGAP was fused to the same antennapedia sequence. This mutant scaffold peptide was shown previously not to bind the portal protein and to compete only inefficiently with portal-scaffold interactions in vitro (25).
CV1 cells were treated with various concentrations of wild-type or mutant scaffold peptides starting at 2 h after infection, and the cells were then infected with 0.01 PFU/cell HSV-1(F). Twenty-four hours after infection, the cells were lysed by freeze-thawing, and the amount of infectious virus was determined by plaque assay. As shown in Fig. 1B, viral titers were reduced by 50% with as little as 5 μM peptide, whereas 50 μM peptide reduced infectious titers by more than 1,000-fold. In contrast, the mutant peptide did not significantly diminish viral replication even when administered at a concentration of 120 μM. Potential toxic effects were also tested by subjecting the cells to a standardized cell proliferation assay (Fig. 1A). The effective concentrations of the scaffold peptide were not toxic to cells inasmuch as only small reductions in cell proliferation were noted at peptide concentrations as high as 400 μM.
To determine the most effective time at which peptide administration decreased viral infectivity, cells with 100 μM peptide were treated (i) with a 100 μM peptide concentration during infection (0-min time point), (ii) with medium containing 0.1% DMSO (no peptide), or (iii) with peptide at various times after infection, as indicated in Materials and Methods. The same medium remained overlying the cells for 24 h, at which time the cells were frozen and thawed and the amount of infectivity was determined by plaque assay. As shown in Fig. 1C, treatment at the time of infection (0 h) was most effective and reduced infectious titers by more than 100,000-fold. Delaying peptide administration relative to the time of infection decreased peptide efficacy such that the peptide decreased viral infectivity by approximately 10,000-fold when added at 30 min postinfection, 1,000-fold when added at 60 min, and approximately 20-fold when added at 180 and 300 min after infection. These data indicate that the peptide was most effective early in infection.
Given that the peptide had antiviral effects, we were interested to determine the mechanism of viral inhibition. As a first step in this regard, the mutant and wild-type peptides were biotinylated and added to wild-type or UL6-null capsids that had been purified and attached to silicon-coated electron microscope grids. Avidin-conjugated colloidal gold was then reacted with the capsids and, after extensive washing, were viewed in a transmission electron microscope.
As shown in Fig. 2, the wild-type peptide bound more than 20% of wild-type capsids. In contrast, the mutant peptide bound significantly fewer (approximately 3%) of wild-type capsids. Capsids lacking portals (Δ6 in Fig. 2) were labeled significantly less than either the wild-type or mutant peptides reacted with wild-type capsids. We conclude that the wild-type peptide can associate with capsids and that this binding was dependent on the presence of the portal protein.
Fig 2.

Wild-type peptide binds to portal-containing capsids in vitro. Portal-containing B capsids (A) purified from HSV-1(F)-infected cells or portal-null B capsids (B) purified from UL6-null infected cells were loaded on carbon-coated grids and incubated with biotin-labeled peptide, and the grids were reacted with 8 to 10 nm colloidal gold beads conjugated to streptavidin. The grids were washed extensively and examined in a transmission electron microscope. (C) Percentage of labeled capsids compared to total capsids counted. GraphPad software was used for statistical analyses using Fisher's exact test. (*, P < 0.01).
To determine whether the peptide inhibited early or late viral protein expression, cells were treated with 20 or 100 μM peptide at the time of infection, and the infected cells were lysed 6 or 24 h later. The lysates were denatured in SDS, electrophoretically separated on an SDS polyacrylamide gel, and transferred to nitrocellulose, and the 6-h lysates were probed with antibodies to ICP4, an early protein, while the 24-h samples were probed with antibody directed against scaffold proteins pUL26 and VP22a, which are expressed late in infection. As shown in Fig. 3, 20 μM wild-type peptide slightly reduced ICP4 expression at 6 h postinfection and VP22a at 24 h after infection. Cells treated with 100 μM peptide showed more dramatic differences inasmuch as ICP4 expression was not detected at 6 h postinfection, whereas VP22a expression was reduced significantly at 24 h. In contrast, administration of the mutant peptide did not significantly diminish ICP4 or VP22a expression compared to the untreated control. We conclude that treatment with wild-type peptide interfered with early-protein expression. The effect on late-gene expression, while significant, may be indirect, because poor early-gene expression can reduce production of late genes (56).
Fig 3.
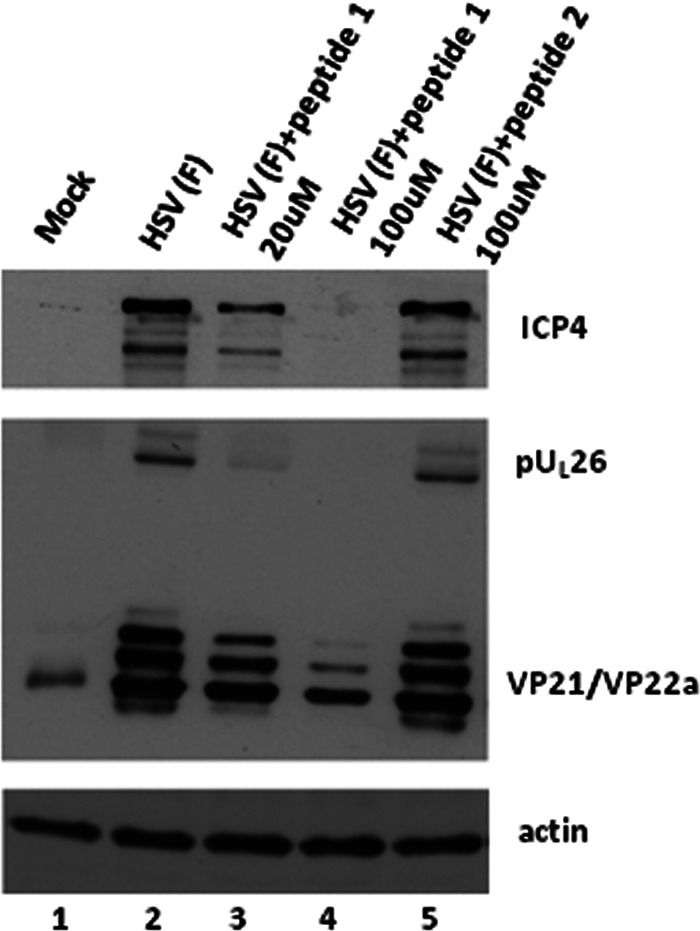
Wild-type peptide inhibits expression of ICP4 of HSV-1. CV1 cells in 6-well plates were infected with HSV-1(F) at an MOI of 10 PFU per cells as described in the legend to Fig. 1B, and the indicated concentrations of peptide 1 or peptide 2 were added to the overlying medium. Six hours or 24 h after infection, total cell lysates were prepared, separated on a denaturing SDS polyacrylamide gel, transferred to nitrocellulose, and reacted with antibodies to ICP4 (6 h postinfection) or ICP35 (24 h postinfection). The blot was stripped and reprobed with anti-actin antibodies as a loading control.
To further characterize the defect in viral replication, cells were infected and treated or left untreated with mutant or wild-type peptides at 30 min after infection in the presence of absence of phosphonoacetic acid (PAA), a potent inhibitor of the viral DNA synthesis (57). Cells were then fixed at 3 h, and DNA was denatured and hybridized with a Cy3-labeled viral DNA probe. As shown in Fig. 4, cells either not treated or treated with mutant peptide were similar in appearance, with large globular regions in the nucleoplasm staining heavily with the DNA probe. In contrast, cells treated with wild-type peptide exhibited substantially less intranuclear staining. Cytoplasmic staining in HSV-1(F)-infected cells treated with the wild-type peptide appeared as faint foci lining the nuclear rim. The position and character of this staining resembled that of cells infected with HSV-1(F) that were treated with PAA. These data suggested that DNA replication and formation of DNA replication compartments were impaired by treatment with wild-type peptide. The cytoplasmic signals likely reflect the staining of input viral DNA. However, given the denaturation of the sample required for proper hybridization, it is unclear whether the viral DNA located at the nuclear rim was present within or outside incoming capsids.
Fig 4.

Reduction of viral DNA replication detected by fluorescence in situ hybridization. CV1 cells were grown on glass coverslips (18 mm by 18 mm) and then infected with HSV-1(F) at an MOI of 20 PFU per cell as described in the legend to Fig. 1B. The cells were untreated or treated with wild-type (WT) or mutant (Mu) scaffold peptide in the presence or absence of phosphonoacetic acid (PAA), a potent viral DNA replication inhibitor. Two and a half hours after onset of the treatments, cells were washed with PBS and then fixed by incubation for 5 min at −20°C with precooled 95% ethanol-5% glacial acetic acid. A Cy3-dCTP-labeled probe containing the BamHI P fragment of the HSV-1 genome was reacted for hybridization, as described in Materials and Methods. After hybridization and washing, the coverslips were counterstained with Hoechst stain, mounted on glass slides, and examined by confocal microscopy. The images were captured and exported as TIFF files and processed with Adobe Photoshop software. The red signal indicates the presence of viral DNA. Globular domains within the nucleus identify the viral DNA replication compartment.
Defects in early-protein expression and formation of the DNA replication compartment could be explained by a failure of capsids to enter the cytosol, a failure to deliver viral DNA into the nucleus, or a defect in early transcription. To distinguish among these possibilities, cells were infected in the presence or absence of wild-type or mutant peptide with a virus that generates capsids with GFP attached to the capsid surface (52). Three hours postinfection, the cells were fixed with 3% paraformaldehyde. Nuclei were stained with Hoechst, and the cells were examined by confocal microscopy. Capsids associated with the nuclear rim were then counted. As shown in Fig. 5, the numbers of capsids associated with the nuclear rim were similar whether the cells were treated with wild-type or mutant peptide or were untreated. These data suggest that neither entry of the virus into the cell nor transport of the capsid to the nucleus were affected by peptide treatment.
Fig 5.

Incoming capsids accumulate near the nuclear rim in the presence of peptide. Confluent CV1 cells on glass coverslips (18 mm by 18 mm) were infected with K26 GFP virus at an MOI of 20 PFU per cell as described in the legend to Fig. 1B, and 100 μm of peptide 1 or 2 was added when the medium was shifted to 37°C. Three hours postinfection, the cells were washed with PBS, fixed with 3% paraformaldehyde, stained with Hoechst to locate cellular nuclei, and viewed by confocal microscopy. Top, example image showing capsids (green dots) on the nuclear rim of cells in the right panel (arrows); bottom, graph shows the number of dots associated with the nuclear rim. The number is an average number of capsids per nucleus from 50 cells viewed for each sample.
It is well established that capsids are transported to the nuclear pore, where they disgorge (or uncoat) their DNA into the nucleus to initiate infection. To determine if the peptide treatment affected uncoating, CV1 cells were infected with 20 PFU/cell HSV-1(F) at 4°C and upon shift to 37°C were untreated or treated with wild-type or mutant peptide. Three hours later, cells were fixed and embedded, and the number and type of capsids within thin sections were determined. As shown in Fig. 6 in untreated cells or cells treated with mutant peptide, approximately 40% of internalized capsids lacked electron-dense cores, indicating a lack of scaffold or DNA within them. These A capsids likely resulted from DNA uncoating events. In contrast, treatment with wild-type peptide significantly decreased the number of empty capsids (to approximately 10% of the total), with a concomitant increase of C capsids. These data indicate that the wild-type peptide was able to partially block generation of empty capsids. This observation suggests that the peptide stabilizes incoming capsids, causing them to retain genomic DNA.
Fig 6.



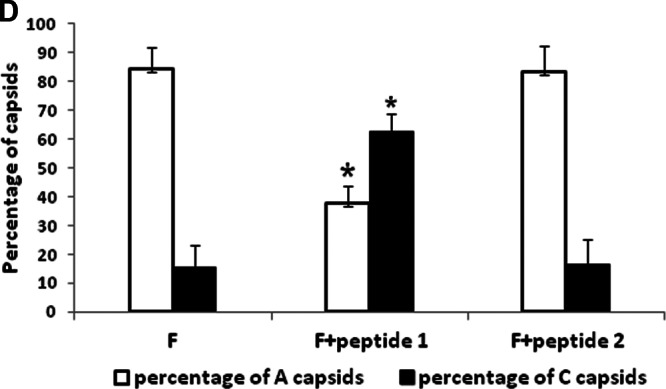
Examination of virus-infected cells in the presence of peptide by electron microscopy. Subconfluent CV1 cells in 6-well plates were infected with HSV-1(F) at an MOI of 20 PFU per cell in the presence or absence of 100 μM wild-type or mutant peptide. Three hours after infection, the cells were fixed and embedded, and thin sections were examined by transmission electron microscopy. Magnifications in the upper panels were ×11,000 for panel A and ×13,000 for panels B and C. Magnification for the selected area (boxed and shown in lower panels) was ×30,000. The images were taken and processed with Adobe Photoshop software. (A) Electron micrograph of untreated HSV-1(F)-infected cells; (B) electron micrograph of cells infected with HSV-1(F) in the presence of wild-type peptide (peptide 1); (C) electron micrograph of cells infected with mutant peptide (peptide 2). Ten sections were examined for each treatment, and the number of DNA-containing capsids (C capsids; open arrowheads) and empty capsids (A capsids; solid arrows) were counted. Approximately 1,000 capsids were counted for each treatment. (D) A histogram of the percentage of A or C capsids relative to total capsids counted is shown. Statistical analyses were performed using Fisher's exact test. *, P < 0.01. A size standard is shown at the lower right of panel A and at the lower left of panels B and C.
DISCUSSION
We report that a scaffold peptide constituting an HSV-1 portal binding domain can interfere with viral replication when added exogenously to cells. The block to viral propagation involves a block in viral DNA replication and formation of the viral replication compartments, a delay and decrease of immediate early and late viral proteins, and a greater than 10,000-fold decrease in production of infectious virus, depending on when the peptide was added after infection.
Early treatment with scaffold peptide was more effective in blocking viral replication than treatment later in infection. Direct addition of antennapedia and other membrane-penetrating peptides either alone or fused to unrelated short peptides can decrease viral entry when added to virions or to cells to which virions had been allowed to attach (58). This mechanism may be due to an induced aggregation of virions or perturbation of the virion envelope. We suspect that this effect was at least partly responsible for the substantial decrease in viral yield when the peptide was present at the time of viral entry (i.e., at the 0-min time point shown in Fig. 1C). However, it is unlikely that this direct effect on viral entry played a significant role in most of the other experiments reported herein, because the wild-type peptide fused to the antennapedia peptide retained effective antiviral activity even when added up to 2 h after viral infection (Fig. 1C). Moreover, virtually no antiviral effect of the mutant scaffold peptide was noted at these times of addition, despite the fact that it was fused to the wild-type antennapedia peptide (Fig. 1B). Thus, the antiviral effect of the scaffold peptide is not likely to be a consequence of a failure of virions to enter the plasma membrane or endosomes, although we cannot exclude minor roles for these effects in some experiments.
High multiplicity of infections were used in the electron microscopy experiments (Fig. 6). This was done to ensure that sufficient numbers of capsids were visible for meaningful statistical analyses after entry in the presence or absence of wild-type or mutant scaffold peptides. Although it is likely that most of these capsids do not represent infectious events inasmuch as many were contained within vesicles and were not docked at nuclear pores, an effect of the peptide was discernible nonetheless. Specifically, more C capsids were present upon wild-type peptide treatment after the virus had entered the plasma membrane than were present in cells treated with mutant peptide or that were untreated. Conversely, more A capsids were observed in cells that were untreated or treated with mutant peptide. Thus, it is likely that the mechanism of peptide interference early in infection involves stabilization of capsids, such that they retain viral DNA. This stabilization may be mediated by direct binding of the peptide to the portal. This binding might block the DNA translocation channel or induce a conformational change that precludes channel patency. These interpretations are supported by the observation that a fusion peptide containing only 2 aa changes that precluded interaction with portal protein also failed to inhibit viral replication (25) (Fig. 1B). It should be noted that under normal conditions, the inhibitory scaffold peptide used in these studies is not present in C capsids inasmuch as the scaffold is replaced with DNA during the DNA packaging reaction. Thus, expulsion of this inhibitory peptide from the capsid may be important for portal function.
If peptide treatment was delayed by 3 h, viral infectivity was reduced only by around 10-fold, indicating a more modest effect of peptide treatment late in infection (Fig. 1C). We speculate that this effect may reflect a role in portal incorporation into the capsid or portal competence in DNA packaging, although further studies will be necessary to test these possibilities.
Use of the scaffold peptide or compounds that mimic it may constitute useful antiviral compounds in the future. The conserved nature of the peptide suggests that such antivirals may have broad activities against other herpesviruses.
ACKNOWLEDGMENTS
We thank Preshant Desai for the K26 GFP virus and Roger Everett for advice on in situ hybridization with DNA probes.
These studies were supported by Public Health Service grant R01 AI 52341 from the National Institutes of Health.
Footnotes
Published ahead of print 10 April 2013
REFERENCES
- 1. Furman PA, de MP, St Clair MH, Elion GB. 1981. Metabolism of acyclovir in virus-infected and uninfected cells. Antimicrob. Agents Chemother. 20:518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elion GB. 1982. Mechanism of action and selectivity of acyclovir. Am. J. Med. 73:7–13 [DOI] [PubMed] [Google Scholar]
- 3. Piret J, Boivin G. 2011. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55:459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson C, Whitley R. 2011. Neonatal herpes simplex virus infections: where are we now? Hot topics in infective immunity in children VII, p 221–230 In Curtis N, Finn A, Pollard AJ. (ed), 697 ed. Springer, New York, NY: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Price NB, Prichard MN. 2011. Progress in the development of new therapies for herpesvirus infections. Curr. Opin. Virol. 1:548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Field HJ, Biswas S. 2011. Antiviral drug resistance and helicase-primase inhibitors of herpes simplex virus. Drug Resist. Updat. 14:45–51 [DOI] [PubMed] [Google Scholar]
- 7. Chou S. 2009. Diverse cytomegalovirus UL27 mutations adapt to loss of viral UL97 kinase activity under maribavir. Antimicrob. Agents Chemother. 53:81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kern ER, Kushner NL, Hartline CB, Williams-Aziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. 2005. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother. 49:1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. 2011. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe 9:103–114 [DOI] [PubMed] [Google Scholar]
- 10. Williams SL, Hartline CB, Kushner NL, Harden EA, Bidanset DJ, Drach JC, Townsend LB, Underwood MR, Biron KK, Kern ER. 2003. In vitro activities of benzimidazole D- and L-ribonucleosides against herpesviruses. Antimicrob. Agents Chemother. 47:2186–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou S, Breitenbach JM, Borysko KZ, Drach JC, Kern ER, Gullen E, Cheng YC, Zemlicka J. 2004. Synthesis and antiviral activity of (Z)- and (E)-2,2-[bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: second-generation methylenecyclopropane analogues of nucleosides. J. Med. Chem. 47:566–575 [DOI] [PubMed] [Google Scholar]
- 12. Bogner E. 2002. Human cytomegalovirus terminase as a target for antiviral chemotherapy. Rev. Med. Virol. 12:115–127 [DOI] [PubMed] [Google Scholar]
- 13. Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anti-cytomegalovirus compound AIC246 inhibits HCMV replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 85:10884–10893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 72:4721–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, Ruebsamen-Schaeff H, Zimmermann H. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 54:1290–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, Haerter M, Buerger I, Trappe J, Herrington JA, Haebich D, Ruebsamen-Waigmann H. 2001. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38–4766): in vitro and in vivo antiviral activity and mechanism of action. J. Antimicrob. Chemother. 48:757–767 [DOI] [PubMed] [Google Scholar]
- 17. Kornbluth RS, Smee DF, Sidwell RW, Snarsky V, Evans DH, Hostetler KY. 2006. Mutations in the E9L polymerase gene of cidofovir-resistant vaccinia virus strain WR are associated with the drug resistance phenotype. Antimicrob. Agents Chemother. 50:4038–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prichard MN, Hartline CB, Harden EA, Daily SL, Beadle JR, Valiaeva N, Kern ER, Hostetler KY. 2008. Inhibition of herpesvirus replication by hexadecyloxypropyl esters of purine- and pyrimidine-based phosphonomethoxyethyl nucleoside phosphonates. Antimicrob. Agents Chemother. 52:4326–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Newcomb WW, Brown JC. 2002. Inhibition of herpes simplex virus replication by WAY-150138: assembly of capsids depleted of the portal and terminase proteins involved in DNA encapsidation. J. Virol. 76:10084–10088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Zijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O'Hara B, Bloom JD, Johann SV. 2000. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J. Virol. 74:9054–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huffman JB, Newcomb WW, Brown JC, Homa FL. 2008. Amino acids 143 to 150 of the herpes simplex virus type 1 scaffold protein are required for the formation of portal-containing capsids. J. Virol. 82:6778–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newcomb WW, Homa FL, Brown JC. 2005. Involvement of the portal at an early step in herpes simplex virus capsid assembly. J. Virol. 79:10540–10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Singer GP, Newcomb WW, Thomsen DR, Homa FL, Brown JC. 2005. Identification of a region in the herpes simplex virus scaffolding protein required for interaction with the portal. J. Virol. 79:132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang K, Baines JD. 2008. Domain within herpes simplex virus 1 scaffold proteins required for interaction with portal protein in infected cells and incorporation of the portal vertex into capsids. J. Virol. 82:5021–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang K, Baines JD. 2009. Proline and tyrosine residues in scaffold proteins of herpes simplex virus 1 critical to the interaction with portal protein and its incorporation into capsids. J. Virol. 83:8076–8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang JT, Schmid MF, Rixon FJ, Chiu W. 2007. Electron cryotomography reveals the portal in the herpesvirus capsid. J. Virol. 81:2065–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newcomb WW, Thomsen DR, Homa FL, Brown JC. 2003. Assembly of the herpes simplex virus capsid: identification of soluble scaffold-portal complexes and their role in formation of portal-containing capsids. J. Virol. 77:9862–9871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rochat RH, Liu X, Murata K, Nagayama K, Rixon FJ, Chiu W. 2011. Seeing the portal in herpes simplex virus type 1 B capsids. J. Virol. 85:1871–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trus BL, Cheng N, Newcomb WW, Homa FL, Brown JC, Steven AC. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78:12668–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Newcomb WW, Homa FL, Thomsen DR, Trus BL, Cheng N, Steven A, Booy F, Brown JC. 1999. Assembly of the herpes simplex virus procapsid from purified components and identification of small complexes containing the major capsid and scaffolding proteins. J. Virol. 73:4239–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trus BL, Booy FP, Newcomb WW, Brown JC, Homa FL, Thomsen DR, Steven AC. 1996. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19C and VP23 in assembly. J. Mol. Biol. 263:447–462 [DOI] [PubMed] [Google Scholar]
- 32. Liu F, Roizman B. 1991. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J. Virol. 65:5149–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu F, Roizman B. 1992. Differentiation of multiple domains in the herpes simplex virus 1 protease encoded by the UL26 open reading frame. Proc. Natl. Acad. Sci. U. S. A. 89:2076–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Preston VG, Rixon F, McDougall IM, McGregor M, Al-Kobaisi MF. 1992. Processing of the herpes simplex virus assembly protein ICP35 near its carboxy-terminal end requires the product of the whole of the UL26 reading frame. Virology 186:87–98 [DOI] [PubMed] [Google Scholar]
- 35. Deckman IC, Hagen M, McCann PJ. 1992. Herpes simplex virus type 1 protease expressed in Escherichia coli exhibits autoprocessing and specific cleavage of the ICP35 assembly protein. J. Virol. 66:7362–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DiIanni CL, Drier DA, Deckman IC, McCann PJ, III, Liu F, Roizman B, Colonno RJ, Cordingley MG. 1993. Identification of the herpes simplex virus-1 protease cleavage sites by direct sequence analysis of autoproteolytic cleavage products. J. Biol. Chem. 268:2048–2051 [PubMed] [Google Scholar]
- 37. Liu F, Roizman B. 1993. Characterization of the protease and other products of amino-terminus-proximal cleavage of the herpes simplex virus 1 UL26 protein. J. Virol. 67:1300–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCann PJ, III, O'Boyle DR, II, Deckman IC. 1994. Investigation of the specificity of the herpes simplex virus type 1 protease by point mutagenesis of the autoproteolysis sites. J. Virol. 68:526–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weinheimer SP, McCann PJ, O'Boyle DR, Stevens JT, Boyd BA, Drier DA, Yamanaka GA, DiIanni CL, Deckman IC, Cordingley MG. 1993. Autoproteolysis of herpes simplex virus type 1 protease releases an active catalytic domain found in intermediate capsid particles. J. Virol. 67:5813–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Desai P, Person S. 1999. Second site mutations in the N terminus of the major capsid protein (VP5) overcome a block at the maturation cleavage site of the capsid scaffold proteins of herpes simplex virus type 1. Virology 261:357–366 [DOI] [PubMed] [Google Scholar]
- 41. Hong Z, Beaudet-Miller M, Durkin J, Zhang R, Kwong AD. 1996. Identification of a minimal hydrophobic domain in the herpes simplex virus type 1 scaffolding protein which is required for interaction with the major capsid protein. J. Virol. 70:533–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kennard J, Rixon FJ, McDougall IM, Tatman JD, Preston VG. 1995. The 25 amino acid residues at the carboxy terminus of the herpes simplex virus type 1 UL26.5 protein are required for the formation of the capsid shell around the scaffold. J. Gen. Virol. 76(Part 7):1611–1621 [DOI] [PubMed] [Google Scholar]
- 43. Warner SC, Desai P, Person S. 2000. Second-site mutations encoding residues 34 and 78 of the major capsid protein (VP5) of herpes simplex virus type 1 are important for overcoming a blocked maturation cleavage site of the capsid scaffold proteins. Virology 278:217–226 [DOI] [PubMed] [Google Scholar]
- 44. Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a ts mutant defective in release of viral DNA and in other stages of viral reproductive cycle. J. Virol. 45:397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Newcomb WW, Booy FP, Brown JC. 2007. Uncoating the herpes simplex virus genome. J. Mol. Biol. 370:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Newcomb WW, Cockrell SK, Homa FL, Brown JC. 2009. Polarized DNA ejection from the herpesvirus capsid. J. Mol. Biol. 392:885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 20:4922–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang K, Wills E, Baines JD. 2009. The putative leucine zipper of the UL6-encoded portal protein of herpes simplex virus 1 is necessary for interaction with pUL15 and pUL28 and their association with capsids. J. Virol. 83:4557–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2:357–364 [DOI] [PubMed] [Google Scholar]
- 51. Patel AH, Rixon FJ, Cunningham C, Davison AJ. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217:111–123 [DOI] [PubMed] [Google Scholar]
- 52. Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deshayes S, Morris MC, Divita G, Heitz F. 2005. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 62:1839–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dorn A, Affolter M, Gehring WJ, Leupin W. 1994. Homeodomain proteins in development and therapy. Pharmacol. Ther. 61:155–184 [DOI] [PubMed] [Google Scholar]
- 55. Thoren PE, Persson D, Karlsson M, Norden B. 2000. The antennapedia peptide penetratin translocates across lipid bilayers—the first direct observation. FEBS Lett. 482:265–268 [DOI] [PubMed] [Google Scholar]
- 56. Roizman B, Sears A. 1996. Herpes simplex viruses and their replication, p 2231–2295 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 3rd ed Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 57. Overby LR, Robishaw EE, Schleicher JB, Rueter A, Shipkowitz NL, Mao JC. 1974. Inhibition of herpes simplex virus replication by phosphonoacetic acid. Antimicrob. Agents Chemother. 6:360–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bultmann H, Brandt CR. 2002. Peptides containing membrane-transiting motifs inhibit virus entry. J. Biol. Chem. 277:36018–36023 [DOI] [PubMed] [Google Scholar]