

Abstract
Human immunodeficiency virus type 1 (HIV-1) antagonizes innate restriction factors in order to infect and persistently replicate in a host. In a previous study, we demonstrated that HIV-1 NL4-3 with a simian immunodeficiency virus mne (SIVmne) vif gene substitution (HSIV-vif-NL4-3) could infect and replicate in pig-tailed macaques (PTM), indicating that APOBEC3 proteins are primary barriers to transmission. Because viral replication was persistent but low, we hypothesized that HSIV-vif-NL4-3 may be suppressed by type I interferons (IFN-I), which are known to upregulate the expression of innate restriction factors. Here, we demonstrate that IFN-α more potently suppresses HSIV-vif-NL4-3 in PTM CD4+ T cells than it does pathogenic SIVmne027. Importantly, we identify a variant (HSIV-vif-Yu2) that is resistant to IFN-α, indicating that the IFN-α-induced barrier can be overcome by HSIV-vif chimeras in PTM CD4+ T cells. Interestingly, HSIV-vif-Yu2 and HSIV-vif-NL4-3 are similarly restricted by PTM BST2/Tetherin, and neither virus downregulates it from the surface of infected PTM CD4+ T cells. Resistance to IFN-α-induced restriction appears to be conferred by a determinant in HSIV-vif-Yu2 that includes env su. Finally, we show that the Yu-2 env su allele may overcome an IFN-α-induced barrier to entry. Together, our data demonstrate that the prototype macaque-tropic HIV-1 clones based on NL4-3 may not sufficiently antagonize innate restriction in PTM cells. However, variants with resistance to IFN-α-induced restriction factors in PTM CD4+ T cells may enhance viral replication by overcoming a barrier early in the viral replication cycle.
INTRODUCTION
Both human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus (SIV) trigger a type I interferon (IFN-I) response during infection of the host (1–4). IFN-I is not induced in productively infected cells. Instead, plasmacytoid dendritic cells (pDCs) release large amounts of IFN-α in part through recognition of HIV-1 RNA by toll-like receptor 7 (TLR7) (5–7). The significance of the IFN response for controlling HIV-1 or SIV infection in vivo has remained unclear because IFN-I expression is elevated during acute infection and high levels of IFN-α during chronic infection typically correlate with high viral loads and rapid disease progression, suggesting that it may hasten disease (1, 4, 8, 9). However, some clinical trials with HIV-1-infected individuals have reported decreases in viral load following treatment with exogenous IFN-α, indicating that IFN-I responses may present a barrier to HIV-1 replication (10–12). Thus, the role of IFN-I responses in HIV-1 infection and disease remains poorly understood.
IFN-I has been shown to interfere with HIV-1 replication at multiple stages of the viral life cycle (13–18). Different interferon-stimulated genes (ISGs) mediate these effects. For example, ISGs have been shown to impede HIV-1 replication by interfering with (i) the entry process (e.g., IFITM2 and IFITM3 [19]); (ii) postentry processes, including disruption of the reverse transcriptase complex and synthesis of the viral cDNA (TRIM5α, APOBEC3G, and SAMHD1) (20–28); (iii) viral gene expression and protein synthesis (TRIM22 and protein kinase R [PKR]) (29–31); (iv) degradation of viral RNA (RNase L) (32); (v) Gag protein production (IFITM1) and assembly (TRIM22 and 2′,3′-cyclic-nucleotide 3′-phosphodiesterase) (19, 33, 34); (vi) virion release from cells (ISG15 and Tetherin) (35–37); and (vii) hypermutation of the viral cDNA (APOBEC3G) (38–40). The effectiveness of the IFN-I response against HIV-1 may depend largely on the combinatorial action of multiple ISGs and on the extent to which the infecting virus can counteract and evade the effect of the individual ISGs, as variant viruses may differ in their ability to antagonize targeted ISGs such as Tetherin or APOBEC3G.
The extent to which IFN-I restricts viral replication in vitro may also depend on the targeted host cell. Both HIV-1 and SIV are inhibited by IFN-I in macrophages and CD4+ T cells, but some studies suggest that inhibition may occur to a lesser extent in CD4+ T cells (4, 9, 13, 15–18, 41–49). The inhibitory effect of IFN-I suggests that HIV-1 and SIV have been under strong selective pressure by the host innate immune response to evolve mechanisms of evasion. Indeed, a gradual decrease in the number of pDCs and their ability to produce interferon occurs during the course of HIV-1 infection (50, 51). Viral infection also disrupts innate antiviral signaling via IFN regulatory factor 3 (IRF-3) by inducing its degradation (52, 53). Finally, viral regulatory proteins actively antagonized innate restriction factors, which are effector proteins of the IFN response. For example, the HIV-1 Vpu and SIV Nef proteins downregulate surface expression of bone marrow stromal cell antigen 2 (BST2/Tetherin/CD317), which would otherwise prevent release of virions from the cell surface (54–57). The importance of overcoming the restrictive activity of Tetherin for replication is highlighted by studies demonstrating that a SIVmac239 mutant with a nef deletion evolves novel compensatory mutations in env, which can neutralize the blocking activity of Tetherin (58). Additionally, Vif degrades the cellular deaminase apolipoprotein B mRNA editing enzyme 3G (APOBEC3G) (59), thereby preventing its incorporation into virions, in which it can associate with the reverse transcription complex in newly infected cells and inhibit viral replication by hypermutation and interfere with reverse transcription.
Recent studies also demonstrate that at least some innate restriction factors (e.g., APOBEC3G, TRIM5α, and Tetherin) may be significant barriers to cross-species transmission of primate lentiviruses, including HIV-1 (reviewed in reference 60). For example, HIV-1 is highly specific to replicating in human cells, and the HIV-1 Vpu and Vif proteins are adapted to antagonizing human Tetherin and APOBEC3G, respectively (54, 61–69). However, they both fail to inhibit Tetherin and APOBEC3G proteins from Old World monkeys. Additionally, TRIM5α from Old World monkeys but not humans restricts HIV-1 infection, but capsid mutations that introduce SIV capsid sequences enable the virus to evade the inhibitory effects of rhesus TRIM5α (28, 70, 71). Moreover, genetic studies of rhesus macaques demonstrate that TRIM5α alleles influence SIV transmission, viral replication, and the rate of progression to AIDS (72–75).
The potency by which these factors inhibit viral replication is further demonstrated by attempts to develop a macaque model of HIV-1 infection and disease. Early experiments showed that commonly used macaque species for AIDS studies are resistant to HIV-1, including rhesus macaques (Macaca mulatta), cynomolgus monkeys (Macaca fascicularis), and pig-tailed macaques (PTMs) (Macaca nemestrina) (76, 77). Of these three species, PTMs are the most susceptible to HIV-1, which can be transiently infected due to the absence of a postentry block to infection (78–81). In contrast to rhesus macaques, PTMs do not express TRIM5 alleles that can inhibit HIV-1 infection (82–87). Recent studies using HIV-1 derivatives with minimal SIV vif substitutions (PTM-tropic HIV-1) further indicate that inhibition of APOBEC3 proteins is central to robust replication in PTM CD4+ T cells in vitro and persistent infection of PTMs (85, 88–90). Other macaque-tropic HIV-1 derivatives with SIV capsid and vif gene substitutions are able to replicate in rhesus and cynomolgus monkey cells in vitro but do not persistently replicate in vivo (90–92). The reason why macaque-tropic HIV-1 clones replicate very well in PTM T cells in vitro but only sustain a low but chronic infection that resembles that of long-term survivors in vivo remains unclear. Although there is some indication from CD8-depletion experiments that cellular immune responses may contribute to control of macaque-tropic HIV-1 in PTMs (88), it is possible that macaque-tropic HIV-1 derivatives do not adequately escape suppression by IFN-I in macaque cells.
In the current study, we investigated the effect of IFN-α on replication of PTM-tropic HIV-1 clones in PTM CD4+ T cells in order to further define the restrictive barriers to HIV-1 replication in macaque cells. We demonstrate that in contrast to a pathogenic SIVmne clone that is adapted to replicate in PTMs, PTM-tropic HIV-1 clones based on NL4-3 (HSIV-vif-NL4-3 and HSIV-vif-NLAD8) are highly restricted by IFN-α in PTM CD4+ T cells, suggesting that the inability of these clones to overcome the IFN-I response may contribute to the attenuated phenotype in vivo. Furthermore, we demonstrate that another variant PTM-tropic HIV-1 based on Bru-Yu2 is able to replicate to high levels during an IFN-α-induced restrictive state, and we show that its envelope (Env) protein confers resistance to IFN-α, indicating the potential for HIV-1 to evade the IFN-α-induced antiviral state in macaque cells.
MATERIALS AND METHODS
Cell lines.
293T and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS), 2 mM glutamine, and p/s (100 U of penicillin per ml and 100 mg of streptomycin per ml) (DMEM complete). TZM-bl cells were obtained from the NIH AIDS Reagent Program and grown in DMEM complete (93, 94). The immortalized PTM CD4+ T cells were obtained from Hans-Peter Kiem (Fred Hutchinson Cancer Research Center) and maintained in Iscove's modified Dulbecco's medium (IMDM) containing 10% HI-FBS, 2 mM glutamine, p/s, and 100 U/ml human interleukin 2 (IL-2) (Roche) (IMDM complete medium) (95).
Tetherin/BST2 constructs.
The human and PTM BST2 cDNAs (huBST2 and PTM BST2) were amplified by reverse transcription (RT)-PCR from mRNA isolated from human and PTM peripheral blood mononuclear cells (PBMCs), respectively, using primers that add the coding sequence for a hemagglutinin (HA) epitope tag at the 5′ end. The forward and reverse primers for huBST2 amplification are 5′-ATTCGGATCCGCCACCATGTACCCATACGACGTCCCAGACTACGCTATGGCATCTACTTCGTAT GAC-3′ (BamHI) and 5′-TAATCTCGAGTCACTGCAGCAGAGCGCTGAG-3′ (XhoI), respectively. The forward and reverse primers for ptBST2 amplification are 5′-ACTCGGATCCGCCACCATGTACCCATACGACGTCCCAGACTACGCTATGGCACCTATTTTGTATG-3′ (BamHI) and 5′-TAATCTCGAGTCACAGCAGCAGAGCGCTCAAG-3′ (XhoI), respectively. The sequences of huBST2 and PTM BST2 were first cloned into TOPO vector pCR 2.1 (Invitrogen) for sequencing and then subcloned into BamHI and XhoI sites of mammalian expression vector pcDNA3.
Proviral clones and generation of virus stocks.
The construction of HIV-1/SIV vif (HSIV-vif) chimeras based on HIV-1 molecular clones NL4-3, Bru-Yu2, and NL-AD8 has been previously described (85). Chimeric viruses of HSIV-vif-NL4-3 and HSIV-vif-Yu2 were generated by using unique restriction enzyme sites in the genome. NL-Yu2 and Bru-NL clones were generated by reciprocal exchange of a BssHII to BstBI gag-pol-vif fragment between HSIV-vif-NL4-3 and HSIV-vif-Yu2. env chimeric viruses were generated by cloning SalI-BamHI, SalI-BsaBI, and BsaBI-BamHI fragments of HSIV-vif-Yu2 into the HSIV-vif-NL4-3 proviral clone, separately. The resulting clones contain the HSIV-vif-Yu2 vpu-env (Yu-envL), vpu-env su (Yu-envS), or env tm (Yu-envT) sequences in the background of HSIV-vif-NL4-3. Bru-nef was generated by cloning the BstBI to BamHI (vif to env tm) fragment of HSIV-vif-NL4-3 into the NL-Yu2 proviral clone. This results in a clone with the nef-LTR sequences of HSIV-vif-Yu2 in the background of HSIV-vif-NL4-3. To generate Env mutant clones HSIV-vif and HSIV-vif-Yu2 and Vpu-corrected clone HSIV-vif-Yu2, EcoRI to BamHI fragments of each of the proviral clones were subcloned into pCR2.1 Topo vector. The start codon of env and vpu were mutated using Quikchange mutagenesis (Agilent), and the mutated SalI and BamHI fragments were cloned back into the respective proviral clones. Virus stocks were generated by transfection of 293T cells. Infectious titers were determined by limiting-dilution infection analysis using TZM-bl indicator cells (93, 94), and the amount of virus in supernatants was measured by HIV-1 p24gag antigen enzyme-linked immunosorbent assay (ELISA) (Advanced Bioscience Laboratories).
Viral replication in PTM CD4+ T cells and human PBMCs.
For viral replication assays, human PBMCs and PTM CD4+ T cells were infected in triplicate cultures using previously described methods (85, 118, 119). Approximately 1 × 106 costimulated human PBMCs in RPMI complete medium were infected at a multiplicity of infection (MOI) of 0.01 in the presence or absence of 200 U/ml IFN-α (Sigma). To compare viral replication in PTM CD4+ T cells, approximately 2.5 × 105 or 5 × 105 cells in IMDM complete were infected with viruses at an MOI of 0.01. After 3 h of incubation, the cells were washed once with phosphate-buffered saline (PBS) and once with complete RPMI medium to remove unbound virus. Infected cells were then resuspended in fresh medium in the presence or absence of 200 U/ml IFN-α. To monitor the replication of HSIV-vif clones or SIVmne027 (120), supernatants were harvested every 2 to 4 days and the amounts of HIV-1 p24gag antigen or SIV p27gag antigen, respectively, were determined using commercially available ELISAs (Advanced Bioscience Laboratories).
Virion release restriction by BST2/Tetherin.
293T cells were seeded in 24-well plates at a density of 2.5 × 105 per well in 0.5 ml medium. The following day, cells were cotransfected with 100 ng of HSIV-vif clones and expression vectors for huBST2 or PTM BST2 (0, 2, 50, 100, and 200 ng) using X-tremeGENE 9 DNA transfection reagents (Roche). Differences in the amount of plasmid DNA in each transfection were offset by the addition of empty Bluescript PKS+ vector or pcDNA3 vector. At 48 after transfection, the amount of virus released into the cell supernatant was measured by HIV-1 p24 antigen ELISA.
BST2/Tetherin downregulation assay.
PTM CD4+ T cells (2.5 × 105 cells per well in 24-well plates) were infected with SIVmne027, HSIV-vif-Yu2, or HSIV-vif-NL4-3 at an MOI of 0.01 or 0.05 in the absence or presence of IFN-α. Every 3 days over a 2-week period, the medium was replaced with new medium lacking or containing IFN-α. At the same time, the cells were collected and stained with phycoerythrin (PE)-conjugated anti-human BST2 antibody (CloneRS38E; Biolegend for 30 min at 4°C. The cells were then washed, fixed in 1% paraformaldehyde PBS, and analyzed on a BD CantoII flow cytometer, and the flow data were processed using FlowJo software.
For 293T or HeLa cells, 2.5 × 105 or 5 × 104 cells, respectively, were seeded into 24-well plates. The following day, 293T cells were cotransfected with 500 ng of proviral plasmids and 50 ng of expression vectors for either hBST2 or ptBST2. HeLa cells were transfected with 500 ng proviral plasmids alone. At 48 h posttransfection, cells were trypsinized and stained by anti-BST2-PE antibody and then analyzed by flow cytometry.
siRNA knockdown of BST2 in PTM CD4+ T cells.
Short interfering RNA (siRNA) oligonucleotides were designed (Dharmacon siDESIGN Center) to target ptBST2 327 to 345 nt (GGCCCAAGGACGAAAGAAA) or a random control sequence. Short hairpin RNA (shRNA) oligonucleotides were then designed from siRNA oligonucleotides as previously described (96). The shRNA oligonucleotide sequences were first cloned into vector PBS-hU6-1 and then subcloned into lentivector FG12 (96).
Pseudotyped lentivirus particles were produced in 293T cells by cotransfection of 293T cells with FG12/shRNA plasmids and a packaging vector (pMDLg/pRRE), vesicular stomatitis virus glycoprotein expression vector (CMV-VSV-G), and Rev expression vector (RSV-Rev). One day before transfections, 8 × 106 293T cells in 10 ml of DMEM without antibiotics were plated in 10-cm culture dish so that the cells would be approximately 90% confluent at the time of transfection. One hour before transfection, the cells were first pretreated by the medium containing 25 μM chloroquine, and then 10 μg of each of the plasmids was cotransfected by a modified calcium phosphate procedure. After 8 h, the medium was replaced with 8 ml of complete medium and cultured in an incubator at 32°C with 5% CO2. The supernatants were collected 48 h and 72 h after transfection, filtered (0.45-μm cellulose acetate membrane filter; VWR), and then used to transduce PTM CD4+ T cells or stored at −80°C until use. The GFP+ cells were analyzed by flow cytometry, and then the cells with the top 20% fluorescence intensity were sorted by flow cytometry (EPICS-Altra; Beckman-Coulter). All postsorting PTM CD4+ T cell pools were approximately 99% GFP+. The levels of ptBST2 downregulation were assessed by flow cytometry using anti-BST2-PE monoclonal antibody (MAb) staining.
Transmission electron microscopy.
PTM CD4+ T cells were infected with SIVmne027, HSIV-vif-NL4-3, or HSIV-vif-Yu2 at an MOI 0.01. After 11 days of infection, cells were treated with 200 U/ml IFN-α. Twenty-four hours posttreatment, the infected cells were pelleted, washed twice in 1× Dulbecco's PBS (DPBS), and fixed overnight in 0.1 M Millonig's phosphate buffer (pH 7.4) containing 2% formaldehyde and 2.5% glutaraldehyde. The pelleted cells were then rinsed 3 times in 0.1 M Millonig's phosphate buffer and postfixed in 0.1 M Millonig's phosphate buffer containing 1% OsO4. Pellets were dislodged from tubes and minced into 1-mm blocks 30 min into the postfix and then allowed to fix another 30 min in the osmium solution. Cell pellets were dehydrated through a gradient series of ethanols (50% to 100%), and en bloc stained with a saturated uranyl acetate solution during the 50% ethanol step. After complete dehydration, the cell pellets were infiltrated in progressively concentrated mixtures of ethanol and Spurr's Low Viscosity resin and left on a shaker in 1 part ethanol to 3 parts resin overnight. Gradual infiltration resumed the following day until the tissue was finally placed in 3 changes of pure resin for 2 h each. After embedding in freshly made resin, the pellet blocks were polymerized at 68°C for 24 h.
Thin sections (65 to 70 nm) were obtained using a Diatome Ultra 45 diamond knife and an RMC MT-6000-XL ultramicrotome and were collected on 150-hexagonal-mesh copper grids. The sections were counterstained with Reynold's lead citrate for 4 min. Dry samples were examined on a Hitachi H7500 transmission electron microscope, and images were captured using a Gatan US1000 digital camera with Digital Micrograph v1.82.366 software.
Virion fusion assay.
The HIV-1 virion fusion assay was performed as previously described (97). To generate HIV-1 particles containing β-lactamase-Vpr (BlaM-Vpr) chimera proteins, 293T cells were cotransfected with 12 μg of proviral DNA plasmids and 4 μg of pCMV-BlaM-Vpr DNA using the calcium phosphate transfection method. Eight hours posttransfection, cells were washed once and then replenished with DMEM complete. After 48 h, the culture supernatants were collected, centrifuged at low speed, clarified by passage through a 0.45-μm filter, and stored at −80°C.
Approximately 2.5 × 105 PTM CD4+ T cells treated with IFN-α for 24 h or untreated cells in 100 μl media (24- or 48-well plate) were infected with equal amounts of virus in the presence of protamine sulfate salt (8 μg/ml). Three hours postinfection, cells were washed with CO2-independent medium (Invitrogen) and loaded with 100 μl CCF2/AM substrate (Invitrogen) for 1 h at room temperature in the dark. The loading solution was prepared by mixing 2 μl of 1 nM CCF2/AM with 8 μl of 0.1% acetic acid containing 100 mg/ml Pluronic-F127 surfactant (solution B, provided by Invitrogen with the CCF2/AM loading kit) and 1 ml of CO2-independent medium. After 1 h, cells were washed with 200 μl of development medium, and the BlaM reaction was developed in 200 μl development medium for 16 h at room temperature in the dark. The development medium was prepared by mixing 10 μl of 250 mM probenecid (one vial in 1 ml Hanks balanced salt solution [HBSS] buffer) with 1 ml CO2-independent medium and 100 μl tetracycline-free FBS. Cells were then washed once with cold 1× PBS containing 2% FBS and fixed with 2% paraformaldehyde. The levels of CCF2/AM and its cleaved products were measured by flow cytometry.
Coreceptor expression on the cell surface of IFN-α-induced PTM CD4+ T cells.
PTM CD4+ T cells (2 × 105 cells per well) in 24-well plates were cultured in the presence of absence of 200 U/ml of IFN-α. At 24 and 48 h, the cells were collected, stained with anti-human CD4-PE (clone RPA-T4), anti-human-CXCR4-APC (Clone 12G5), or anti-human CCR5-PE (clone 3A9), and analyzed by flow cytometry. Anti-human-IG1-PE and anti-human IG1-APC (clone MOPC-21) were used as isotype staining controls. All antibodies were purchased from BD Pharmingen.
IFN-α ELISA.
The amount of IFN-α in the cell culture supernatant collected from HSIV-vif-infected cells was determined using the high-sensitivity VerikineTM IFN-α ELISA kit (PBL Interferon Source) according to the manufacturer's protocol.
Statistical analysis.
One-way analysis of variance (ANOVA) models with virus type as the fix factor were used to compare the mean IFN-α-induced inhibition values among virus types. As the significant unequal variance among virus types, weighted analysis was used. The inverse of the within-group variance is used as weight. After the overall test, the comparisons for each variant to the wild-type virus were made while adjusting the multiple comparisons by Dunnett's procedure. Calculations were performed using SAS 9.3.
RESULTS
Inhibition of HIV-1 replication in PTM T cells by IFN-α.
In previous studies, we showed that a PTM-tropic HIV-1, HSIV-vif-NL4-3, could replicate in PTM PBMCs in vitro (85). While it also persistently replicated in PTMs in vivo, viral loads were controlled during the later stages of infection and remained low, with intermittent blips. To determine if IFN-α may have a role in limiting replication of a PTM-tropic HIV-1, we compared the replication of HSIV-vif-NL4-3 with that of a pathogenic SIVmne variant, SIVmne027 (120), in PTM CD4+ T cells in the presence and absence of IFN-α (Fig. 1A and B). In the SIVmne027-infected cultures, high levels of SIV p27gag antigen accumulated rapidly in the supernatants through 7 days postinfection, demonstrating rapid replication kinetics. The level of virus decreased thereafter in association with virus-induced cell killing. IFN-α reduced replication of SIVmne027 by only approximately 25%. In contrast, accumulation of HIV p24gag in HSIV-vif-NL4-3-infected cultures was delayed, peaking at 20 days postinfection. Quite strikingly, HSIV-vif-NL4-3 was significantly reduced by IFN-α. These data suggest that a major phenotypic difference between pathogenic SIVmne027 and the PTM-tropic HIV-1 clone, HSIV-vif-NL4-3, is sensitivity to inhibition by IFN-α. Thus, the suppressive effects of IFN-α may contribute to the control of HSIV-vif-NL4-3 in the PTM host.
Fig 1.

Replication of HSIV-vif clones in PTM CD4+ T cells in the presence or absence of IFN-α. PTM CD4+ T cell cultures were infected with the indicated viruses at an MOI of 0.01 and cultured in the presence or absence of 200 U/ml IFN-α. Viral replication was measured by assaying culture supernatants for SIV p27 ELISA for SIVmne027 (A) or HIV p24 ELISA for HSIV-vif-NL4-3 (B), HSIV-vif-Yu2 (C), and HSIV-vif-NLAD8 (D). The average SIV p27 or HIV p24 values ± standard deviations (SD) are shown. Similar results were observed in two additional experiments.
To determine if other HSIV-vif clones are potently restricted by IFN-α, we also examined the effects of IFN-α on chimeric viruses based on the CCR5-tropic HIV-1 Bru-Yu2 (HSIV-vif-Yu2) and NL-AD8 (HSIV-vif-NLAD8) clones in PTM CD4+ T cells. Interestingly, the HSIV-vif-Yu2 clone replicated to higher levels and with more rapid kinetics than HSIV-vif-NL4-3 in the absence of IFN-α (Fig. 1C). Furthermore, it demonstrated resistance to the inhibitory effects of IFN-α, achieving a level of replication that was about 65% of the level observed in the absence of IFN-α. In contrast, HSIV-vif-NLAD8 demonstrated a phenotype similar to that of HSIV-vif-NL4-3, showing slower replication kinetics and greater sensitivity to inhibition by IFN-α (Fig. 1D). The data indicate that while IFN-α can potently inhibit HIV-1 replication in PTM CD4+ T cells, variant viruses may have the capacity to evade the effects of the IFN response. Additionally, the data also indicate that CCR5 tropism is not sufficient for evasion of IFN-α-induced restriction.
To determine if the slow replication kinetics of HSIV-vif-NL4-3 compared to that of HSIV-vif-Yu2 in the absence of exogenously added IFN-α in PTM CD4 T cells was due to induction of IFN-α by viral infection, we assayed culture supernatants by a sensitive IFN-α ELISA that can detect IFN levels as low as 12.5 pg/ml. At both early and late time points postinfection, supernatants from cells infected with either virus were negative for IFN-α (data not shown). Thus, any difference in the replication kinetics of HSIV-vif and HSIV-vif-Yu2 viruses in PTM CD4 T cells is not the result of induction of detectable levels of IFN-α.
Env of HIV-1 Yu-2 confers resistance to IFN-α in PTM CD4+ T cells.
To map the IFN-α resistance determinant, we generated chimeric viruses between the IFN-α-sensitive virus, HSIV-vif-NL4-3, and the IFN-α-resistant virus, HSIV-vif-Yu2 (Fig. 2A) and examined the replication of each chimera in the presence or absence of IFN-α in PTM CD4+ T cells. Similar to what was observed in the experiment illustrated in Fig. 1, replication of HSIV-vif-NL4-3 (NL4-3) was inhibited by IFN-α (Fig. 2B) more than HSIV-vif-Yu2 (Bru-Yu2) (Fig. 2C). Interestingly, the chimera that included the gag-pol region of Bru-Yu2 in the background of NL4-3 (Bru-NL) was strongly inhibited by IFN-α (Fig. 2D). In contrast, the reciprocal chimera (NL-Yu2) was more resistant to IFN-α, like the parental HSIV-vif-Yu2 clone (Fig. 2E), suggesting that the IFN-α resistance determinant is located within the 3′ half of the genome.
Fig 2.

Mapping the IFN-α resistance determinant. (A) Diagram of HSIV-vif chimeric clones. (B to I) Replication of each chimeric virus in PTM CD4+ T cells in the presence or absence of IFN-α. Infections were carried out as described for Fig. 1. Viral replication was measured by assaying culture supernatants for HIV p24 by ELISA. The average HIV p24 values ± SD are shown and are representative of three different experiments.
As neither of the HSIV-vif clones used in these experiments express the Vpr protein (data not shown) and the HSIV-vif-Yu2 clone contains the vpu allele from HIV-1 Yu2, which has a natural mutation in the initiation codon, we examined whether the Yu2 env or nef genes play a role in conferring resistance to IFN-α. When the complete env gene of Yu2 was inserted into the background of HSIV-vif-NL4-3, the virus (Yu-envL) showed a level of resistance to IFN-α that was similar to that of the HSIV-vif-Yu2 parent virus (Fig. 2C and F). We therefore subdivided the Yu2 env, inserting either the env su region or the env tm region alone into the background of HSIV-vif-NL4-3 (Yu-envS or Yu-envT, respectively), and tested the resulting chimeras for IFN-α resistance in the PTM CD4+ T-cell lines. Although there was a reduction in the level of replication in the presence of IFN-α compared to either the parental HSIV-vif-Yu2 or a chimera containing the full-length Yu2 env gene, env su of Yu2 (Yu-envS chimera) was sufficient for overcoming the inhibitory effects of IFN-α (Fig. 2G). On the other hand, the chimera with just the env tm of Yu2 (Yu-envT) was almost completely inhibited by the addition of IFN-α (Fig. 2H). Replacement of the nef allele of the HSIV-vif NL4-3 chimera with that from HIV-1 Bru-Yu2 (Bru-nef) also did not enhance replication in the presence of IFN-α (Fig. 2I). Interestingly, the Yu-2 env-containing determinant also enhanced virus production in IFN-α-treated costimulated human PBMCs, suggesting that its effect may not be species-specific (data not shown).
Because Env expression can be altered by mutations that affect vpu translation (98, 99), we restored the vpu start codon in the HSIV-vif-Yu2 clone to determine if it would affect IFN-α resistance. However, replication of HSIV-vif-Yu2 was not affected by the vpu mutation in either the presence or the absence of IFN-α (Fig. 3). Passaging HSIV-vif-Yu2 in PTM CD4+ T cells also did not reverse the vpu start codon mutation (data not shown). Finally, restoring Vpr expression in either HSIV-vif parental clone did not enhance viral replication in IFN-α-treated PTM CD4+ T cells (data not shown). Together, these data indicate that the Env SU protein of HIV-1 Yu2 supports evasion of the IFN-α response in PTM CD4+ T cells.
Fig 3.
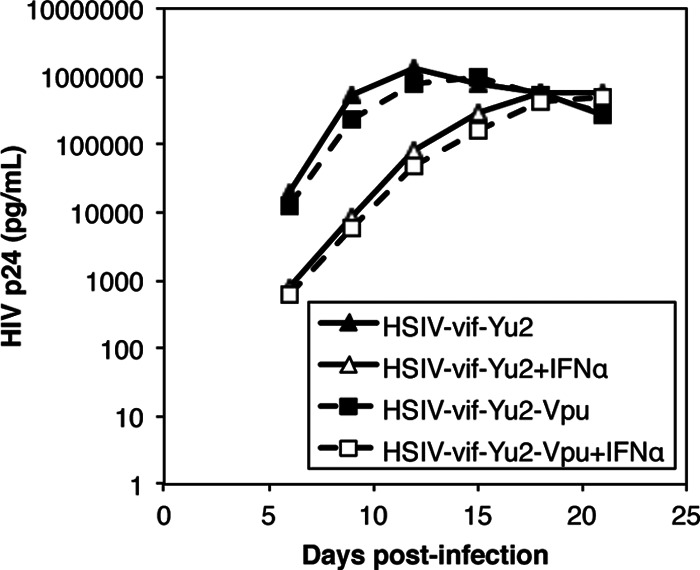
Replication of HSIV-vif-Yu2 and a vpu reversion mutant in pig-tailed macaque CD4+ T cells. PTM CD4+ T cells were infected with the indicated viruses at an MOI of 0.01 and cultured in the presence or absence of 200 U/ml IFN-α. Viral replication was measured by assaying culture supernatants by HIV p24 ELISA. The average HIV p24 values ± SD are shown. Similar results were observed in two additional experiments.
HSIV-vif virions accumulate at the surface of IFN-α-treated PTM CD4+ T cells.
Previous studies indicated that a major block to HIV-1 replication induced by IFN-α occurred at the stage of virion release and caused accumulation of virions at the surface of cells (55). Additionally, studies indicated that the HIV-2Rod env allele promotes virion release (100, 101). Thus, we examined whether IFN-α differentially affected virion release of HSIV-vif-NL4-3 and HSIV-vif-Yu2 by electron microscopy. Figure 4 shows some accumulation of both HSIV-vif-NL4-3 and HSIV-vif-Yu2 at the surface of infected PTM CD4+ T cells after treatment with IFN-α compared to the untreated control cells. In contrast, increased tethering of virions was not observed on cells infected with SIVmne027. These data suggest that enhanced replication of HSIV-vif-Yu2 in the presence of IFN-α may not be due to escape from virion-tethering mechanisms.
Fig 4.
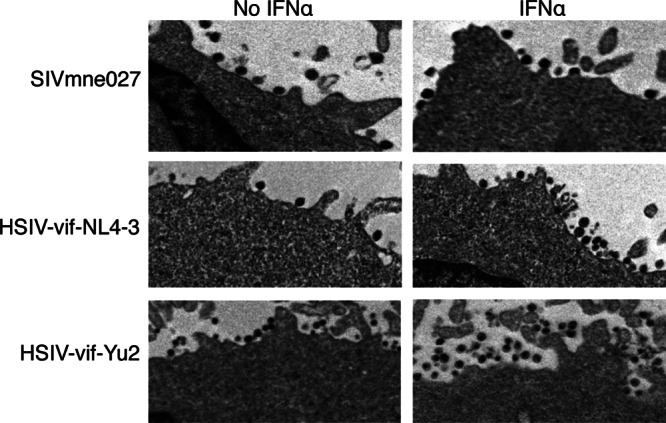
Accumulation of HSIV-vif and HSIV-vif-Yu2 virions at the surface of PTM CD4+ T cells. PTM CD4+ T cells were infected with SIVmne027, HSIV-vif-NL4-3, or HSIV-vif-Yu2 at an MOI of 0.01. At 11 days postinfection, infected cells were treated with IFN-α for 24 h, collected, fixed, and analyzed by transmission electron microscopy.
HSIV-vif clones are restricted by PTM BST2/Tetherin.
As BST2/Tetherin is known to mediate IFN-induced accumulation of virions at the surface of cells (35, 56), we first examined the effect of PTM BST2 on the release of both HSIV-vif-NL4-3 and HSIV-vif-Yu2 in 293T cells. Each proviral clone was cotransfected with increasing amounts of a PTM BST2 expression vector (ptBST2), and the results were compared with those from inhibition by human BST2 (hBST2). As expected, HSIV-vif-NL4-3 was more resistant to inhibition by hBST2 than was HSIV-vif-Yu2, which has a natural mutation in the vpu start codon (Fig. 5A). Interestingly, virion release of the two HSIV-vif clones was inhibited to similar levels by ptBST2, which was approximately 100-fold at the highest level of ptBST2 used in the assay compared to the maximal release in the absence of ptBST2. Additionally, neither virus downregulated BST2 surface expression in infected PTM CD4+ T cells (Fig. 5B) or when cotransfected with PTM BST2 into 293T cells (data not shown). Thus, HSIV-vif-NL4-3 and HSIV-vif-Yu2 demonstrated similar susceptibilities to restriction by ptBST2.
Fig 5.

Interactions of HSIV-vif clones and PTM BST2/Tetherin. (A) Suppression of HSIV-vif-NL4-3 and HSIV-vif-Yu2 release by PTM and human BST2. 293T cells were cotransfected with either of the HSIV-vif proviral clones and increasing amounts of a human or PTM BST2 expression vector. Virus production was measured by assaying supernatants by HIV p24 ELISA. (B) Surface expression of BST2 on infected PTM CD4+ T cells was examined by flow cytometry at peak virus production using an anti-BST2-PE MAb or isotypic control MAb.
Because the HSIV-vif-Yu2 clone also escapes suppression by IFN-α in human PBMCs even though it does not express Vpu, we compared the ability of the different HSIV-vif clones to downregulate endogenous human BST2/Tetherin and enhance virion release in HeLa cells. Cell surface downregulation of BST2 was observed only with proviral clones that expressed Vpu (NL4-3, HSIV-vif-NL4-3, HSIV-vif-NL4-3ΔEnv, and HSIV-vif-Yu2-Vpu) but not with proviral clones that did not express Vpu (NL4-3 ΔVpu, HSIV-vif-Yu2, HSIV-vif-Yu2-ΔEnv, and Yu-envL) (Fig. 6A). Importantly, we did not observe any notable difference in the cell surface downregulation of BST2 or virion release between wild-type HSIV-vif clones and their env deletion mutants (HSIV-vif-NL4-3 versus HSIV-vif-NL4-3ΔEnv, and HSIV-vif-Yu2 versus HSIV-vif-Yu2-ΔEnv), indicating that HIV-1 Env may not be playing an appreciable role in antagonizing BST2/Tetherin activity for enhanced virion release (Fig. 6A and B). Furthermore, virion release was enhanced only by Vpu and not by Env. Together, these data indicate that the Env-mediated IFN-α resistance of HSIV-vif-Yu2 is not due to an anti-BST2/Tetherin activity.
Fig 6.

Downregulation of BST-2/Tetherin and release of HSIV-vif clones in HeLa cells. HeLa cells were transfected with the indicated proviral clones, and 48 h posttransfection, cells and culture supernatants were collected and analyzed for surface expression of BST2 by flow cytometry (A) and HIV p24 by ELISA (B), respectively.
Finally, to further examine the role of BST2 on the replication of HSIV-vif clones in PTM CD4+ T cells, we compared viral replication in cells transduced with vectors expressing control or PTM BST2-targeting shRNAs. Although the BST2 shRNA downregulated the surface expression of PTM BST2 compared to what was seen in control transduced cells (Fig. 7A), we did not observe a significant difference in the replication of HSIV-vif-NL4-3 or HSIV-vif-Yu2 in the BST2 knockdown or control PTM CD4 T cells in the presence or absence of IFN-α (Fig. 7B). These data further suggest that the IFN-α resistance exhibited by HSIV-vif-Yu2 virus in PTM CD4+ T cells may not be due to inhibition of BST2 function.
Fig 7.
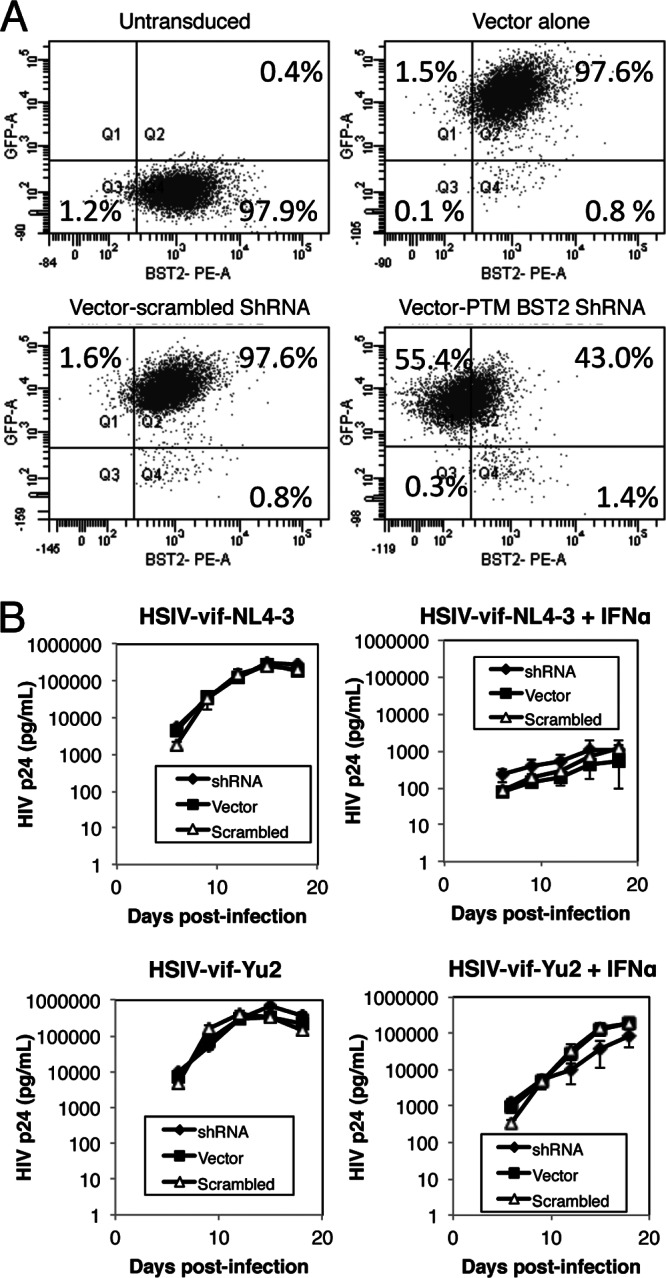
Knockdown of pig-tailed macaque BST2/Tetherin and replication of HSIV-vif clones. (A) PTM CD4 T cells were transduced with lentiviral vectors encoding BST2 shRNA, scrambled shRNA control, or control empty vector and the GFP+ cells enriched by cell sorting. The levels of PTM BST2 downregulation were assessed by flow cytometry and compared to those of untransduced cells. (B) Transduced PTM CD4+ T cells were infected with HSIV-vif-NL4-3 and HSIV-vif-Yu2 viruses at an MOI of 0.01 in the presence and absence of IFN. Viral supernatants collected at various days postinfection were assayed by HIV-1 p24 ELISA.
IFN-α does not affect coreceptor expression.
Since HSIV-vif-NL4-3 and HSIV-vif-Yu2 use CXCR4 and CCR5, respectively, as coreceptors for entry, we asked whether IFN-α could be downregulating expression of CXCR4 but not CCR5. Both PTM CD4+ T cells and human PBMCs were incubated in the presence or absence of IFN-α for 48 h, and surface expression of CXCR4 and CCR5 was examined by flow cytometry. Consistent with previous studies (16), we did not observe a decrease in expression of either coreceptor on PTM CD4+ T cells or human T cells following incubation with IFN-α (Fig. 8 and data not shown), suggesting that susceptibility or resistance to IFN-α treatment exhibited by HSIV-vif clones is not due to altered coreceptor expression.
Fig 8.

IFN-α does not affect CD4 or coreceptor expression on pig-tailed macaque CD4+ T cells. Cells were treated with IFN for 24 h, and then the cell surface expression of CD4, CXCR4, and CCR5 was determined by flow cytometry.
HSIV-vif-NL4-3 is restricted at entry by IFN-α.
Because the IFN-α resistance determinant mapped to a fragment containing the env of Yu2, we next determined whether there was any difference in the relative IFN-α-induced inhibition of entry of PTM CD4+ T cells between the parental and chimeric HSIV-vif clones by using the Blam-Vpr fusion assay. Figure 9 shows the relative decrease in infectivity induced by IFN-α in PTM CD4+ T cells for each virus. Compared to infection by HSIV-vif-NL4-3, which was reduced by approximately 39%, infections by all the clones carrying the Yu2 env su (HSIV-vif-Yu2, NL-Yu2, Yu-envL, and Yu-envS) were inhibited by an average of 10% or less by IFN-α (Fig. 9A and B). These results were statistically highly significant. Clones encoding the 3′ half of NL4-3, the Yu2 env tm, or Bru-nef were not significantly different from HSIV-vif-NL4-3. These data are in agreement with the replication data in Fig. 2 and suggest that the Yu-2 env determinant may enhance IFN-α resistance in PTM CD4+ T cells by overcoming a factor that limits entry.
Fig 9.

Effects of IFN-α on early events in HSIV-vif infection of PTM CD4+ T cells. (A and B) PTM CD4+ T cells, treated with IFN-α for 24 h or mock treated, were infected with Blam-Vpr fusion protein-containing viruses. After 3 h of infection, cells were incubated with the CCF2/AM substrate for 1 h, and the BlaM reaction was developed for 16 h. The levels of CCF2/AM and its cleaved products were measured by flow cytometry to enumerate cells that virus had entered. (A) Data from a representative infection of PTM CD4+ T cells with Blam-Vpr-HSIV-vif-NL4-3 and Blam-Vpr-HSIV-vif-Yu2. (B) The decrease in viral infectivity of each Blam-Vpr-HSIV-vif clone due to IFN-α is shown relative to its infectivity in the absence of IFN-α. The averages ± standard deviations of three to five independent experiments using the indicated Blam-Vpr-HSIV-vif clones are shown. (C) Inhibition of infection of VSV-G-NL4-3-luc by IFN-α. PTM CD4+ T cells were treated with IFN-α 24 h prior to infection or 3 h or 48 h postinfection. Infection was determined by luciferase expression. Averages of three independent infections ± standard deviations are shown.
To examine whether IFN-α might also suppress other early stages of infection in PTM CD4+ T cells in a manner that is independent of the HIV-1 Env protein, we infected cells with VSV-G pseudotyped NL4-3-luc either before or after treatment with IFN-α. Treatment of the PTM CD4+ T cells with IFN-α prior to infection and at 3 h postinfection reduced luciferase expression by 74 and 79%, respectively (Fig. 9C), while addition of IFN-α at 48 h postinfection reduced luciferase activity by 47%. These data suggest that in addition to inducing partial inhibition at entry of some HIV-1 Env variants, IFN-α could be interfering with other early postentry steps leading to and including integration and gene expression.
DISCUSSION
In recent studies, we and others have tested the hypothesis that counteracting innate restriction barriers is necessary for cross-species transmission of primate lentiviruses by engineering novel HIV-1 chimeras with minimal SIV gene substitutions that enable the virus to counteract or evade specific restriction factors and by testing their replication capacity in macaque T cells (85, 88–91, 121). Some of these clones were designed to counteract macaque APOBEC3 proteins with a SIV vif substitution and evade rhesus or cynomolgus TRIM5α-mediated restriction with a CA mutation. Alternatively, other clones included only the SIV vif substitution, and pig-tailed macaque T cells were used because they do not express TRIM5α protein or TRIM5-cyclophilin A (TRIM-cyp) fusion protein, proteins that inhibit HIV-1. While each virus persistently infects macaque T cells in vitro, when a macaque host is infected, replication of these viruses is eventually controlled. Importantly, none of these previous studies evaluated whether macaque-tropic HIV-1 clones remain sensitive to restriction by IFN-α in macaque cells. In the present study, we demonstrate that our prototype PTM-tropic HIV-1 clone based on NL4-3, HSIV-vif-NL4-3, is suppressed by IFN-α, in contrast to the pathogenic SIVmne027, in PTM CD4+ T cells, suggesting that HIV-1 may be highly sensitive to IFN-α in a nonnatural host, and perhaps explaining why it is easily controlled in vivo. These data confirm a recent study by Bitzegeio et al., which demonstrated that HIV-1 and SIVmac have become adapted to overcome the IFN response in their natural host cells but are sensitive to IFN-induced restriction factors in the nonnatural hosts (102). However, we also show that a novel variant PTM-tropic HIV-1 is capable of overcoming inhibition by IFN-α in PTM CD4+ T cells. Furthermore, IFN-α resistance of this virus is conferred by a determinant within the env sequence, which may counteract a factor hindering an early stage of the viral life cycle.
In our previous study, we had constructed HSIV-vif clones based on HIV-1 Bru-Yu2 (HSIV-vif-Yu2) and NL-AD8 (HSIV-vif-NLAD8) in addition to NL4-3 (HSIV-vif-NL4-3) (85). Here, we present evidence that the HSIV-vif-Yu2 clone is resistant to the IFN-α-induced antiviral state in PTM CD4+ T cells compared to HSIV-vif-NL4-3 or HSIV-vif-AD8. Because HSIV-vif-Yu2 does not express either Vpu or Vpr, we could rule out a role for these accessory proteins in enabling viral replication in IFN-α-treated PTM CD4+ T cells. Restoring Vpr expression in either HSIV-vif clone did not increase viral replication, further indicating that Vpr is not necessary for overcoming the suppressive activity of IFN-α. Interestingly, HSIV-vif-Yu2 and the parental HIV-1 Bru-Yu2 clones are also resistant to the suppressive effects of IFN-α in human PBMCs, indicating that Bru-Yu2 may encode unique determinants for escaping the inhibitory effects of IFN-α independent of Vpu.
In the HIV-1 genome, the 3′ end of the vpu gene overlaps the env gene, and both genes are expressed from a bicistronic mRNA via leaky scanning by ribosomes (103, 104). It has been reported that the mutations that disrupt Vpu expression result in increased translation of env (98, 99), which can enhance viral infectivity. Additionally, Vpu-defective viruses appear to spread more efficiently in T cells via cell-to-cell transfer (105, 106). Interestingly, we found that passaging HSIV-vif-Yu2 in either the presence or the absence of IFN-α in PTM CD4+ T cells did not correct for the mutation in the vpu start codon. Furthermore, reversion of the start codon mutation by site-directed mutagenesis did not have an effect on viral replication. While we cannot rule out a potential role for efficient cell-to-cell transfer of virus as a significant mechanism of replication of the HSIV-vif-Yu2 clone, these data suggest that Vpu may be unnecessary for replication of HSIV-vif-Yu2 in PTM CD4+ T cells in vitro and that a Vpu-mediated effect on Env probably does not play a role in IFN-α resistance.
It is possible that greater expression of Env could enhance production, assembly, and budding of virions. The Env protein may saturate the effect of a restriction factor that might block the release of progeny virions. However, we found that deletion of the env gene from HSIV-vif-Yu2 did not reduce virion release from HeLa cells and the addition of the Yu-2 env to HSIV-vif-NL4-3 did not enhance its release.
An earlier study of SIVmac demonstrated that novel mutations evolve in env to counteract BST2/Tetherin when its primary antagonist, Nef, is deleted (58), indicating the importance of antagonizing Tetherin for viral replication and escaping the antiviral effect of IFN-α. Another recent study showed that some HIV-1 vpu alleles may counteract macaque Tetherin and enhance replication and pathogenesis of SHIV chimeras in vivo (107). We show that the resistance of HSIV-vif-Yu2 to IFN-α does not appear to be due to antagonizing BST2/Tetherin. Several lines of evidence support this conclusion. First, IFN-α treatment increased the amount of virions attached to the surface of PTM CD4+ T cells infected with either HSIV-vif-Yu2 or HSIV-vif-NL4-3. Second, similar to HSIV-vif-NL4-3, release of HSIV-vif-Yu2 was restricted when cotransfected with PTM Tetherin. Third, HSIV-vif-Yu2 did not downregulate Tetherin from the surface of PTM or human cells. Not surprisingly, given that it does not express Vpu, HSIV-vif-Yu2 was also suppressed by human Tetherin to a greater extent than HSIV-vif-NL4-3 and reversion of the vpu start codon was required to downregulate human Tetherin and enhance virion release from HeLa cells. Fourth, knockdown of Tetherin did not increase replication of either HSIV-vif-NL4-3 or HSIV-vif-Yu2 in PTM CD4+ T cells. Interestingly, a recent report also showed that deletion of nef, which is required for counteraction of PTM BST2, does not increase sensitivity of SIVmac to IFN-α in PBMCs from PTMs, suggesting that Tetherin may not be the major effector of the IFN-induced antiviral state in PTM cells against simian-tropic HIV-1 (102). Furthermore, incorporation of vpu alleles from SIVden and SIVgsn71, which are known to antagonize macaque Tetherin, did not reduce the sensitivity of simian-tropic HIV-1 clones to an IFN-induced antiviral state in PTM PBMCs. Thus, our data imply that the increased capacity of HSIV-vif-Yu2 to replicate in T cells during an IFN-α restrictive state may result from greater capacity to complete another part of the viral life cycle. Additionally, because HSIV-vif-Yu2 replicates to higher levels than HSIV-vif-NL4-3, it may saturate the antiviral effect of Tetherin, thereby facilitating particle release. This possibility is consistent with a recent report showing that a high level of expression of HIV-1 virus-like particles overcomes Tetherin restriction (108).
By creating chimeric viruses between the IFN-α-sensitive clone HSIV-vif-NL4-3 and the IFN-α-resistant clone HSIV-vif-Yu2, we narrowed the determinant of IFN-α resistance to a portion of the 3′ half of the virus that includes env su. Although the env tm of HSIV-vif-Yu2 alone was not sufficient to enhance viral replication in the presence of IFN-α, our data suggest that it may contribute to IFN-α resistance because the complete env conferred greater replication than env su alone in IFN-α-treated PTM CD4+ T cells. Additionally, because HSIV-vif-Yu2 is partially restricted at the stage of release by IFN-α and Tetherin and because the IFN-α resistant env chimeric clones are isogenic with HSIV-vif-NL4-3, the data also suggest that the Yu-2 env determinant may enable progression through the early part of the viral life cycle. However, because the fragment also encodes both exons of tat and rev, we cannot rule out that the Yu2 alleles of either of these genes may antagonize the activities of ISGs that inhibit HSIV-vif replication in PTM CD4+ T cells at other stages of the viral life cycle. Although it seems unlikely that Rev could be involved in escape from restriction by IFN-α, previous studies have shown that Tat can downregulate protein kinase R (PKR) and prevent its induction by IFN treatment, thereby limiting PKR's ability to restrict HIV replication (109–111). Whether there are differences in the functional activity of the Yu2 and NL4-3 Tat proteins is unknown. Additional studies should address the contribution of the Yu2 Tat to HSIV-vif-Yu2 replication in PTM CD4+ T cells during the IFN-α-induced antiviral state.
We demonstrate that the env determinant enables HSIV-vif-Yu2 to overcome an IFN-α-induced barrier to entry. Using the Blam-Vpr fusion assay, we found that entry of each HSIV-vif clone expressing the env determinant of Yu2 had a higher relative infectivity in IFN-α-treated PTM CD4+ T cells than the parent virus HSIV-vif-NL4-3, indicating a differential effect of Env on entry in the presence of IFN-α. These data are consistent with earlier studies that demonstrated an IFN-α-induced block to HIV-1 entry (19, 112–114). On the other hand, Goujon and Malim showed that IFN induces postentry blocks at the level of viral cDNA synthesis with substantial decreases (sometimes >99%) in nascent cDNA accumulation (16). It is important to consider that these other studies were done in human cells and there may be species-specific effects of IFN-α. Indeed, it was demonstrated that the sensitivity of simian-tropic HIV-1 viruses to IFN-α in PTM PBMCs is not due to the restriction factors TRIM5, SAMHD1, APOBEC3, or Tetherin and that other IFN-α-induced restriction factors may inhibit replication at early stages of the life cycle in this species (102). In agreement with these findings, we also found early postentry blocks to VSV-G pseudotyped HIV-1 infection of PTM CD4+ T cells induced by IFN-α. Other studies have also suggested the existence of HIV restriction factors in human and rhesus macaque cells, termed LV2 and LV3, respectively, that impede postentry events in replication in an Env-dependent manner (115–117). Based on these data, it can be hypothesized that the decrease in entry of HSIV-vif clones combined with a decrease in cDNA synthesis and other unknown IFN-induced restriction factors could potentially have a significant effect on the replication of HSIV-vif variants in PTM CD4+ T cells and that escape from any one of these factors could result in enhanced viral replication. We propose that mutations in the Yu-2 Env may enable escape from an IFN-α-induced factor that impedes an early step in viral replication.
Finally, downregulation of CD4 and coreceptors' expression does not explain the early-stage interference in HSIV-vif-NL4-3 replication in PTM CD4+ T cells. As reported in human T cells (16), we did not observe a decrease in surface CD4, CCR5, or CXCR4 by IFN-α in PTM CD4+ T cells. Possible ISGs that might reduce entry of HIV-1 have been recently reported. For example, IFITM2 and IFITM3 have been shown to interfere with entry of HIV-1 into human cells (19). We have recently found that IFN-α induces high-level expression of IFITM3 in PTM CD4+ T cells (unpublished observations), and therefore, it would be interesting to test whether IFITM3 differentially affects entry of HSIV-vif clones expressing the NL4-3 or Yu-2 Env proteins.
It is possible that HIV-1 Yu-2 may have evolved an alternative mechanism to overcome IFN-α-induced restriction in the absence of Vpu. From our findings, we hypothesize that the virus compensated for the absence of Vpu with enhanced entry. Perhaps by increasing viral infectivity and entry, the virus is able to overcome not only the IFN-α-induced barrier to virion release and entry but also restriction by saturating postentry blocks that limit reverse transcription. The significance of this effect for persistent replication in vivo is unknown but may be tested by infection of pig-tailed macaques with the HSIV-vif-Yu2 clone.
ACKNOWLEDGMENTS
We thank Celeste Alavarez for assistance with cloning, Ben Hornstein for assistance with flow cytometry, Anisha Misra for critical comments, and Xiaoying Yu of the Baylor-UT Houston CFAR Design and Analysis Core for help with statistical analysis. NL4-3, NL-AD8, and TZM-bl cells were obtained from M. Martin, E. Freed, and J. Kappes, respectively, via the NIH AIDS Reagent and Reference Program.
This work was supported by NIH grants AI083095 and AI099007 to J.T.K. and in part by the Baylor-UT Houston CFAR (AI036211). It was also supported by grants from the Chinese National Science Foundation (number 31170871) and National Science and Technology Major Project (number 2012ZX10001-008) to P.Z.
Footnotes
Published ahead of print 3 April 2013
REFERENCES
- 1. Abel K, Alegria-Hartman MJ, Rothaeusler K, Marthas M, Miller CJ. 2002. The relationship between simian immunodeficiency virus RNA levels and the mRNA levels of alpha/beta interferons (IFN-alpha/beta) and IFN-alpha/beta-inducible Mx in lymphoid tissues of rhesus macaques during acute and chronic infection. J. Virol. 76:8433–8445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abel K, Rocke DM, Chohan B, Fritts L, Miller CJ. 2005. Temporal and anatomic relationship between virus replication and cytokine gene expression after vaginal simian immunodeficiency virus infection. J. Virol. 79:12164–12172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, Self SG, Borrow P. 2009. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 83:3719–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. von Sydow M, Sonnerborg A, Gaines H, Strannegard O. 1991. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res. Hum. Retroviruses 7:375–380 [DOI] [PubMed] [Google Scholar]
- 5. Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. 2005. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 115:3265–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fonteneau JF, Larsson M, Beignon AS, McKenna K, Dasilva I, Amara A, Liu YJ, Lifson JD, Littman DR, Bhardwaj N. 2004. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 78:5223–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smed-Sorensen A, Lore K, Vasudevan J, Louder MK, Andersson J, Mascola JR, Spetz AL, Koup RA. 2005. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J. Virol. 79:8861–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rotger M, Dang KK, Fellay J, Heinzen EL, Feng S, Descombes P, Shianna KV, Ge D, Gunthard HF, Goldstein DB, Telenti A. 2010. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog. 6:e1000781 doi: 10.1371/journal.ppat.1000781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stylianou E, Aukrust P, Bendtzen K, Muller F, Froland SS. 2000. Interferons and interferon (IFN)-inducible protein 10 during highly active anti-retroviral therapy (HAART)-possible immunosuppressive role of IFN-alpha in HIV infection. Clin. Exp. Immunol. 119:479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aguilar Marucco D, Veronese L, de Requena DG, Bonora S, Calcagno A, Cavecchia I, Sinicco A, De Rosa FG, Cariti G, Di Perri G. 2007. Antiretroviral activity of pegylated interferon alfa-2a in patients co-infected with HIV/hepatitis C virus. J. Antimicrob. Chemother. 59:565–568 [DOI] [PubMed] [Google Scholar]
- 11. Hatzakis A, Gargalianos P, Kiosses V, Lazanas M, Sypsa V, Anastassopoulou C, Vigklis V, Sambatakou H, Botsi C, Paraskevis D, Stalgis C. 2001. Low-dose IFN-alpha monotherapy in treatment-naive individuals with HIV-1 infection: evidence of potent suppression of viral replication. J. Interferon Cytokine Res. 21:861–869 [DOI] [PubMed] [Google Scholar]
- 12. Tavel JA, Huang CY, Shen J, Metcalf JA, Dewar R, Shah A, Vasudevachari MB, Follmann DA, Herpin B, Davey RT, Polis MA, Kovacs J, Masur H, Lane HC. 2010. Interferon-alpha produces significant decreases in HIV load. J. Interferon Cytokine Res. 30:461–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agy MB, Acker RL, Sherbert CH, Katze MG. 1995. Interferon treatment inhibits virus replication in HIV-1- and SIV-infected CD4+ T-cell lines by distinct mechanisms: evidence for decreased stability and aberrant processing of HIV-1 proteins. Virology 214:379–386 [DOI] [PubMed] [Google Scholar]
- 14. Bednarik DP, Mosca JD, Raj NB, Pitha PM. 1989. Inhibition of human immunodeficiency virus (HIV) replication by HIV-trans-activated alpha 2-interferon. Proc. Natl. Acad. Sci. U. S. A. 86:4958–4962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gendelman HE, Baca LM, Turpin J, Kalter DC, Hansen B, Orenstein JM, Dieffenbach CW, Friedman RM, Meltzer MS. 1990. Regulation of HIV replication in infected monocytes by IFN-alpha. Mechanisms for viral restriction. J. Immunol. 145:2669–2676 [PubMed] [Google Scholar]
- 16. Goujon C, Malim MH. 2010. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 84:9254–9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meylan PR, Guatelli JC, Munis JR, Richman DD, Kornbluth RS. 1993. Mechanisms for the inhibition of HIV replication by interferons-alpha, -beta, and -gamma in primary human macrophages. Virology 193:138–148 [DOI] [PubMed] [Google Scholar]
- 18. Shirazi Y, Pitha PM. 1993. Interferon alpha-mediated inhibition of human immunodeficiency virus type 1 provirus synthesis in T-cells. Virology 193:303–312 [DOI] [PubMed] [Google Scholar]
- 19. Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J. Virol. 85:2126–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bishop KN, Holmes RK, Malim MH. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 80:8450–8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4:e1000231 doi: 10.1371/journal.ppat.1000231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. 2006. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 80:11710–11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35:7096–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 81:7099–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650 [DOI] [PubMed] [Google Scholar]
- 28. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 29. Kajaste-Rudnitski A, Marelli SS, Pultrone C, Pertel T, Uchil PD, Mechti N, Mothes W, Poli G, Luban J, Vicenzi E. 2011. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 85:5183–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagai K, Wong AH, Li S, Tam WN, Cuddihy AR, Sonenberg N, Mathews MB, Hiscott J, Wainberg MA, Koromilas AE. 1997. Induction of CD4 expression and human immunodeficiency virus type 1 replication by mutants of the interferon-inducible protein kinase PKR. J. Virol. 71:1718–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tissot C, Mechti N. 1995. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J. Biol. Chem. 270:14891–14898 [DOI] [PubMed] [Google Scholar]
- 32. Maitra RK, Silverman RH. 1998. Regulation of human immunodeficiency virus replication by 2′,5′-oligoadenylate-dependent RNase L. J. Virol. 72:1146–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barr SD, Smiley JR, Bushman FD. 2008. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 4:e1000007 doi: 10.1371/journal.ppat.1000007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson SJ, Schoggins JW, Zang T, Kutluay SB, Jouvenet N, Alim MA, Bitzegeio J, Rice CM, Bieniasz PD. 2012. Inhibition of HIV-1 particle assembly by 2′,3′-cyclic-nucleotide 3′-phosphodiesterase. Cell Host Microbe 12:585–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430 [DOI] [PubMed] [Google Scholar]
- 36. Okumura A, Lu G, Pitha-Rowe I, Pitha PM. 2006. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U. S. A. 103:1440–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pincetic A, Kuang Z, Seo EJ, Leis J. 2010. The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol. 84:4725–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809 [DOI] [PubMed] [Google Scholar]
- 39. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103 [DOI] [PubMed] [Google Scholar]
- 40. Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baca-Regen L, Heinzinger N, Stevenson M, Gendelman HE. 1994. Alpha interferon-induced antiretroviral activities: restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus type 1-infected monocytes. J. Virol. 68:7559–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fernie BF, Poli G, Fauci AS. 1991. Alpha interferon suppresses virion but not soluble human immunodeficiency virus antigen production in chronically infected T-lymphocytic cells. J. Virol. 65:3968–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hansen BD, Nara PL, Maheshwari RK, Sidhu GS, Bernbaum JG, Hoekzema D, Meltzer MS, Gendelman HE. 1992. Loss of infectivity by progeny virus from alpha interferon-treated human immunodeficiency virus type 1-infected T cells is associated with defective assembly of envelope gp120. J. Virol. 66:7543–7548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ho DD, Hartshorn KL, Rota TR, Andrews CA, Kaplan JC, Schooley RT, Hirsch MS. 1985. Recombinant human interferon alfa-A suppresses HTLV-III replication in vitro. Lancet i:602–604 [DOI] [PubMed] [Google Scholar]
- 45. Kornbluth RS, Oh PS, Munis JR, Cleveland PH, Richman DD. 1989. Interferons and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J. Exp. Med. 169:1137–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kornbluth RS, Oh PS, Munis JR, Cleveland PH, Richman DD. 1990. The role of interferons in the control of HIV replication in macrophages. Clin. Immunol. Immunopathol. 54:200–219 [DOI] [PubMed] [Google Scholar]
- 47. Michaelis B, Levy JA. 1989. HIV replication can be blocked by recombinant human interferon beta. AIDS 3:27–31 [PubMed] [Google Scholar]
- 48. Poli G, Orenstein JM, Kinter A, Folks TM, Fauci AS. 1989. Interferon-alpha but not AZT suppresses HIV expression in chronically infected cell lines. Science 244:575–577 [DOI] [PubMed] [Google Scholar]
- 49. Shirazi Y, Pitha PM. 1992. Alpha interferon inhibits early stages of the human immunodeficiency virus type 1 replication cycle. J. Virol. 66:1321–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Donaghy H, Pozniak A, Gazzard B, Qazi N, Gilmour J, Gotch F, Patterson S. 2001. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood 98:2574–2576 [DOI] [PubMed] [Google Scholar]
- 51. Soumelis V, Scott I, Gheyas F, Bouhour D, Cozon G, Cotte L, Huang L, Levy JA, Liu YJ. 2001. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood 98:906–912 [DOI] [PubMed] [Google Scholar]
- 52. Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr 2009. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83:10395–10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. 2008. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5:e1000429 doi: 10.1371/journal.ppat.1000429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. 2007. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Serra-Moreno R, Jia B, Breed M, Alvarez X, Evans DT. 2011. Compensatory changes in the cytoplasmic tail of gp41 confer resistance to tetherin/BST-2 in a pathogenic Nef-deleted SIV. Cell Host Microbe 9:46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407 [DOI] [PubMed] [Google Scholar]
- 60. Thippeshappa R, Ruan H, Kimata JT. 2012. Breaking barriers to an AIDS model with macaque-tropic HIV-1 derivatives. Biology 1:134–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. 2004. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad. Sci. U. S. A. 101:3770–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, Krijnse-Locker J, Banting G, Krausslich HG, Fackler OT, Keppler OT. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285–297 [DOI] [PubMed] [Google Scholar]
- 63. Mangeat B, Turelli P, Liao S, Trono D. 2004. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J. Biol. Chem. 279:14481–14483 [DOI] [PubMed] [Google Scholar]
- 64. Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31 [DOI] [PubMed] [Google Scholar]
- 65. McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. 2009. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5:e1000300 doi: 10.1371/journal.ppat.1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schrofelbauer B, Chen D, Landau NR. 2004. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. U. S. A. 101:3927–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. 2004. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. U. S. A. 101:5652–5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang SJ, Lopez LA, Hauser H, Exline CM, Haworth KG, Cannon PM. 2010. Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology 7:13 doi: 10.1186/1742-4690-7-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. U. S. A. 102:2832–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stremlau M, Perron M, Welikala S, Sodroski J. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J. Virol. 79:3139–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462 doi: 10.1371/journal.pbio.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lim SY, Rogers T, Chan T, Whitney JB, Kim J, Sodroski J, Letvin NL. 2010. TRIM5alpha modulates immunodeficiency virus control in rhesus monkeys. PLoS Pathog. 6:e1000738 doi: 10.1371/journal.ppat.1000738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Reynolds MR, Sacha JB, Weiler AM, Borchardt GJ, Glidden CE, Sheppard NC, Norante FA, Castrovinci PA, Harris JJ, Robertson HT, Friedrich TC, McDermott AB, Wilson NA, Allison DB, Koff WC, Johnson WE, Watkins DI. 2011. The TRIM5{alpha} genotype of rhesus macaques affects acquisition of simian immunodeficiency virus SIVsmE660 infection after repeated limiting-dose intrarectal challenge. J. Virol. 85:9637–9640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yeh WW, Rao SS, Lim SY, Zhang J, Hraber PT, Brassard LM, Luedemann C, Todd JP, Dodson A, Shen L, Buzby AP, Whitney JB, Korber BT, Nabel GJ, Mascola JR, Letvin NL. 2011. The TRIM5 gene modulates penile mucosal acquisition of simian immunodeficiency virus in rhesus monkeys. J. Virol. 85:10389–10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ambrose Z, KewalRamani VN, Bieniasz PD, Hatziioannou T. 2007. HIV/AIDS: in search of an animal model. Trends Biotechnol. 25:333–337 [DOI] [PubMed] [Google Scholar]
- 77. Heeney JL, Dalgleish AG, Weiss RA. 2006. Origins of HIV and the evolution of resistance to AIDS. Science 313:462–466 [DOI] [PubMed] [Google Scholar]
- 78. Agy MB, Frumkin LR, Corey L, Coombs RW, Wolinsky SM, Koehler J, Morton WR, Katze MG. 1992. Infection of Macaca nemestrina by human immunodeficiency virus type-1. Science 257:103–106 [DOI] [PubMed] [Google Scholar]
- 79. Bosch ML, Schmidt A, Agy MB, Kimball LE, Morton WR. 1997. Infection of Macaca nemestrina neonates with HIV-1 via different routes of inoculation. AIDS 11:1555–1563 [DOI] [PubMed] [Google Scholar]
- 80. Gartner S, Liu Y, Lewis MG, Polonis V, Elkins WR, Zack PM, Miao J, Hunter EA, Greenhouse J, Eddy GA. 1994. HIV-1 infection in pigtailed macaques. AIDS Res. Hum. Retroviruses 10(Suppl 2):S129–S133 [PubMed] [Google Scholar]
- 81. Kimball LE, Bosch ML. 1998. In vitro HIV-1 infection in Macaca nemestrina PBMCs is blocked at a step beyond reverse transcription. J. Med. Primatol. 27:99–103 [DOI] [PubMed] [Google Scholar]
- 82. Brennan G, Kozyrev Y, Hu SL. 2008. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. U. S. A. 105:3569–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Brennan G, Kozyrev Y, Kodama T, Hu SL. 2007. Novel TRIM5 isoforms expressed by Macaca nemestrina. J. Virol. 81:12210–12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O'Neil SP, Johnson W. 2008. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog. 4:e1000003 doi: 10.1371/journal.ppat.1000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thippeshappa R, Polacino P, Yu Kimata MT, Siwak E, Anderson D, Wang W, Sherwood L, Arora R, Wen M, Zhou P, Hu SL, Kimata JT. 2011. Vif substitution enables persistent infection of pig-tailed macaques by human immunodeficiency virus type 1. J. Virol. 85:3767–3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T. 2008. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. U. S. A. 105:3563–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wilson SJ, Webb BL, Ylinen LM, Verschoor E, Heeney JL, Towers GJ. 2008. Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc. Natl. Acad. Sci. U. S. A. 105:3557–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hatziioannou T, Ambrose Z, Chung NP, Piatak M, Jr, Yuan F, Trubey CM, Coalter V, Kiser R, Schneider D, Smedley J, Pung R, Gathuka M, Estes JD, Veazey RS, KewalRamani VN, Lifson JD, Bieniasz PD. 2009. A macaque model of HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 106:4425–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Igarashi T, Iyengar R, Byrum RA, Buckler-White A, Dewar RL, Buckler CE, Lane HC, Kamada K, Adachi A, Martin MA. 2007. Human immunodeficiency virus type 1 derivative with 7% simian immunodeficiency virus genetic content is able to establish infections in pig-tailed macaques. J. Virol. 81:11549–11552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kamada K, Igarashi T, Martin MA, Khamsri B, Hatcho K, Yamashita T, Fujita M, Uchiyama T, Adachi A. 2006. Generation of HIV-1 derivatives that productively infect macaque monkey lymphoid cells. Proc. Natl. Acad. Sci. U. S. A. 103:16959–16964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hatziioannou T, Princiotta M, Piatak M, Jr, Yuan F, Zhang F, Lifson JD, Bieniasz PD. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314:95. [DOI] [PubMed] [Google Scholar]
- 92. Saito A, Nomaguchi M, Iijima S, Kuroishi A, Yoshida T, Lee YJ, Hayakawa T, Kono K, Nakayama EE, Shioda T, Yasutomi Y, Adachi A, Matano T, Akari H. 2011. Improved capacity of a monkey-tropic HIV-1 derivative to replicate in cynomolgus monkeys with minimal modifications. Microbes Infect. 13:58–64 [DOI] [PubMed] [Google Scholar]
- 93. Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74:8358–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Munoz NM, Trobridge GD, Kiem HP. 2009. Ex vivo expansion and lentiviral transduction of Macaca nemestrina CD4+ T cells. J. Med. Primatol. 38:438–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Qin XF, An DS, Chen IS, Baltimore D. 2003. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. U. S. A. 100:183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cavrois M, Neidleman J, Bigos M, Greene WC. 2004. Fluorescence resonance energy transfer-based HIV-1 virion fusion assay. Methods Mol. Biol. 263:333–344 [DOI] [PubMed] [Google Scholar]
- 98. Schubert U, Bour S, Willey RL, Strebel K. 1999. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J. Virol. 73:887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stephens EB, McCormick C, Pacyniak E, Griffin D, Pinson DM, Sun F, Nothnick W, Wong SW, Gunderson R, Berman NE, Singh DK. 2002. Deletion of the vpu sequences prior to the env in a simian-human immunodeficiency virus results in enhanced Env precursor synthesis but is less pathogenic for pig-tailed macaques. Virology 293:252–261 [DOI] [PubMed] [Google Scholar]
- 100. Bour S, Schubert U, Peden K, Strebel K. 1996. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: a Vpu-like factor? J. Virol. 70:820–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bour S, Strebel K. 1996. The human immunodeficiency virus (HIV) type 2 envelope protein is a functional complement to HIV type 1 Vpu that enhances particle release of heterologous retroviruses. J. Virol. 70:8285–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bitzegeio J, Sampias M, Bieniasz PD, Hatziioannou T. 2013. Adaptation to the IFN-induced antiviral state by human and simian immunodeficiency viruses. J. Virol. 87:3549–3560 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 103. Schwartz S, Felber BK, Fenyo EM, Pavlakis GN. 1990. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J. Virol. 64:5448–5456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Schwartz S, Felber BK, Pavlakis GN. 1992. Mechanism of translation of monocistronic and multicistronic human immunodeficiency virus type 1 mRNAs. Mol. Cell. Biol. 12:207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gummuluru S, Kinsey CM, Emerman M. 2000. An in vitro rapid-turnover assay for human immunodeficiency virus type 1 replication selects for cell-to-cell spread of virus. J. Virol. 74:10882–10891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jolly C, Booth NJ, Neil SJ. 2010. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 84:12185–12199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shingai M, Yoshida T, Martin MA, Strebel K. 2011. Some human immunodeficiency virus type 1 Vpu proteins are able to antagonize macaque BST-2 in vitro and in vivo: Vpu-negative simian-human immunodeficiency viruses are attenuated in vivo. J. Virol. 85:9708–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yadav SS, Wilson SJ, Bieniasz PD. 2012. A facile quantitative assay for viral particle genesis reveals cooperativity in virion assembly and saturation of an antiviral protein. Virology 429:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Brand SR, Kobayashi R, Mathews MB. 1997. The Tat protein of human immunodeficiency virus type 1 is a substrate and inhibitor of the interferon-induced, virally activated protein kinase, PKR. J. Biol. Chem. 272:8388–8395 [DOI] [PubMed] [Google Scholar]
- 110. Cai R, Carpick B, Chun RF, Jeang KT, Williams BR. 2000. HIV-I TAT inhibits PKR activity by both RNA-dependent and RNA-independent mechanisms. Arch. Biochem. Biophys. 373:361–367 [DOI] [PubMed] [Google Scholar]
- 111. Roy S, Katze MG, Parkin NT, Edery I, Hovanessian AG, Sonenberg N. 1990. Control of the interferon-induced 68-kilodalton protein kinase by the HIV-1 tat gene product. Science 247:1216–1219 [DOI] [PubMed] [Google Scholar]
- 112. Rogez C, Martin M, Dereuddre-Bosquet N, Martal J, Dormont D, Clayette P. 2003. Anti-human immunodeficiency virus activity of tau interferon in human macrophages: involvement of cellular factors and beta-chemokines. J. Virol. 77:12914–12920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shirazi Y, Pitha PM. 1998. Interferon downregulates CXCR4 (fusin) gene expression in peripheral blood mononuclear cells. J. Hum. Virol. 1:69–76 [PubMed] [Google Scholar]
- 114. Vieillard V, Lauret E, Rousseau V, De Maeyer E. 1994. Blocking of retroviral infection at a step prior to reverse transcription in cells transformed to constitutively express interferon beta. Proc. Natl. Acad. Sci. U. S. A. 91:2689–2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pineda MJ, Orton BR, Overbaugh J. 2007. A TRIM5alpha-independent post-entry restriction to HIV-1 infection of macaque cells that is dependent on the path of entry. Virology 363:310–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Reuter S, Kaumanns P, Buschhorn SB, Dittmar MT. 2005. Role of HIV-2 envelope in Lv2-mediated restriction. Virology 332:347–358 [DOI] [PubMed] [Google Scholar]
- 117. Schmitz C, Marchant D, Neil SJ, Aubin K, Reuter S, Dittmar MT, McKnight A. 2004. Lv2, a novel postentry restriction, is mediated by both capsid and envelope. J. Virol. 78:2006–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Arora R, Bull L, Siwak EB, Thippeshappa R, Arduino RC, Kimata JT. 2010. Dendritic cell-mediated HIV-1 infection of T cells demonstrates a direct relationship to plasma viral RNA levels. J. Acquir. Immune Defic. Syndr. 54:115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Biesinger T, Yu Kimata MT, Kimata JT. 2008. Changes in simian immunodeficiency virus reverse transcriptase alleles that appear during infection of macaques enhance infectivity and replication in CD4+ T cells. Virology 370:184–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kimata JT, Wilson JM, Patel PG. 2004. The increased replicative capacity of a late-stage simian immunodeficiency virus mne variant is evident in macrophage- or dendritic cell-T-cell cocultures. Virology 327:307–317 [DOI] [PubMed] [Google Scholar]
- 121. Humes D, Overbaugh J. 2011. Adaptation of subtype A human immunodeficiency virus type 1 envelope to pig-tailed macaque cells. J. Virol. 85:4409–4420 [DOI] [PMC free article] [PubMed] [Google Scholar]