

Abstract
Murine leukemia virus (MLV) can efficiently spread in tissue cultures by polarizing assembly to virological synapses. The viral envelope glycoprotein (Env) establishes cell-cell contacts and subsequently recruits Gag by a process that depends on its cytoplasmic tail. MLV Gag is recruited to virological synapses through the matrix domain (MA) (J. Jin, F. Li, and W. Mothes, J. Virol. 85:7672–7682, 2011). However, how MA targets Gag to sites of cell-cell contact remains unknown. Here we report that basic residues within MA are critical for directing MLV Gag to virological synapses. Alternative membrane targeting domains (MTDs) containing multiple basic residues can efficiently substitute MA to direct polarized assembly. Similarly, mutations in the polybasic cluster of MA that disrupt Gag polarization can be rescued by N-terminal addition of MTDs containing basic residues. MTDs containing basic residues alone fail to be targeted to the virological synapse. Systematic deletion experiments reveal that domains within Gag known to mediate Gag multimerization are also required. Thus, our data predict the existence of a specific “acidic” interface at virological synapses that mediates the recruitment of MLV Gag via the basic cluster of MA and Gag multimerization.
INTRODUCTION
Retroviral assembly is driven by the viral precursor polyprotein Gag (1). For C-type retroviruses, such as the human immunodeficiency virus (HIV) and murine leukemia viruses (MLV), viral assembly is initiated at the plasma membrane in most cell types (2–4). Membrane binding of most retroviral Gag proteins is mediated by two signals within the matrix (MA) domain: the myristate moiety at the N terminus and a conserved polybasic cluster (5–8). Both signals are required for Gag plasma membrane localization. Neutralization of the basic residues in the polybasic cluster leads to Gag intracellular relocalization and severely reduces viral particle production (9–15).
The membrane targeting of cellular N-myristoylated proteins is described by the two-signal model (16). According to this theory, myristoylation is necessary but not sufficient to anchor a protein to the membrane. A second signal is required and has been defined as either a polybasic cluster or a palmitate moiety (16). Within the family of Src tyrosine kinases, Src contains a polybasic cluster, whereas Lck features palmitates as the second signal. Fyn contains both a polybasic cluster and palmitates (16).
The polybasic cluster is thought to contribute to membrane binding through electrostatic interactions with acidic phospholipids (16–20). Acidic phospholipids, especially phosphatidylserine (PS) and phosphatidylinositol phosphates (PIPs), are concentrated at the cytosolic surface of cellular membranes (21, 22). An important function of them is to target and retain cationic proteins to specific subcellular membrane compartments through direct protein-lipid interactions (21, 23–25). The localization of specific PIPs is determined by a concerted action of spatially regulated kinases and phosphatases (21). PS and several PIPs, including PI(4,5)P2 and PI(3,4,5)P3, are mostly enriched at the inner leaflet of the plasma membrane (21, 22).
Strong evidence that the membrane binding of retroviral Gag is also facilitated by interactions between a polybasic cluster within MA and acidic phospholipids has accumulated (15, 26, 27). Some of them, including HIV, MLV, equine infectious anemia virus (EIAV), and Mason-Pfizer monkey virus (MPMV), exhibit specificity for PI(4,5)P2 (11, 28–33). In the case of MLV, PS participates in Gag localization to the plasma membrane by increasing the specificity for PI(4,5)P2 (11). Consistent with this notion, PI(4,5)P2 has been shown to be enriched in MLV and HIV virions (28). HIV carrying a deletion within the basic patch of MA fails to incorporate PI(4,5)P2 (28). EIAV has recently been reported to use PI(3)P and PI(3,5)P2 for targeting to its sites of assembly (34).
Besides interactions between MA and acidic lipids, Gag multimerization also plays an important role in targeting Gag to specific microdomains in the plasma membrane (35–39). Three regions within retroviral Gag proteins have been shown to contribute to multimerization, the capsid (CA), nucleocapsid (NC), and MA domains. While the C-terminal-domain (CTD) and NTD helices of CA enable dimerization and hexamerization within the lattice of the capsid, respectively (40, 41), NC is thought to contribute to Gag multimerization through binding to RNA (42–44). In polarized T cells, NC-mediated oligomerization is critical for targeting of HIV Gag to the cytoplasmic face of the plasma membrane at the uropod (38). MA assembles into trimers in solution and in three-dimensional crystals (45, 46). MA can also organize into hexamers or trimers of hexamers on lipid monolayers (47, 48).
Viruses exploit and manipulate cell-cell contacts to facilitate viral spreading by efficiently coordinating virus assembly and entry at the sites of cell-cell contacts, designated virological synapses (49–58). Similar cell-cell contacts have been observed for MLV in which virus assembly is polarized to the contact sites (2, 59–61). MLV synapses are the consequence of interactions between the viral envelope glycoprotein (Env), expressed by infected cells, and the viral receptor mCAT-1, expressed on the uninfected target cells (2, 60, 61). Viral Env and receptor mCAT-1 both accumulate at the cell-cell interface (2, 61). Subsequent recruitment of MLV Gag to Env and receptor accumulation at the cell-cell interface depend on the C-terminal residues of the cytoplasmic tail, designated the R peptide (2, 59, 61). Recruitment of MLV Gag to virological synapses requires MA (59). The molecular basis of the specific recruitment of Gag by MA and whether it is mediated by a direct or indirect interaction between MA and the cytoplasmic tail of Env are unknown.
Here, we present evidence for an indirect recruitment of Gag by Env to virological synapses that is mediated by basic residues in the membrane targeting domain (MTD) and requires the initiation of Gag multimerization. MA plays no direct structural role, as it is entirely dispensable for this step. Our data point to the existence of a specific “acidic” interface at virological synapses that mediates the recruitment of MLV Gag via the basic cluster of MA and Gag multimerization.
MATERIALS AND METHODS
Plasmids and reagents.
Proviral pLRB303 derived MLV Gag-GFP (ΔPol), Moloney MLV Gag-YFP, MLV GagPol, Friend MLV EnvΔH8, and EnvΔH8ΔR expression vectors were previously described (2, 59, 62). PLCδ PH, Akt PH, Btk PH, and TAPP1 PH were kindly provided by Mark Lemmon (University of Pennsylvania School of Medicine, Philadelphia, PA) and Cornelis Weijer (University of Dundee, Dundee, United Kingdom) (63). The C2 domain of lactadherin (Lact C2) conjugated to green fluorescent protein (Lact C2-GFP) was provided by Sergio Grinstein, University of Toronto. Src-GFP was kindly provided by Marc Johnson, University of Missouri. Specific domains of Gag protein were deleted or replaced by standard overlapping PCR. HEK293 and XC cells stably expressing mCherry-tagged mCAT-1 were previously described (2, 59).
Visualizing virus cell-to-cell transmission.
HEK293 donor cells were transfected with the indicated plasmids by FuGENE 6 and cocultured with XC target cells expressing mCAT-1-mCherry at 4 h posttransfection as previously described (2, 59). The formation of MLV synapses was reproducibly observed 2 to 6 h after initiation of cocultures (2, 59). Imaging of fixed samples and time-lapse video microscopy in living cells were performed using a spinning disc confocal microscope as previously described (2). The extent of polarization of assembly to the MLV synapse in fixed images was quantified using Volocity (PerkinElmer) as described previously (59). Briefly, a polarization index (PI) was determined based on the ratio of Gag fluorescence intensity in the cell-cell interface (identified by the accumulation of mCAT-1) compared to that outside the contact zone. Mean PI values were calculated from 10 MLV virological synapses. The accumulation of Env/receptor complexes at the cell-cell interface was also quantified by using intensity profiles along the lines drawn across the center of the MLV synapse.
VLP (virus-like particle) budding assay.
HEK293 cells were transfected with various MLV Gag Pol or Gag GFP constructs. At 24 h after transfection, VLPs were pelleted by microcentrifugation of 1 ml 0.45-μm-filtered culture supernatants at 14,000 rpm for 2 h over a 15% sucrose cushion. Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer. VLP and cell lysates were separated on polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes, followed by Western blotting using anti-p30 (Invitrogen, CA) primary antibody and peroxidase-conjugated secondary antibodies.
RESULTS
MLV MA can be replaced with MTDs containing basic residues to promote polarized assembly.
We have previously shown that targeting of Gag to MLV synapses depends on MA (59). This evidence was based on several independent observations: first, the N-terminal myristoylation-mediated membrane binding of MA was a prerequisite for targeting to the cell-cell interface. Second, replacing the entire MA domain with either a constitutive MTD from Lck kinase (LckΔMA) or a PH domain specific for PI(4,5)P2 prevented Gag recruitment to sites of cell-cell contact (59). Third, generating MA domain chimeras between MLV (polarized to MLV synapses) and HIV (not polarized to MLV synapses) demonstrated that the ability of Gag to polarize to MLV synapses mapped to MLV MA. Collectively, these data indicated that MLV MA carries specific signals that are required for the polarized assembly of MLV (59).
To learn more about the role of MLV MA and the effects of various MTDs in membrane targeting and polarized assembly of MLV, we further replaced MA with the MTDs from Src and Fyn. Lck contains two palmitates as a second membrane-binding signal in addition to the N-terminal myristate. In contrast, Src contains an additional polybasic cluster and Fyn contains both an additional polybasic cluster and two palmitates (16) (Fig. 1A). To measure polarized MLV assembly to virological synapses, we applied a quantitative visual assay that allows the assessment of Gag polarization to the cell-cell interface (59). Briefly, HEK293 donor cells expressing wild-type or mutant MLV Gag-GFP (Fig. 1A) and Env were cocultured for 2 to 6 h with XC target cells expressing viral receptor mCAT1-mCherry. Cells were fixed and virological synapses between donor cells (marked with D) and target XC cells (marked with T) were identified based on the strong accumulation of mCAT1-mCherry in the cell-cell interface. The MLV synapse is endocytically active, which leads to the internalization of mCAT1-mCherry into the donor cell (2, 61). The extent of Gag polarization to the Env/receptor interface can be displayed using fluorescence intensity profiles and can be quantified by determining the polarization index (PI) of Gag (59). PI is defined as the ratio of Gag fluorescence intensity generated by viral particles within the contact zone (marked by the accumulation of mCAT-1) compared to that outside the contact zone (see reference 59 for a detailed description). A polarization index of 1 indicates random assembly. Using various controls previously described, we determined that a mean polarization index larger than 2.5 would indicate clear polarization of MLV Gag (59). For comparison and in agreement with previous results (59), the behavior of wild-type (wt) Gag, LckΔMA, and LckGag in this assay is presented in Fig. 1B. Wt Gag strongly polarized to the virological synapses (PI = 7.35). The polarization was lost when MA was replaced with the MTD of Lck (PI = 1.16) but restored when MA was reintroduced into the construct (PI = 6.10) (Fig. 1B). Surprisingly, when MA was replaced with the MTD of Src or Fyn (SrcΔMA and FynΔMA), strong recruitment of Gag was still observed, indicating that MA is dispensable under these conditions (PI of 6.62 and 4.95, respectively) (Fig. 1B). The preferential recruitment of SrcΔMA to the cell-cell contact sites was confirmed by time-lapse video microscopy (see Video S1 in the supplemental material). It should be noted that the deletion of MA in the MLV full-length Gag-GFP constructs prevented virus assembly and release (Fig. 2) (59). The resulting prevention of transmission of virus variants lacking MA can cause an accumulation of Gag in intracellular compartments (Fig. 1B and 2). While SrcΔMA, FynΔMA, and LckΔMA, like wild-type MLV Gag, all exhibited some perinuclear and endosomal localization, regardless of whether the contact was present (data not shown), this perinuclear localization was more evident in the case of LckΔMA. Nevertheless, the local enrichment of SrcΔMA and FynΔMA, but not LckΔMA, at the sites of cell-cell contact was evident, and we observed no correlation between intracellular localization of Gag and its ability to polarize to the virological synapse (Fig. 1B). As expected, when MA was reintroduced back into these ΔMA constructs (SrcGag, FynGag, and LckGag), Gag again fully assembled and was transmitted to the target cells (Fig. 1B). These data indicate that the small MTDs of Src and Fyn carry signals that mimic the role of MA in polarized assembly.
Fig 1.

MLV matrix domain (MA) is dispensable for Gag polarization. (A) Wild-type (wt) and mutant forms of MLV Gag tested. The MTD of Lck, Src, or Fyn replaced the entire MA domain (left) or only the first 2 amino acids of MA (right). All mutations were made from a full-length virus construct in which GFP is fused to the C terminus of Gag (59). Basic residues (K and R) and cysteines (C) that are palmitoylated, contributing to the binding of respective MTDs to membranes, are in bold. (B) HEK293 cells transfected with the viral constructs expressing wt or respective mutants of MLV Gag-GFP (green) and MLV Env were cocultured with XC cells expressing mCAT-1-mCherry (red). At 2 to 6 h after initiation of coculture, cells were fixed. A spinning disc confocal microscope was used to visualize the virological synapses between donor (D) and target (T) cells. The images of the resulting 3-μm z stack were merged into a single extended focus view displayed here. Intensity profiles along the indicated lines of the displayed images and the mean polarization index (PI) of 10 images were determined to quantify the accumulation of Gag-GFP (green) in the area of Env/receptor complexes (red) at the site of cell-cell contact (59). Scale bar, 10 μm.
Fig 2.
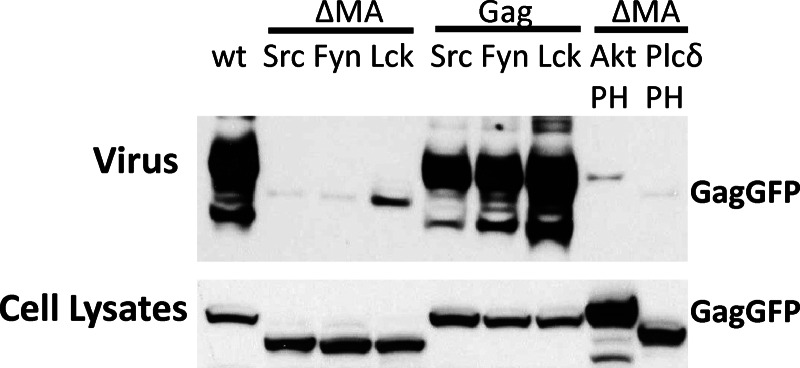
MA is required for VLP release in the full-length MLV Gag-GFP context. To measure VLP release, full-length MLV Gag-GFP mutant constructs were used to transfected HEK293 cells. At 24 h posttransfection, cells and viral culture supernatant were collected, followed by Western blotting and anti-p30 CA staining.
The ability of Src and Fyn MTD to compensate for MA is dependent on their basic residues.
In contrast to Lck, both MTDs from Src and Fyn contain polybasic clusters (Fig. 1A). Since the common feature of MLV MA and MTDs from Src and Fyn are basic residues, we tested if the ability of Src and Fyn MTDs to compensate for the MA deletion lay in their basic residues. SrcNΔMA and FynNΔMA were generated by substituting the basic residues with the neutral residue asparagine (Fig. 3A). Consistent with the two-signal model for N-myristoylated proteins, removing the basic residues from Src weakened the membrane targeting capabilities and increased the intracellular pool of SrcNΔMA Gag-GFP (Fig. 3B). However, the removal of basic residues in Fyn was tolerated and still allowed membrane targeting most likely because Fyn contains three signals (myristate, palmitate, and basic residues). Importantly, FynNΔMA and the plasma membrane-associated pool of SrcNΔMA failed to be recruited to the contact sites, indicating that the basic residues in the MTDs are required to direct ΔMA Gag to the contact sites. We also tested if the lysines are specifically required by replacing them with arginines. The resulting SrcRΔMA was still able to polarize to MLV synapses, indicating that it is the positive charge that matters (Fig. 3B). Last, replacing cysteine with serine in FynΔMA and LckΔMA (FynSΔMA and LckSΔMA) did not affect their polarization, indicating that palmitoylation is unlikely to play a role in targeting ΔMA Gag to the contact sites.
Fig 3.

The basic residues within the MTD of Src and Fyn are essential for their ability to compensate for the deletion of matrix domain. (A) MLV Gag mutants tested. The basic residues within Src and Fyn were replaced with the neutral residue Asn (N) to generate SrcNΔMA and FynNΔMA. SrcRΔMA was created by substituting Lys (K) with the basic Arg (R). Elimination of palmitylation sites within the MTD of Fyn and Lck was achieved by replacement of the Cys with Ser. The nonpalmitylated mutants were named FynSΔMA and LckSΔMA. (B) The experiment was carried out as described for Fig. 1B for the indicated Gag mutants (Gag-GFP, green; mCAT1-mCherry, red). Scale bar, 10 μm.
MA lacking the polybasic region fails to be targeted to virological synapses but is rescued by N-terminal MTDs from Src or Fyn.
The ability of MTDs carrying basic residues to compensate for the absence of MA suggests that the only MA-specific feature required for polarized assembly is its basic properties. The conserved polybasic region of MLV MA is known to be required for plasma membrane binding and efficient viral particle production (10, 11, 15). To test if the MLV polybasic region is required for MLV polarized assembly, we generated a basic patch mutant (bm) of Gag in which the polybasic region in the MA was neutralized by replacing two basic residues with acidic residues (K31KRR34 to EKER) as previously described by Hamard-Peron and coworkers (11) (Fig. 4A). We confirmed that the bm Gag mutant was localized intracellularly (Fig. 4B) and was impaired in virus release (Fig. 4C). N-terminally adding the MTDs of Src, Fyn, and Lck in front of MA (Srcbm, Fynbm, and Lckbm) restored both plasma membrane localization and virus release (Fig. 4). However, the polarized assembly of MLV was rescued only by the basic-residue-containing Src and Fyn MTD, not by Lck (Fig. 4B). The mild PI of 2.08 observed for Lckbm is below the previously determined cutoff PI of 2.5 for polarized assembly (59). Essentially identical results were obtained when the polybasic region of MA was replaced with neutral residues (K31KRR34 to NNNN or AAAR) (data not shown). The ability of Src and Fyn MTDs to rescue the polarized assembly of bm Gag again lay in their basic residues. Substitution of the basic residues in both MTDs with neutral residues (SrcNbm and FynNbm) disrupted their ability to rescue polarized assembly of bm Gag (Fig. 5A and B). Mutating the palmitoylation sites in Fyn and Lck MTD (FynSbm and LckSbm) did not affect their ability to rescue bm Gag (Fig. 5A and B). Notably, both FynS and FynN rescued membrane binding and viral particle production of the bm mutants to a similar extent, but only the basic residue containing FynSbm polarized (Fig. 5). These data again indicate a clear separation of the roles for basic residues in membrane targeting and polarized assembly. As expected, the corresponding mutants with a wild-type MA (SrcNGag, FynNGag, FynSGag, and LckSGag) were all targeted to the sites of cell-cell contact (data not shown). This indicates that the basic residues in the Src or Fyn MTD and basic residues in the MA polybasic region are functionally interchangeable. The behavior of all MLV Gag mutants studied thus far is summarized in Table 1. Basic residues, irrespective of whether they are contributed by the endogenous polybasic region of MA or come from heterologous MTDs, are strictly required for the polarized recruitment of Gag to the sites of cell-cell contact.
Fig 4.

The polybasic cluster within MA is required to target MLV Gag to the virological synapse, and if mutated, can be compensated by N-terminal Src and Fyn MTDs. (A) The basic cluster mutant (bm) of Gag was created by substituting two basic residues with acidic residues, resulting in a neutral charge (K31KRR34 to EKER) as previously described (11). SrcbmMA, FynbmMA, and LckbmMA were generated by the N-terminal addition of the respective MTD to the bm mutant of Gag. The mutants were constructed using Gag-YFP as a parental plasmid (62). (B) Gag-YFP mutants were tested as described for Fig. 1B. The donor HEK293 cells were transfected with respective Gag-YFP mutants and MLV Env (Gag-YFP, green; mCAT-1-mCherry, red). Scale bar, 10 μm. (C) To measure VLP release, mutants were transferred into the context of a MLV GagPol expression construct. At 24 h after transfection in HEK293 cells, cells and viral culture supernatant were collected, followed by Western blotting and anti-p30 CA staining.
Fig 5.

Rescue of polarized assembly of bm Gag is dependent on the basic residues within the N-terminal MTDs. (A) SrcNbm and FynNbm Gag were created by replacing the basic residues of SrcbmMA and FynbmMA with the neutral Asn (N). Cys (C) residues were replaced with Ser (S) in FynSbm and LckSbm to delete cysteine-based palmitoylation sites. (B) The respective bmGag mutants were analyzed as described for Fig. 3B (Gag-YFP, green; mCAT-1-mCherry, red). Scale bar, 10 μm. (C) VLP budding assay using respective Gag-YFP constructs followed by Western blotting and staining with antibodies against p30.
Table 1.
Summary of polarization and other features of MLV mutants
| MLV | Myristoylation | Palmitoylation | Presence of: |
Membrane localization | Polarization index |
Polarization | ||
|---|---|---|---|---|---|---|---|---|
| MLV basic patch | MTD basic residues | Mean | SEM | |||||
| Wt | + | − | + | − | +++ | 7.35 | 1.04 | + |
| G2A | − | − | + | − | − | 0.91 | 0.12 | − |
| SrcΔMA | + | − | − | + | +++ | 6.62 | 0.80 | + |
| SrcNΔMA | + | − | − | − | + | 1.26 | 0.20 | − |
| SrcRΔMA | + | − | − | + | +++ | 7.12 | 0.93 | + |
| FynΔMA | + | + | − | + | +++ | 4.95 | 0.68 | + |
| FynNΔMA | + | + | − | − | ++ | 1.13 | 0.15 | − |
| FynSΔMA | + | − | − | + | ++ | 4.45 | 0.56 | + |
| LckΔMA | + | + | − | − | +++ | 1.16 | 0.12 | − |
| LckSΔMA | + | − | − | − | + | 0.89 | 0.13 | − |
| bm | + | − | − | − | + | 0.66 | 0.11 | − |
| Srcbm | + | − | − | + | +++ | 8.37 | 0.84 | + |
| SrcNbm | + | − | − | − | + | 0.63 | 0.11 | − |
| Fynbm | + | + | − | + | +++ | 5.97 | 0.75 | + |
| FynNbm | + | + | − | − | ++ | 0.78 | 0.10 | − |
| FynSbm | + | − | − | + | ++ | 4.26 | 0.42 | + |
| Lckbm | + | + | − | − | +++ | 2.08 | 0.23 | − |
| LckSbm | + | − | − | − | + | 1.66 | 0.19 | − |
| SrcGag | + | − | + | + | +++ | 6.53 | 0.60 | + |
| SrcNGag | + | − | + | − | +++ | 10.18 | 1.33 | + |
| FynGag | + | + | + | + | +++ | 7.25 | 0.79 | + |
| FynNGag | + | + | + | − | +++ | 3.92 | 0.43 | + |
| FynSGag | + | − | + | + | +++ | 6.31 | 0.74 | + |
| LckGag | + | + | + | − | +++ | 6.10 | 0.70 | + |
| LckSGag | + | − | + | − | +++ | 5.88 | 0.68 | + |
Targeting of Gag through basic residues is dependent on the cytoplasmic tail of Env.
The biogenesis of the MLV synapses is driven by Env-receptor interactions (61). Assembly of MLV is subsequently polarized to the synapse in a process that is dependent on the R peptide within the cytoplasmic tail of Env (2, 59). We therefore asked if the identified role for basic residues in the targeting of MLV Gag to the MLV synapse is similarly dependent on the R peptide. To suppress the high fusogenicity of the R-peptide deletion mutant (Env ΔR), we deleted histidine 8 (2, 64). For comparison and consistency with previous results (59), the behavior of Env ΔR and wt Env is shown at the top of Fig. 6. While synapses were still established, MLV Gag failed to be recruited to cell-cell contact sites in the absence of the R peptide. This resulted in a mean polarization index close to 1, the expected theoretical value for nonpolarized random assembly (59) (Fig. 6). Importantly, the ability of SrcΔMA and Srcbm Gag to polarize to the MLV synapse was also dependent on the R peptide (Fig. 6). Both SrcΔMA and Srcbm failed to be recruited to the cell-cell contact established by Env ΔR, resulting in mean polarization indexes of 1.32 and 1.85, respectively. In contrast, Lckbm Gag exhibited a modest PI below the cutoff value of 2.5 for both wt Env and Env ΔR (PI of 2.08 and 2.21, respectively).
Fig 6.

Targeting of Gag through basic residues is downstream of the cytoplasmic tail of Env. Indicated Gag-YFP-mutants were transfected together with either wt MLV Env or Env ΔR, and polarized assembly was analyzed as described for Fig. 1B (Gag-GFP, green; mCAT-1-mCherry, red). Scale bar, 10 μm.
To gain further insights into the kinetics of MLV polarized assembly and to better understand the nature of the borderline PI value observed for Lckbm Gag, we recorded multiple time-lapse videos for wt Gag, Srcbm, and Lckbm Gag, and the PI was calculated every 30 min and plotted over time. Figure 7 depicts the quantification process for a single time-lapse movie for the polarized assembly of wt Gag (Fig. 7A and B; also, see Video S2 in the supplemental material). To quantify visual data, the results from 5 time-lapse videos were normalized by setting the time when viral assembly initiated to zero (Fig. 7C). Because de novo assembly of MLV is polarized (2, 61), the initiation of MLV assembly is already polarized. Such polarization was further enhanced with the additional accumulation of viral Gag-GFP particles in the cell-cell interface (59). The PI peaked about 2 h after initiation of assembly and finally declined somewhat as MLV assembly occurred increasingly all over the plasma membrane (Fig. 7C). Importantly, Srcbm Gag phenocopied the behavior of wt Gag. The polarization of both wt Gag and Srcbm Gag was also entirely dependent on the presence of the R peptide of Env (Fig. 7C). These data indicate that the basic residues in the Src MTD confer the same polarization kinetics to Gag as the native polybasic region of MA and they are both downstream of the R peptide of MLV Env. In contrast, Lckbm Gag that lacks basic residues started as more nonpolarized, and only a minor increase of the PI was observed over time. This steady slow increase was independent of the R peptide. This suggests that a moderate accumulation of some viral particles at the adhesive cell-cell interface can occur late and independently of polarized assembly. This process may involve surface diffusion of viral particles.
Fig 7.

The kinetics of polarized assembly for wt and mutant MLV Gag illustrates the importance of the basic residues. (A) A 3-h time-lapse movie monitoring polarized assembly of wt Gag (Gag-YFP, green; mCAT-1-mCherry, red) was analyzed every 30 min. The polarization indexes (PI) of single frames at various times are presented at the upper right corners of the images. Scale bar, 10 μm. (B) Polarization indexes over time for the time-lapse movie frames shown in panel A. (C) Polarization indexes over time for wt and the indicated Gag mutants in the presence of wt Env or Env ΔR. The time of the movies was set to zero when viral assembly was initiated. Error bars represent the standard errors of the means for 5 time-lapse movies.
The acidic lipids phosphatidylinositol (3,4,5)-triphosphate and phosphatidylserine are enriched at the MLV synapse.
Basic residues within MTDs are believed to mediate membrane binding through interaction with acidic lipids such as phosphoinositides (PIPs) or phosphatidylserine (PS). We therefore sought to test the possible model that these acidic lipids could play an exclusive role in the targeting of MLV Gag to virological synapses. Such a model would make several predictions. First, specific acidic lipids should be locally enriched at the MLV synapse. Second, the localization of these lipids to the MLV synapse should be downstream of the R peptide of MLV Env. Third, fusion of a PH or C2 domain with specificity for these lipids should direct Gag lacking MA to the MLV synapse. Finally, interfering with the biosynthesis of these lipids should disrupt polarized assembly. To address the first point, we took advantage of well-studied and widely used reporters that label the cellular distribution of the acidic lipids (21, 25). These reporters are GFP proteins fused to the PH domains of Akt, TAPP1, Btk, and PLCδ and the C2 domain of lactadherin, which specifically interact with defined PIPs or PS, respectively (Fig. 8A). To identify acidic lipids specifically localized to the MLV synapse formed by MLV Env and receptor mCAT-1, we cotransfected the donor cells with each of these reporters together with MLV Env. These donor cells were cocultured with mCAT-1-mCherry-expressing target cells. Interestingly, these results revealed that the PH domain, recognizing PI(3,4,5)P3, and Lact C2, recognizing PS, were enriched at the contact sites (Fig. 8B). In contrast, PI(4,5)P2 and PI(3,4)P2 were localized homogeneously to the plasma membrane (Fig. 8B). However, the acidic markers PI(3,4,5)P3 and PS were not exclusively localized to the MLV synapse but were also enriched at some other regions along the plasma membrane (Fig. 8B). Second, PI(3,4,5)P3 and PS were also recruited to the cell-cell contacts established by Env ΔR, indicating that the enrichment of both lipids was not downstream of the cytoplasmic tail of Env (Fig. 8B). Third, MLV Gag lacking MA was directed to the MLV synapse by the N-terminal PH domain from Akt [recognizing PI(3,4,5)P3 and PI(3,4)P2] but not by the PH domain of PLCδ [recognizing PI(4,5)P2] (Fig. 9). However, Akt PHΔMA Gag was not exclusively concentrated at synapses but was also localized to a few additional regions at the plasma membrane. Moreover, neither the labeling of the virological synapse by these PI(3,4,5)P3 and PS markers nor the recruitment of Akt PHΔMA Gag was downstream of the R peptide (Fig. 9). Last, MEF IPMK−/− cells, in which the PI(3,4,5)P3 is reduced by half, showed no difference compared to parental wild-type cell lines in their ability to support polarized virus assembly (data not shown) (65). Thus, while PI(3,4,5)P3 and PS are biomarkers for specific regions of the plasma membrane that include but are not restricted to MLV synapses, the localization of these markers likely plays no functional role in polarized assembly. While acidic lipids may be involved in the upstream biogenesis of MLV synapses, they do not appear to play an exclusive role in the recruitment of MLV Gag to the sites of cell-cell contact. It remains unknown what acidic interface specifically generated at the virological synapse is recognized by MLV Gag.
Fig 8.

The acidic phospholipids PI(3,4,5)P3 and PS are enriched at the virological synapse independently of the cytoplasmic tail of Env. (A) Probes used and their specificity for acidic lipids. (B) Constructs expressing the indicated probes (green) were cotransfected together with wt Env or Env ΔR, and the localization of the probes to MLV synapses formed by Env and mCAT-1-mCherry (red) was tested in an coculture experiment as described for Fig. 1B. Scale bar, 10 μm.
Fig 9.

MLV Gag lacking MA was directed to cell-cell interface by the N-terminal PH domain of Akt but not by the PH domain of PLC. (A) MLV Gag lacking MA carrying lipid binding PH domains of Akt [recognizing PI(3,4)P2 and PI(3,4,5)P3] or PLCδ [recognizing PI(4,5)P2]. (B) Gag-GFP constructs were assayed in the presence of wt Env or Env ΔR, as described for Fig. 1B, and the localization of Gag-GFP to the MLV synapse, characterized by the accumulation of mCAT-1-mCherry, was quantified.
Gag multimerization is required for targeting to the MLV synapse.
We have shown that basic residues, provided either by MA or by heterologous MTDs, target MLV Gag to the virological synapse. Interestingly, basic residues, while required, were not sufficient, as the MTD of Src fused to GFP failed to be targeted to MLV synapses (Fig. 10A and B). Our finding that SrcΔMA Gag localized to the MLV synapses indicated that a second signal within Gag must also be required. To identify this signal, we deleted the p12-CA or NC domains within the context of SrcΔMA, respectively (SrcΔMAΔp12ΔCA and SrcΔMAΔNC) (Fig. 10A). These experiments revealed that NC was required and sufficient to localize SrcΔMA Gag to contact sites (Fig. 10B). The inability of SrcΔMAΔNC (or Src-p12CA) to localize to MLV synapses was restored by introducing the trimeric leucine zipper domain from the yeast transcription factor GCN4 (66) (SrcΔMAΔNC-LZ or Src-p12CA-LZ) at the position of NC. These data indicate that Gag-mediated multimerization likely constitutes a second signal required for polarized assembly. Three domains within retroviral Gag proteins contribute to Gag-Gag interactions, CA, NC, and MA (40, 42–44, 47, 48, 67, 68). A model in which several domains within Gag can contribute a multimerization feature is supported by our previous observation that NC is dispensable for synapse location in the context of Gag containing MA and CA (GagΔNC) (Fig. 10A) (59). However, NC is the only single domain strong enough to target the Src MTD to synapses. MA, CA, and LZ in isolation all failed to target the Src MTD to the MLV synapse (Fig. 10; also data not shown). However, combinations of two of these three domains permitted targeting to sites of cell-cell contact in an NC-independent manner (Fig. 10B). Taken together, our results suggest that Gag-mediated higher-order multimerization is required for localization to the contact sites.
Fig 10.

Several domains within Gag contribute to the targeting of MLV Gag to virological synapses. (A) Wild-type (wt) and various Gag deletion constructs. Gag mutants lacking MA were targeted to membrane and the virological synapse by the MTD of Src. LZ represents the trimeric leucine zipper domain and was used to replace NC. *, MLV Gag lacking NC was previously described (59). (B) Localization of indicated Gag-GFP mutants (green) within HEK293 cells cocultured with XC cells expressing mCAT-1-mCherry (red) as described for Fig. 1B. Scale bar, 10 μm.
DISCUSSION
MLV can enhance the efficiency of viral spreading by polarizing assembly to sites of cell-cell contact. Here we have shown that the recruitment of MLV Gag to virological synapses is dependent on basic residues within MA and Gag domains that mediate Gag-Gag interactions. A requirement for the basic cluster in MA- and Gag-mediated multimerization, described here for MLV, resembles the mechanism of cytoplasmic targeting of HIV Gag to the uropod in polarized T cells (38, 69). They likely represent basic principles of how retroviral Gag proteins achieve polarity on the cytoplasmic phase of membranes.
However, beyond these general similarities, the MLV MA appears to fundamentally differ from HIV MA. The HIV MA domain placed at the N terminus of MLV Gag cannot promote polarized assembly to virological synapses generated by MLV Env and its receptor mCAT-1 (59). HIV MA specifically binds PI(4,5)P2 (30, 41, 70), and the PI(4,5)P2 specific PH domain of PLCδ can functionally replace HIV MA (3). In contrast, neither the HIV MA nor the PI(4,5)P2-specific PH domain can polarize MLV Gag to virological synapses. Rather, MLV MA appears to mimic the biology of Src and Fyn MTDs that contain basic residues. In fact, the role of MLV MA domain in Gag polarization is dispensable as long as it is replaced by Src- and Fyn-like MTDs. Similarly, mutations in the polybasic cluster of MA disrupt Gag polarization and can be restored by N-terminal addition of MTD containing basic residues. A previous report demonstrated that MLV MA behaves like a bona fide PI(4,5)P2-specific MTD (11). While we confirm that PI(4,5)P2-specific PH domains can mediate MLV Gag localization to membranes, our present study suggests that MLV MA must recognize an acidic environment at cell-cell contacts that is distinct from PI(4,5)P2-enriched microenvironment. In contrast to PI(4,5)P2-specific domains, PI(3,4,5)P3- and PS-specific domains exhibited specificity for the MLV synapse. However, the labeling was never exclusive for the synapses. The recruitment of PI(3,4,5)P3 and PS to the MLV synapse was also not downstream of the C tails of MLV Env, and cells with reduced PIP3 levels still promoted MLV synapse formation.
We have studied the recruitment of MLV Gag to the MLV synapse established by MLV Env binding to the receptor mCAT-1. Interestingly, a similar role for multiple domains in MLV Gag was recently described for the specific incorporation of MLV Env into budding MLV particles (71). Potentially, direct or indirect interactions between the cytoplasmic tail of Env and MLV Gag depend on multimerization in both cases. Our current working hypothesis is that MA targets Gag to specific microdomains at the plasma membrane. Gag multimerization creates a basic surface that can interact with an acidic interface that is downstream of the cytoplasmic tail of MLV Env. The identity of the specific acidic interface remains elusive. Future work should concentrate on the underlying cell biology to understand MLV Gag targeting to virological synapses.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Sergio Grinstein, Marc C Johnson, Mark Lemmon, Cornelis Weijer, and Pietro De Camilli for reagents. We thank Luis Agosto for critical reading of the manuscript.
This research was supported by grant R01 CA098727 from the National Institutes of Health to W.M. We declare that no competing interests exist.
Footnotes
Published ahead of print 24 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03263-12.
REFERENCES
- 1. Morita E, Sundquist WI. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20:395–425 [DOI] [PubMed] [Google Scholar]
- 2. Jin J, Sherer NM, Heidecker G, Derse D, Mothes W. 2009. Assembly of the murine leukemia virus is directed towards sites of cell-cell contact. PLoS Biol. 7:e1000163 doi: 10.1371/journal.pbio.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435 doi: 10.1371/journal.pbio.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Krausslich HG. 2007. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3:e36 doi: 10.1371/journal.ppat.0030036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murray PS, Li Z, Wang J, Tang CL, Honig B, Murray D. 2005. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure 13:1521–1531 [DOI] [PubMed] [Google Scholar]
- 6. Spearman P, Wang JJ, Vander Heyden N, Ratner L. 1994. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 68:3232–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou W, Parent LJ, Wills JW, Resh MD. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68:2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou W, Resh MD. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 70:8540–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Freed EO, Orenstein JM, Bucklerwhite AJ, Martin MA. 1994. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 68:5311–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Granowitz C, Goff SP. 1994. Substitution mutations affecting a small region of the Moloney murine leukemia virus MA Gag protein block assembly and release of virion particles. Virology 205:336–344 [DOI] [PubMed] [Google Scholar]
- 11. Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, Darlix JL, Picart C, Muriaux D. 2010. Targeting of murine leukemia virus Gag to the plasma membrane is mediated by PI(4,5)P-2/PS and a polybasic region in the matrix. J. Virol. 84:503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hermida-Matsumoto L, Resh MD. 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 74:8670–8679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 gag targeting to the plasma membrane. Mol. Biol. Cell 15:122a–123a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soneoka Y, Kingsman SM, Kingsman AJ. 1997. Mutagenesis analysis of the murine leukemia virus matrix protein: Identification of regions important for membrane localization and intracellular transport. J. Virol. 71:5549–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Resh MD. 1999. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451:1–16 [DOI] [PubMed] [Google Scholar]
- 17. McLaughlin S, Aderem A. 1995. The myristoyl-electrostatic switch—a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20:272–276 [DOI] [PubMed] [Google Scholar]
- 18. Murray D, Matsumoto LH, Buser CA, Tsang J, Sigal CT, Ben-Tal N, Honig B, Resh MD, McLaughlin S. 1998. Electrostatics and the membrane association of Src: theory and experiment. Biochemistry 37:2145–2159 [DOI] [PubMed] [Google Scholar]
- 19. Reuther GW, Buss JE, Quilliam LA, Clark GJ, Der CJ. 2000. Analysis of function and regulation of proteins that mediate signal transduction by use of lipid-modified plasma membrane-targeting sequences. Methods Enzymol. 327:331–350 [DOI] [PubMed] [Google Scholar]
- 20. Sigal CT, Zhou WJ, Buser CA, Mclaughlin S, Resh MD. 1994. Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc. Natl. Acad. Sci. U. S. A. 91:12253–12257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657 [DOI] [PubMed] [Google Scholar]
- 22. Leventis PA, Grinstein S. 2010. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39:407–427 [DOI] [PubMed] [Google Scholar]
- 23. Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. 2006. PI(3,4,5)P-3 and PI(4,5)P-2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314:1458–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McLaughlin S, Murray D. 2005. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438:605–611 [DOI] [PubMed] [Google Scholar]
- 25. Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. 2008. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319:210–213 [DOI] [PubMed] [Google Scholar]
- 26. Hamard-Peron E, Muriaux D. 2011. Retroviral matrix and lipids, the intimate interaction. Retrovirology 8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ono A. 2009. HIV-1 assembly at the plasma membrane: Gag trafficking and localization. Future Virol. 4:241–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. 2008. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J. Virol. 82:11228–11238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen K, Bachtair I, Piszczek G, Bouamr F, Carter C, Tjandra N. 2008. Solution NMR characterizations of oligomerization and dynamics of equine infectious anemia virus matrix protein and its interaction with PIP2. Biochemistry 47:1928–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 101:14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saad JS, Ablan SD, Ghanam RH, Kim A, Andrews K, Nagashima K, Soheilian F, Freed EO, Summers MF. 2008. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J. Mol. Biol. 382:434–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U. S. A. 103:11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stansell E, Apkarian R, Haubova S, Diehl WE, Tytler EM, Hunter E. 2007. Basic residues in the Mason-Pfizer monkey virus Gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J. Virol. 81:8977–8988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fernandes F, Chen K, Ehrlich LS, Jin J, Chen MH, Medina GN, Symons M, Montelaro R, Donaldson J, Tjandra N, Carter CA. 2011. Phosphoinositides direct equine infectious anemia virus Gag trafficking and release. Traffic 12:438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding LM, Derdowski A, Wang JJ, Spearman P. 2003. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J. Virol. 77:1916–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Halwani R, Khorchid A, Cen S, Kleiman L. 2003. Rapid localization of Gag/GagPol complexes to detergent-resistant membrane during the assembly of human immunodeficiency virus type 1. J. Virol. 77:3973–3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindwasser OW, Resh MD. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75:7913–7924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Llewellyn GN, Hogue IB, Grover JR, Ono A. 2010. Nucleocapsid promotes localization of HIV-1 gag to uropods that participate in virological synapses between T cells. PLoS Pathog. 6:e1001167 doi: 10.1371/journal.ppat.1001167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ono A, Freed EO. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U. S. A. 98:13925–13930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ehrlich LS, Agresta BE, Carter CA. 1992. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 66:4874–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853 [DOI] [PubMed] [Google Scholar]
- 42. Burniston MT, Cimarelli A, Colgan J, Curtis SP, Luban J. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol. 73:8527–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dawson L, Yu XF. 1998. The role of nucleocapsid of HIV-1 in virus assembly. Virology 251:141–157 [DOI] [PubMed] [Google Scholar]
- 44. Lapadat-Tapolsky M, De Rocquigny H, Van Gent D, Roques B, Plasterk R, Darlix JL. 1993. Interactions between HIV-1 nucleocapsid protein and viral DNA may have important functions in the viral life cycle. Nucleic Acids Res. 21:831–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U. S. A. 93:3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U. S. A. 101:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alfadhli A, Barklis RL, Barklis E. 2009. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alfadhli A, Huseby D, Kapit E, Colman D, Barklis E. 2007. Human immunodeficiency virus type 1 matrix protein assembles on membranes as a hexamer. J. Virol. 81:1472–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hope TJ. 2007. Bridging efficient viral infection. Nat. Cell Biol. 9:243–244 [DOI] [PubMed] [Google Scholar]
- 50. Hubner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FY, Li XD, Asmuth DM, Huser T, Chen BK. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716 [DOI] [PubMed] [Google Scholar]
- 52. Johnson DC, Huber MT. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 76:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jolly C, Sattentau QJ. 2004. Retroviral spread by induction of virological synapses. Traffic 5:643–650 [DOI] [PubMed] [Google Scholar]
- 54. McDonald D, Wu L, Bohks SM, KewalRamani VN, Unutmaz D, Hope TJ. 2003. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300:1295–1297 [DOI] [PubMed] [Google Scholar]
- 55. Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Phillips DM. 1994. The role of cell-to-cell transmission in HIV infection. AIDS 8:719–731 [DOI] [PubMed] [Google Scholar]
- 57. Rudnicka D, Feldman J, Porrot F, Wietgrefe S, Guadagnini S, Prevost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J. Virol. 83:6234–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826 [DOI] [PubMed] [Google Scholar]
- 59. Jin J, Li F, Mothes W. 2011. Viral determinants of polarized assembly for the murine leukemia virus. J. Virol. 85:7672–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat. Commun. 3:1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 9:310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, Mothes W. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785–801 [DOI] [PubMed] [Google Scholar]
- 63. Dormann D, Weijer G, Dowler S, Weijer CJ. 2004. In vivo analysis of 3-phosphoinositide dynamics during Dictyostelium phagocytosis and chemotaxis. J. Cell Sci. 117:6497–6509 [DOI] [PubMed] [Google Scholar]
- 64. Zavorotinskaya T, Qian Z, Franks J, Albritton LM. 2004. A point mutation in the binding subunit of a retroviral envelope protein arrests virus entry at hemifusion. J. Virol. 78:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Maag D, Maxwell MJ, Hardesty DA, Boucher KL, Choudhari N, Hanno AG, Ma JF, Snowman AS, Pietropaoli JW, Xu RS, Storm PB, Saiardi A, Snyder SH, Resnick AC. 2011. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc. Natl. Acad. Sci. U. S. A. 108:1391–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Accola MA, Strack B, Gottlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74:5395–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee PP, Linial ML. 1994. Efficient particle formation can occur if the matrix domain of human immunodeficiency virus type 1 Gag is substituted by a myristylation signal. J. Virol. 68:6644–6654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang CT, Zhang Y, McDermott J, Barklis E. 1993. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J. Virol. 67:7067–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Llewellyn GN, Grover JR, Olety B, Ono A. 2013. HIV-1 Gag associates with specific uropod-directed microdomains in a manner dependent on its MA highly basic region. J. Virol. 87:6441–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of HIV-1 Gag. J. Virol. 85:3802–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gregory DA, Lyddon TD, Johnson MC. 2012. Multiple Gag domains contribute to selective recruitment of murine leukemia virus (MLV) Env to MLV virions. J. Virol. 87:1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.