

Abstract
Chronic hepatitis B virus (HBV) infection, a serious public health problem leading to cirrhosis and hepatocellular carcinoma, is currently treated with either pegylated alpha interferon (pegIFN-α) or one of the five nucleos(t)ide analogue viral DNA polymerase inhibitors. However, neither pegIFN-α nor nucleos(t)ide analogues are capable of reliably curing the viral infection. In order to develop novel antiviral drugs against HBV, we established a cell-based screening assay by using an immortalized mouse hepatocyte-derived stable cell line supporting a high level of HBV replication in a tetracycline-inducible manner. Screening of a library consisting of 26,900 small molecules led to the discovery of a series of sulfamoylbenzamide (SBA) derivatives that significantly reduced the amount of cytoplasmic HBV DNA. Structure-activity relationship studies have thus far identified a group of fluorine-substituted SBAs with submicromolar antiviral activity against HBV in human hepatoma cells. Mechanistic analyses reveal that the compounds dose dependently inhibit the formation of pregenomic RNA (pgRNA)-containing nucleocapsids of HBV but not other animal hepadnaviruses, such as woodchuck hepatitis virus (WHV) and duck hepatitis B virus (DHBV). Moreover, heterologous genetic complementation studies of capsid protein, DNA polymerase, and pgRNA between HBV and WHV suggest that HBV capsid protein confers sensitivity to the SBAs. In summary, SBAs represent a novel chemical entity with superior activity and a unique antiviral mechanism and are thus warranted for further development as novel antiviral therapeutics for the treatment of chronic hepatitis B.
INTRODUCTION
Hepatitis B virus (HBV) infection is one of the major public health challenges worldwide. While the availability of a vaccine has reduced the number of new HBV infections, the vaccine does not benefit the 350 million people already chronically infected by the virus (1). In fact, approximately one-third of these chronically infected individuals will die from serious liver diseases, such as cirrhosis, hepatocellular carcinoma (HCC), and liver failure, if left untreated (2, 3). Currently, seven drugs have been approved for the treatment of chronic hepatitis B, which include two formulations of alpha interferon (standard and pegylated [IFN-α and pegIFN-α, respectively]) that enhance the host antiviral immune response and five nucleos(t)ide analogues (lamivudine, adefovir, entecavir, telbivudine, and tenofovir) that inhibit HBV DNA polymerase with various potencies and barriers to resistance (4, 5). At present, the preferred first-line treatment choices are pegylated alpha 2a interferon (pegIFN-α), entecavir, or tenofovir, based on their superior antiviral efficacy and/or high resistance barrier (6). However, even with the first-line treatment options, pegIFN-α is effective in achieving sustained virological response, defined as hepatitis e antigen (HBeAg) seroconversion and/or hepatitis B virus DNA levels below 20,000 copies/ml at 6 months after completion of the therapy, in only 30% of HBeAg-positive and 40% of HBeAg-negative cases (7–9). Moreover, pegIFN-α treatment is usually associated with severe side effects (7). On the other hand, the nucleos(t)ide analogues are well tolerated and potently suppress HBV replication in the vast majority of treated patients. It is generally acknowledged that the degrees of histological, biochemical, and serological improvement and reduction of HCC morbidity and mortality are correlated with the degree of viral replication suppression by the therapy (10). However, despite significant inhibition of viral replication, even the most potent nucleos(t)ide analogues, such as entecavir and tenofovir, fail to reliably clear HBV infection. Moreover, long-term, and possibly life-long, treatment is required to continuously suppress HBV replication, which is essential to achieve the clinical benefits of the therapy (10). Hence, there is a pressing need for the introduction of therapeutic regimens that are safer, more effective in achieving clinical endpoints, not hampered by resistance, and capable of inducing a more durable suppression of HBV replication following cessation of treatment.
To address these unmet medical needs, we set out to identify small-molecule inhibitors of HBV replication steps other than viral DNA synthesis. Ideally, these would develop into drug candidates. In eventual clinical use, such novel antiviral medications would offer the following therapeutic advantages. First, they may complement the current treatment regimen by providing additional options for a subpopulation of patients that do not tolerate or benefit from pegylated alpha 2a interferon and nucleos(t)ide analogues (11–14). Second, due to their distinct antiviral mechanisms, these novel antiviral drugs may be effective against HBV variants that are resistant to the nucleoside analogue viral DNA polymerase inhibitors (5). Third, similar to the highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV) infection (15), novel antiviral drugs used in combination with the current DNA polymerase inhibitors may synergistically suppress HBV replication and prevent the emergence of drug resistance, offering a more effective, and possibly curative, treatment for chronic hepatitis B (16).
HBV is the prototype member of Hepadnaviridae family, which also includes several animal hepatitis viruses, such as woodchuck hepatitis virus (WHV) and duck hepatitis B virus (DHBV). The genome of HBV is a relaxed circular partially double-stranded DNA (rcDNA; 3.2 kb in length) (17–19). Upon entry into a hepatocyte, the nucleocapsid delivers the genomic rcDNA into the nucleus, where the rcDNA is converted into covalently closed circular DNA (cccDNA). The cccDNA exists as an episomal minichromosome and serves as the template for the transcription of viral RNAs (20). The viral pregenomic RNA (pgRNA) is translated to produce both the core protein and DNA polymerase (21). The DNA polymerase binds to the epsilon sequence within the 5′ portion of pgRNA to prime viral DNA synthesis and initiate nucleocapsid assembly (22, 23). The encapsidated pgRNA is then reverse transcribed into minus-strand viral DNA, which serves as a template for the subsequent synthesis of plus-strand DNA by viral polymerase. The nucleocapsid matures as rcDNA is formed and can be enveloped and secreted out of the cell as a virion particle; alternatively, the nucleocapsid may deliver its rcDNA into the nucleus to amplify the nuclear cccDNA pool (24–26).
In order to discover novel drugs that inhibit HBV replication, we took advantage of an immortalized murine hepatocyte (AML12)-derived stable cell line that supports HBV pgRNA transcription and subsequent DNA replication in a tetracycline (Tet)-dependent manner and developed a convenient cell-based assay for the discovery of compounds that inhibit HBV replication steps between the biosynthesis of HBV proteins and reverse transcription of pgRNA into minus-strand viral DNA (27). Our high-throughput screening campaign has yielded a series of structurally related sulfamoylbenzamide (SBA) derivatives that potently inhibit the formation of pgRNA-containing nucleocapsids of HBV but not WHV and DHBV. Genetic complementation studies further demonstrated that HBV core proteins are the molecular targets of the SBAs. Not surprisingly, SBAs efficiently inhibited the replication of HBV mutants that are resistant to nucleos(t)ide analogues. Hence, the SBAs represent a novel chemical entity with a unique antiviral mechanism and favorable pharmacological properties and are thus warranted for further development as novel antiviral therapeutics for the treatment of chronic hepatitis B.
MATERIALS AND METHODS
Cell culture.
HepG2 cells were maintained in Dulbecco's modified Eagle's medium and nutrient mixture F-12 (DMEM/F-12) (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. AML12HBV10 and HepDES19 cells are, respectively, immortalized mouse hepatocyte (AML12)- and human hepatoma cell (HepG2)-derived stable cell lines supporting replication of an stably transfected envelope protein-deficient HBV genome in a tetracycline-inducible manner (27, 28). These two stable cell lines were maintained in DMEM/F-12 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml tetracycline, and 400 μg/ml G418. To initiate HBV pgRNA transcription and DNA replication in these cell lines, tetracycline was withdrawn from the culture medium.
Plasmids.
pCMVHBV, pCMVWHV, and pCMVDHBV that express the pgRNA of HBV, WHV, and DHBV, respectively, under the control of the cytomegalovirus immediate early (CMV IE) promoter have been described previously (29–31). The plasmids pTREHBV, pTREHBV/rtL180M/rtM204V (pTREHBV expressing mutations in the HBV reverse transcriptase [rt]), pTREHBV/rtA181V, pTREHBV/rtN236T, pTREHBV/rtM204I, and pTREHBV/rtL180M/rtM204V/rtT184G/rtS202I were reported previously (28, 32).
An HBV core-deficient (C−) plasmid, pCMVHBV/C−, was constructed by changing the start codon of the HBV core protein to CTG in the plasmid pCMVHBV with a QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's directions. The primers used for the mutagenesis are 5′-CTTGGGTGGCTTTGGGGCCTGGACATCGACCC-3′ (sense) and 5′-GGGTCGATGTCCAGGCCCCAAAGCCACCCAAG-3′ (antisense). Taking the same strategy, a WHV polymerase-deficient (P−) plasmid, pCMVWHV/P−, was constructed by introducing two tandem stop codons in the 5′ portion of the WHV polymerase gene with primers 5′-TTTGGTTCATCTAATGAGTTGTTGAATTTTCTTCC-3′ (sense) and 5′-TCAACAACTCATTAGATGAACCAAATTCTTTATAGGG-3′ (antisense). All the designed mutations were confirmed in the resulting plasmids by DNA sequencing.
For the construction of a plasmid expressing the WHV core protein, the core protein coding region (nucleotides 1997 to 2644) and its downstream sequence (nucleotides 901 to 1938) of the WHV genome (GenBank accession number J04514) were amplified by PCR with pCMVWHV as a template. The primers for the amplification of the core coding region are 5′-CGCGGATCCTGTGCCTTGGATGGCTTTG-3′ (sense) and 5′-TAAATGGCGGTAAGATGCTCGCAGCTTGGTTAGAGTAAAGACC TG-3′ (antisense). The primers for the amplification of the core fragment downstream sequence (nucleotides 901 to 1938) are 5′-GAGCATCTTACCGCCATTTATTCCC-3′ (sense) and 5′-GCTCTAGAGATACATGGTTACAGAAGTCGCATG-3′ (antisense). The two PCR fragments were merged by overlapping PCR with primers 5′-CGCGGATCCTGTGCCTTGGATGGCTTTG-3′ (sense) and 5′-GCTCTAGAGATACATGGTTACAGAAGTCGCATG-3′ (antisense). The resulting PCR product was digested with BamH I and XbaI and inserted into pcDNA3 digested with the same restriction enzymes. Similarly, the HBV core protein coding region (nucleotides 1902 to 2604) was amplified by using the primers 5′-ACCATGGACATCGACCCTTATAAAGAATTTG-3′ and 5′-CGGACCGAGTGGGCCTACAAACTGTTCACAT-3′ (the restriction endonuclease RsrII recognition site is underlined). The region coding the 3′ portion of HBV pgRNA (nucleotides 1573 to 1926) was amplified with primers 5′-CGGACCGTGTGCACTTCGCTTCACCTCT-3′ and 5′-GAGTAACTCCACAGTAGCTCCAAA-3′ (the RsrII recognition site is underlined). The aforementioned two PCR fragments were inserted into the pcDNA3.1/V5-His-TOPO vector in a head-to-tail manner after being cut by RsrII.
High-throughput screening assay.
AML12HBV10 cells were seeded into 96-well plates at a density of 2 × 104 cells per well and cultured in DMEM/F-12 medium with 10% fetal bovine serum in the absence of tetracycline. One day after seeding, cells in columns 1 and 12 of the plates were treated with medium containing 0.1% dimethyl sulfoxide (DMSO; mock-treated controls) and 1 μg/ml tetracycline, respectively. Each of the remaining 80 wells in columns 2 to 11 of the plates were treated with a library compound at a concentration of 10 μM. The library contains 26,900 drug-like small molecular compounds collected from multiple commercial providers (33). Forty-eight hours later, cells were lysed by the addition to each well of 100 μl of lysis buffer containing 10 mM Tris-HCl (pH 7.6), 1 mM EDTA, 100 mM NaCl, and 1% NP-40 and incubated at 37°C for 30 min. Half the amount (50 μl) of the cell lysate from each well was combined with an equal volume of denaturing solution containing 1N NaOH and 1.5 M NaCl. After a 5-min incubation, 100 μl of neutralization solution (1 M Tris-HCl, pH 7.4, 1.5 M NaCl) was added into each well. The denatured cell lysates were applied onto nylon membranes using a 96-well dot blot manifold (Bio-Rad). HBV DNA amounts in the cell lysates were determined by dot blot hybridization with an [α-32P]UTP-labeled riboprobe specific for HBV minus-strand DNA. The compounds that reduced the amount of HBV core DNA to less than 30% of that of the mock-treated controls were scored as primary “hits.”
To confirm the primary hits and determine their antiviral potency, AML12HBV10 cells were seeded into 96-well plates at a density of 2 × 104 cells per well and cultured in the absence of tetracycline. One day after seeding, cells were left untreated or treated with a serial dilution of testing compounds, ranging from 50 μM to 0.39 μM, for 48 h. Intracellular HBV core DNA amounts were determined by dot blot hybridization as described above. The antiviral efficacy of a compound was expressed as the effective concentration that reduced the amount of HBV DNA by 50% (EC50) compared to the levels of the untreated controls.
Cytotoxicity of the hit compounds was determined by plating and treating the AML12HBV10 cells in 96-well plates under exactly identical conditions used for the antiviral efficacy assay described above. The cell viability was measured by an MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay, following the procedure provided by the manufacturer (Promega). The cytotoxicity of a compound was expressed as the concentration of compound that reduced the viability of the cells by 50% (CC50) compared to the levels of the untreated controls.
Determination of antiviral efficacy and cytotoxicity in human hepatoma cells.
To further confirm the antiviral activity of the compounds against HBV in human hepatocyte-derived cells, HepDES19 cells were seeded into 12-well plates at a density of 5 × 105 cells per well and cultured in DMEM/F-12 medium with 1 μg/ml tetracycline. Two days after seeding, the cells were mock treated or treated with a serial dilution of compounds, ranging from 10 μM to 0.018 μM, for 6 days in the absence of tetracycline. The amounts of cytoplasmic HBV core-associated HBV DNA were extracted and determined by Southern blot hybridization (27). The antiviral efficacy of a compound was expressed as the concentration that reduced the amount of HBV DNA by 50% (EC50) in comparison with the levels of the mock-treated controls.
To determine the cytotoxicity of compounds in HepDES19 cells, the cells were seeded into 96-well plates at a density of 6 × 104 cells per well and cultured in DMEM/F-12 medium with 10% fetal bovine serum in the absence of tetracycline. One day after seeding, cells were left untreated or treated with a serial dilution of testing compounds, ranging from 50 μM to 0.39 μM, for 6 days. The cell viability was measured by an MTT assay, following a procedure provided by the manufacturer (Promega). The cytotoxicity of a compound was expressed as the concentration of compound that reduced the viability of the cells by 50% (CC50).
HBV RNA analysis.
Total cellular RNA was extracted with TRIzol reagents (Invitrogen). Five micrograms of total RNA was resolved in a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto Hybond-XL membrane in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer. Encapsidated HBV RNA was extracted as previously described (34). One half of the RNA sample from each well of the 12-well plates was resolved in a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto Hybond-XL membrane in 20× SSC buffer. For the detection of HBV RNA, membranes were probed with an [α-32P]UTP (800 Ci/mmol; PerkinElmer)-labeled plus-strand-specific full-length HBV riboprobe as described previously. The membrane was exposed to a phosphorimager screen, and hybridization signals were quantified with QuantityOne software (Bio-Rad).
Particle gel assay.
HBV capsids and associated viral DNA were analyzed by a native agarose gel electrophoresis-based assay (28). Briefly, AML12HBV10 or HepDES19 cells cultured in 12-well plates were lysed by the addition of 300 μl of buffer containing 10 mM Tris-HCl (pH 7.6), 100 mM NaCl, 1 mM EDTA, and 0.1% NP-40 per well. Cell debris was removed by centrifugation at 5,000 × g for 10 min. Ten microliters of the clarified cell lysates was fractionated by electrophoresis through nondenaturing 1% agarose gels and transferred to a nitrocellulose filter by blotting with TNE buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 1 mM EDTA). HBV capsids were detected by probing the membrane with an antibody against HBV core protein (Dako). Bound antibody was revealed by IRDye secondary antibodies and visualized by a Li-COR Odyssey system. To detect capsid-associated HBV DNA, the membranes were treated with a denaturing solution containing 0.5 N NaOH and 1.5 M NaCl for 5 min, followed by neutralization with a buffer containing 1 M Tris-HCl and 1.5 M NaCl for 5 min. The viral DNA was detected by hybridization with an [α-32P]UTP (800 Ci/mmol; PerkinElmer)-labeled minus-strand-specific full-length HBV riboprobe.
Determination of antiviral spectrum against hepadnaviruses.
HepG2 cells were seeded into six-well plates at a density of 1.2 × 106 cells per well and cultured in antibiotic-free complete DMEM/F-12 medium. Six hours after seeding, each well was transfected with 4 μg of pCMVHBV, pCMVWHV, or pCMVDHBV with Lipofectamine 2000 (Invitrogen) according to the manufacturer's directions. Six hours later, the transfected cells were mock treated or treated with indicated concentrations of compounds, as specified in the legend to Fig. 5, for 3 days. Intracellular HBV core DNA was extracted and analyzed by Southern blot hybridization (28, 34).
Fig 5.
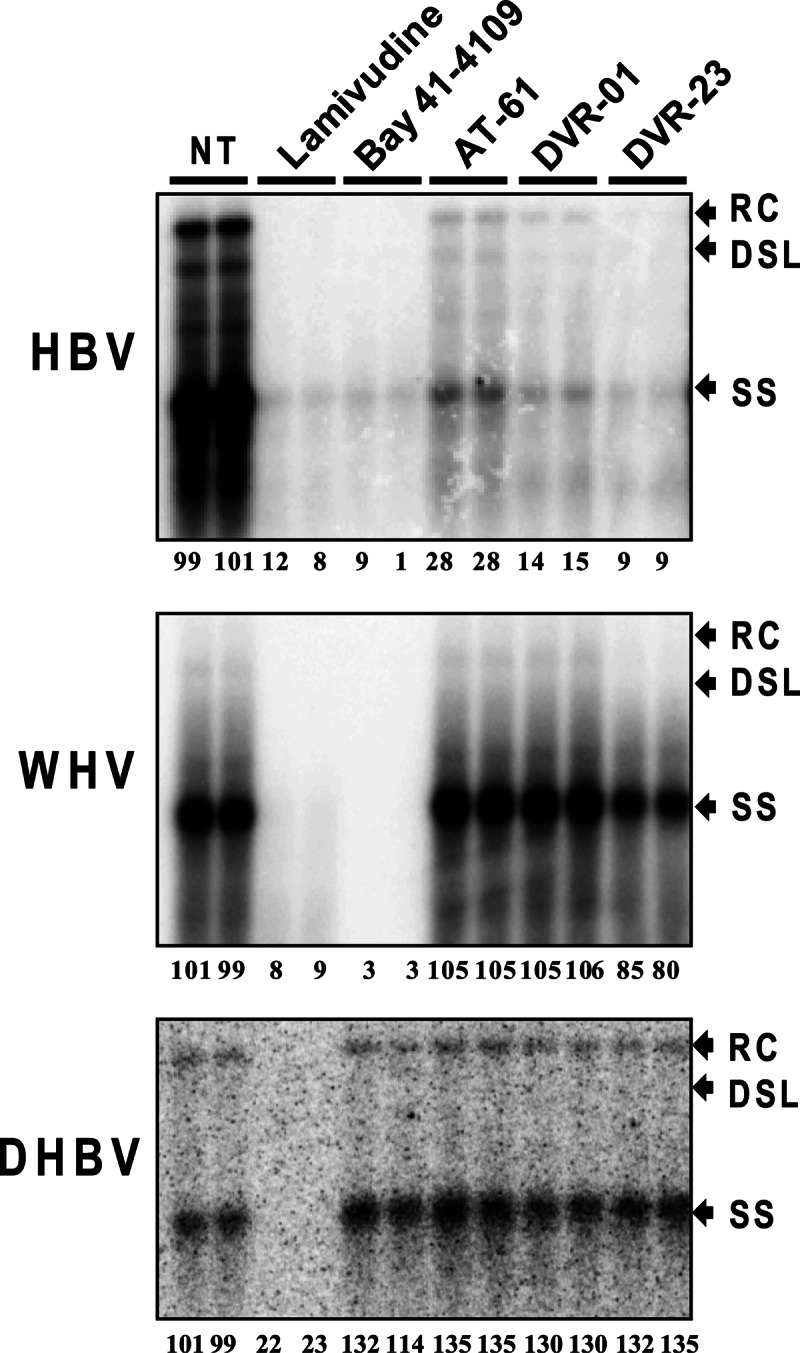
Antiviral spectrum of sulfamoylbenzamide derivatives against hepadnaviruses. HepG2 cells were transfected with plasmids pCMV-HBV, pCMV-WHV, and pCMV-DHBV. Six hours after transfection, the cells were mock treated or treated with 10 μM lamivudine, 2.5 μM Bay 41-4109, 25 μM AT-61, 10 μM DVR-01, or 5 μM DVR-23 for 3 days. Cytoplasmic core-associated viral DNA replication intermediates were extracted and determined by Southern blot hybridizations with a 32P-labeled full-length riboprobe specific to the minus strand of HBV, WHV, and DHBV. Relative viral DNA level in each sample is expressed as the percentage of the average level of viral DNA in the two mock-treated controls (NT) and is presented underneath each of the blots. RC, DSL, and SS indicate relaxed circular, double-stranded linear, and single-stranded viral DNA, respectively.
Determination of antiviral efficacy against nucleoside analogue-resistant HBV.
HepG2 cells were seeded into 24-well plates at a density of 2 × 105 cells per well and cultured in antibiotic-free complete DMEM/F-12 medium. Twenty-four hours after seeding, each well was cotransfected with 0.4 μg of pTREHBV or one of the pTREHBV-derived plasmids containing a drug resistance mutation(s) and 0.4 μg of pTet-off with Lipofectamine 2000 (Invitrogen) according to the manufacturer's directions. Six hours later, the transfected cells were mock treated or treated with six 1:4 dilutions of the compounds specified in Table 3 at concentrations ranging from 10 to 0.0097 μM for 2 days. Intracellular HBV core DNA was extracted as described previously (28). HBV core DNA was quantified by a real-time PCR assay using a LightCycler 480 SYBR green I Master PCR kit (Roche) with primers 5′-GGCTTTCGGAAAATTCCTATG-3′ (sense) and 5′-AGCCCTACGAACCACTGAAC-3′ (antisense). The PCR was carried out as follows: denaturing at 95°C for 5 min, followed by 40 cycles of amplification at 95°C for 15 s and at 60°C for 30 s.
Table 3.
Antiviral activity of representative SBA compounds against drug-resistant HBV mutants
| Compound | EC50 (μM) against the indicated HBVa |
|||||
|---|---|---|---|---|---|---|
| wt | rtL180 M/rtM204V | rtA181V | rtN236T | rtM204I | rtL180 M/rtM204V/rtT184G/rtS202I | |
| Lamivudine | 0.354 (±0.006) | >10 | 0.090 (±0.001) | 0.346 (±0.015) | >10 | >10 |
| Entecavir | 0.022 (±0.002) | 0.778 (±0.405) | 0.041 (±0.000) | 0.018 (±0.003) | 2.027 (±0.035) | >10 |
| Adefovir | 0.349 (±0.002) | 0.862 (±0.064) | 0.360 (±0.004) | 0.710 (±0.005) | 0.440 (±0.002) | 1.520 (±0.036) |
| DVR-01 | 2.899 (±0.140) | 3.273 (±0.107) | 3.180 (±0.347) | 2.752 (±0.173) | 2.567 (±0.057) | 2.403 (±0.569) |
| DVR-23 | 0.779 (±0.008) | 0.568 (±0.025) | 0.159 (±0.031) | 0.858 (±0.121) | 0.149 (±0.010) | 1.753 (±0.303) |
Mean values (± standard derivations) are presented from duplicate experiments.
Genetic complementation assay.
HepG2 cells were seeded into six-well plates at a density of 1.2 × 106 cells per well and cultured in antibiotic-free complete DMEM/F-12 medium. Six hours after seeding, each well was cotransfected with 2 μg of each of the indicated plasmids with Lipofectamine 2000 (Invitrogen) according to the manufacturer's directions. Six hours later, the transfected cells were mock treated or treated with the indicated concentrations of compounds, as specified in the legend to Fig. 6, for 3 days. Intracellular HBV or WHV core DNA was extracted and analyzed by Southern blot hybridization with an [α-32P]UTP (800 Ci/mmol; PerkinElmer)-labeled minus-strand-specific full-length HBV or WHV riboprobe (28, 34).
Fig 6.

Identification of the molecular target of sulfamoylbenzamide derivatives by genetic complementation assays between HBV and WHV. HepG2 cells were cotransfected with plasmids pCMVHBV/C− and pcDNA3/HBVcore (A), pCMVHBV/C− and pcDNA3/WHVcore (B), or pCMVHBV/C− and pCMVWHV/P− (C and D). Six hours after transfection, the cells were mock treated or treated with 10 μM lamivudine, 2.5 μM Bay 41-4109, 25 μM AT-61, or 10 μM DVR-01 for 3 days. Cytoplasmic core-associated viral DNA replication intermediates were extracted and determined by Southern blot hybridizations with a 32P-labeled full-length riboprobe specific to the minus strand of HBV (A, B, and C) and WHV (D). RC and SS indicate relaxed circular and single-stranded HBV DNA, respectively. The sources of the capsid protein, polymerase, pgRNA, and probe for hybridization for each of the experiments are presented below each blot.
RESULTS
The AML12HBV10 cell line is suitable for screening compounds that inhibit HBV replication.
Discovery of novel antiviral compounds against HBV is hampered by a lack of cell-based assays suitable for high-throughput screening of compound libraries. Currently, the antiviral activity of compounds against HBV is usually assayed in human hepatoma (HepG2)-derived HBV replicating cell lines, such as HepG2.2.15 (35), HepAD38 (36), and HepDES19 (28), which typically require more than 6 days of incubation with drugs due to the slow dynamics of HBV replication in these cell lines (29, 37, 38). Recently, we established an immortalized mouse hepatocyte (AML12)-derived stable cell line (AML12HBV10) that supported HBV pgRNA transcription and subsequent DNA replication in a tetracycline (Tet)-dependent manner (27). In the presence of Tet, the transcription of viral pgRNA is suppressed. However, upon removal of Tet from the culture medium, HBV pgRNA is transcribed and accumulates to a detectable level at 24 h, reaching a peak level at 96 h. As expected, following pgRNA transcription, HBV DNA replication intermediates can be detected at 48 h and increase 50- to 100-fold, reaching steady-state level at 96 h after Tet removal (27). The robustness and great dynamic range of HBV replication in this cell line between 48 to 96 h after Tet removal allow the measurement of the antiviral effects of compounds following only 2 days of treatment (27), which is ideal for an anti-HBV drug screening assay.
The feasibility of the AML12HBV10 cell-based antiviral assay in a 96-well plate format was evaluated with three antiviral compounds that inhibit distinct steps of HBV replication, namely, lamivudine, Bay 41-4109, and AT-61 (Fig. 1A). Briefly, AML12HBV10 cells maintained in Tet-free medium for 24 h were seeded into 96-well plates. One day after seeding, cells were left untreated or treated with either 1 μg/ml of Tet to shut off pgRNA transcription or a serial dilution of lamivudine, Bay 41-4109, or AT-61 for 48 h. Cells were then lysed, and HBV DNA amounts in the cell lysates were determined by dot blot hybridization assays. Under this condition, the signal-to-noise ratio of HBV DNA signals between mock- and tetracycline-treated cultures was about 100:1, and the Z′ value, a measurement of statistical reproducibility and variation of the assay, was 0.56 to 0.7 (Fig. 1B and data not shown). As shown in Fig. 1, all three antiviral drugs dose dependently reduced the amounts of HBV DNA. However, while the two HBV capsid assembly effectors, Bay 41-4109 and AT-61, demonstrated similar antiviral potencies in AML12HBV10 cells as that reported in human hepatoma cells (39, 40), the HBV DNA polymerase inhibitor lamivudine only modestly inhibited HBV DNA replication in the mouse hepatocytes, most likely due to inefficient phosphorylation of the compound in rodent hepatocytes (41). Hence, the AML12HBV10 cell-based assay is suitable for screening compounds other than nucleoside analogues for their ability to inhibit any step of HBV replication between the translation of viral proteins and synthesis of viral DNA inside nucleocapsids.
Fig 1.
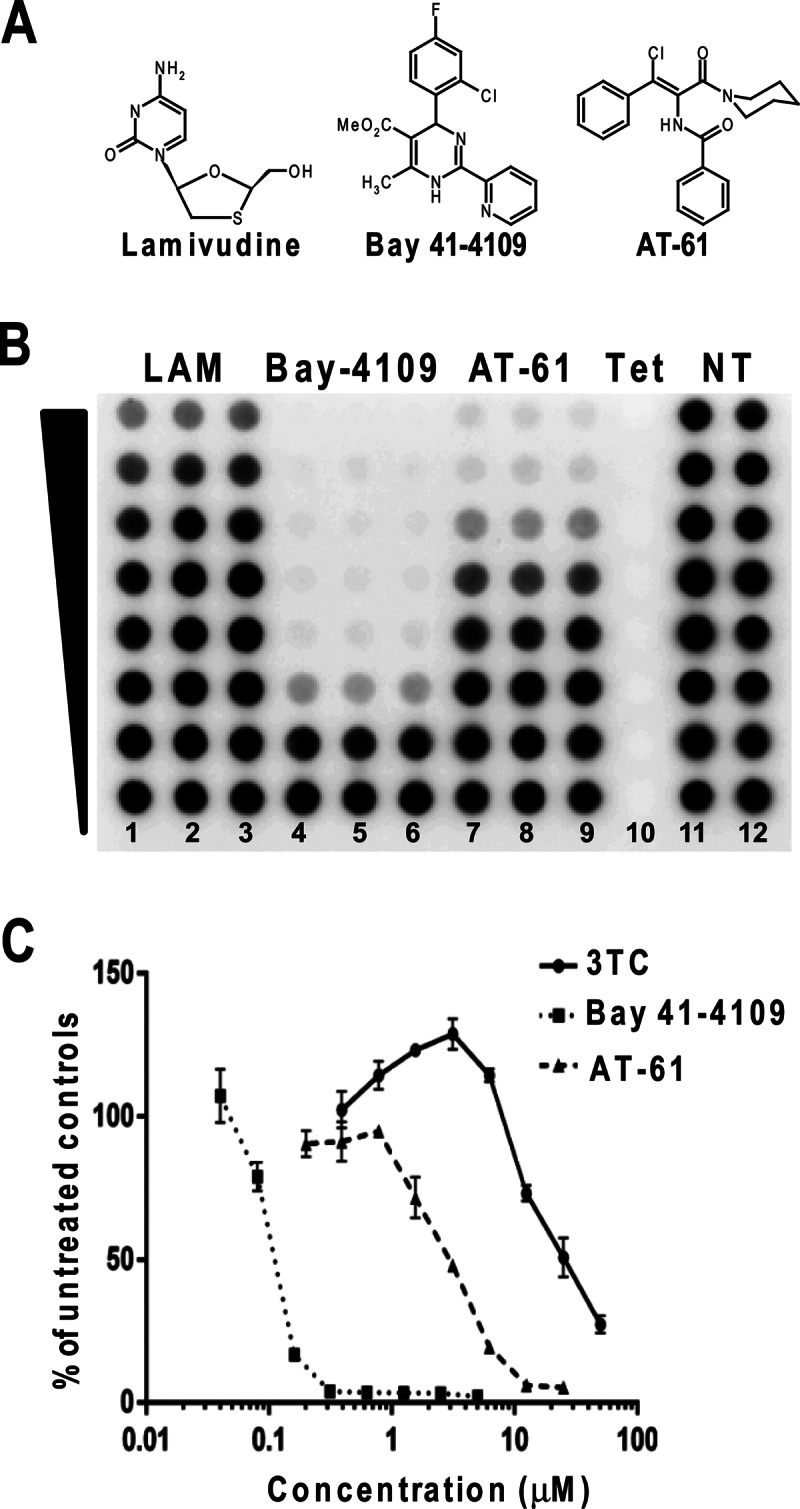
Experimental demonstration of the utility of the AML12HBV10 cell-based assay for the discovery of antiviral drugs against HBV. (A) Structures of three known anti-HBV compounds. (B) AML12HBV10 cells were seeded into a 96-well plate and cultured in the absence of Tet for 24 h. Cells were then left untreated (control, columns 11 and 12) or treated with 1 μg/ml Tet (column 10), serial 2-fold dilutions of lamivudine (100 μM to 0.78 μM; columns 1 to 3), Bay 41-4109 (5 μM to 0.039 μM; columns 4 to 6), and AT-61 (25 μM to 0.39 μM; columns 7 to 9) for 2 days. Cell lysates were blotted onto a nylon membrane and hybridized with a 32P-labeled full-length riboprobe specific to the minus strand of HBV. The HBV DNA hybridization signals were revealed by a phosphorimager and quantified with QuantityOne software (Bio-Rad). (C) The HBV DNA amounts in the drug-treated wells were expressed as percentages of the values of the mock-treated controls. The means and standard deviations (n = 3) were plotted.
Discovery of sulfamoylbenzamide derivatives as HBV replication inhibitors.
The primary screening of 26,900 compounds at a 10 μM concentration in a 96-well plate format identified 120 compounds that reduced the amount of HBV core DNA by more than 70% compared to levels in the mock-treated controls. All the primary hits were further tested in AML12HBV10 cells at an increased range of concentrations (0.39 to 50 μM) to determine antiviral activity (EC50) by dot blot hybridization and cellular toxicity (CC50) by an MTT assay. A total of 40 compounds were confirmed to selectively inhibit HBV replication with EC50s of less than 10 μM and without measurable cytotoxicity up to 50 μM. Among the 40 confirmed hits, 36 compounds share a sulfamoylbenzamide pharmacophore moiety with functionality at the sulfonamide, amide, and the middle phenyl sites (Fig. 2). Another four compounds are benzamide derivatives, which will be reported elsewhere. Based on the different substitution patterns at the amide linker, the sulfamoylbenzamide (SBA) derivatives can be categorized into two groups, group 1 and group 2. Structures of representative compounds, DVR-01 and DVR-23, from each group are presented in Fig. 2.
Fig 2.

Structures of representative hits from the high-throughput screening. General formulas of the two classes of sulfamoylbenzamides and representative hit compounds of each class are presented.
Structure-activity relationship (SAR).
Upon discovering SBA derivatives that inhibited HBV replication, a structure similarity search of our in-house chemical libraries consisting of 87,000 drug-like molecules revealed a total of 591 SBA derivatives, in which 12 and 579 compounds were categorized into group 1 and group 2, respectively. Study of the 12 group 1 SBA derivatives in the AML12HBV10 and HepDES19 cells reveals that the Rx position (Fig. 2) can accommodate cycloalkyl and open-ring alkyl structures. However, when Rx is replaced with morpholine moiety, the activity is totally lost. Halide substitution at the R7 and R9 positions proves to be beneficial to the activity. The compound DVR-01 with a chlorine atom at the R9 and a cycloheptyl ring at the Rx position displayed the best activity, with EC50s of 1.7 and 1.6 μM in AML12HBV10 and HepDES19 cells, respectively (Fig. 3 and data not shown). The compound has no measurable toxicity in either cell line at 50 μM.
Fig 3.
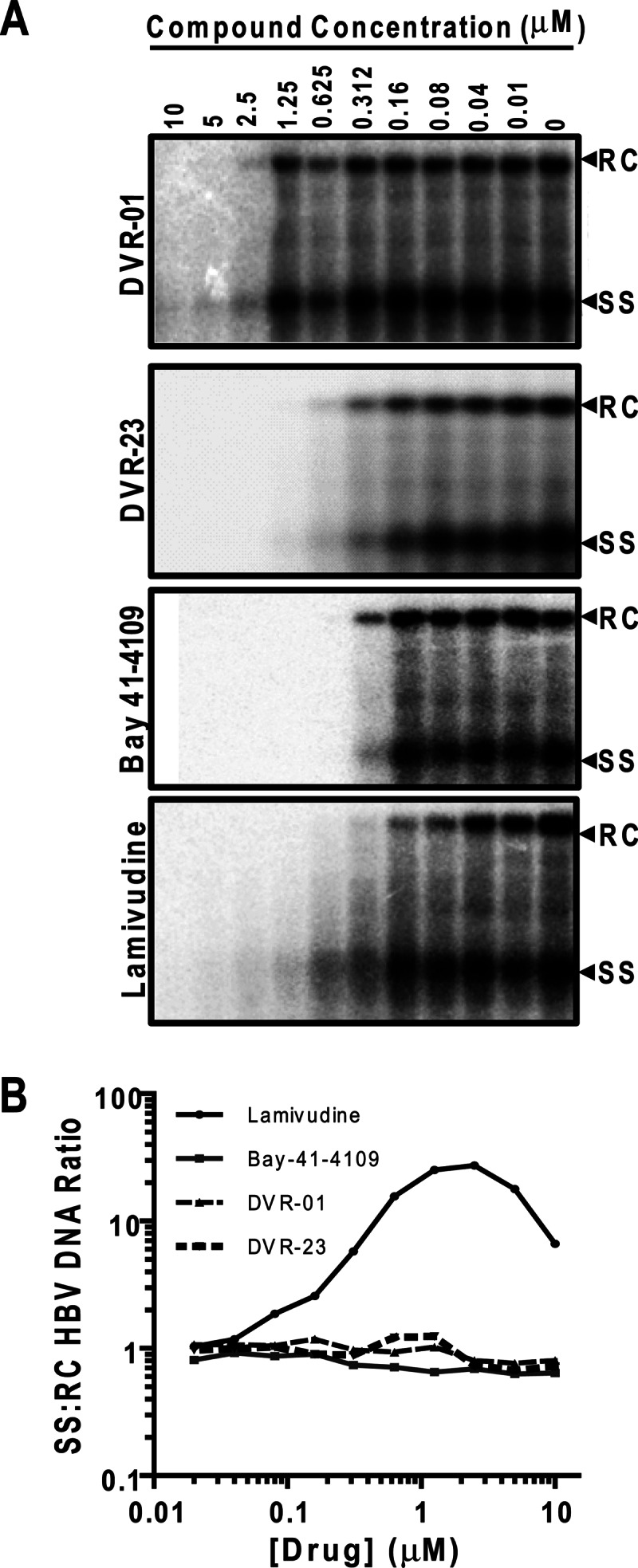
The antiviral activity of two representative sulfamoylbenzamide compounds against HBV in human hepatoma cells. (A) HepDES19 cells were left untreated (0) or treated with the indicated concentrations (μM) of DVR-01, DVR-23, lamivudine, or Bay 41-4109 for 4 days. Cytoplasmic core-associated HBV DNA replication intermediates were extracted and determined by Southern blot hybridizations. (B) The ratios of single-stranded (SS) to relaxed circular (RC) HBV DNA were determined.
Study of the 579 group 2 SBA derivatives showed that the most potent compounds in this series, DVR-23, DVR-43, and DVR-56, displayed sub- or low-micromolar activity in both AML12HBV10 and HepDES19 cells (Fig. 2 and Table 1). All three compounds have been resynthesized to gram quantity in-house. A side-by-side comparison demonstrated that the antiviral potency in human hepatocyte-derived cells of the representative analog DVR-23 is similar to that of two well-known HBV inhibitors, lamivudine and Bay 41-4109 (Fig. 3A). DVR-23, DVR-43, and DVR-56 share common structural features, with fluorine substitution at R2, R3, and R7 and alkyl substitution at the Rx site. This could be the best combination for optimal activity. Based on analysis of the biological data for the 579 compounds, small cycloalkyl or open alkyl substitutions at Rx confer the most potent antiviral activity (Table 1). However, the R2, R3, R4, R7, R9, and Rx sites can also tolerate other substitution patterns for activity. Further lead optimization efforts are being carried out, and the findings will be reported elsewhere.
Table 1.
Structure and activity relationship of SBA compounds against HBV in murine and human hepatocytes
| Compound | RX | Antiviral activity by cell line |
|||
|---|---|---|---|---|---|
| AML12HBV10 |
HepDES19 |
||||
| EC50 (μM)a | CC50 (μM) | EC50 (μM)a | CC50 (μM) | ||
| DVR-23 | 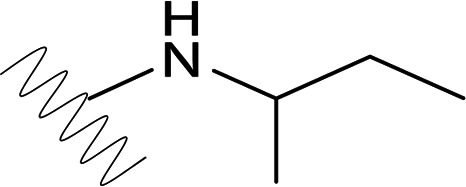 |
0.3 (±0.05) | >50 | 0.1 (±0.01) | >50 |
| DVR-25 | 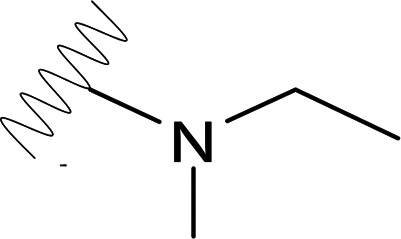 |
1.07 (±0.35) | >50 | 2.58 (±1.70) | >50 |
| DVR-34 | 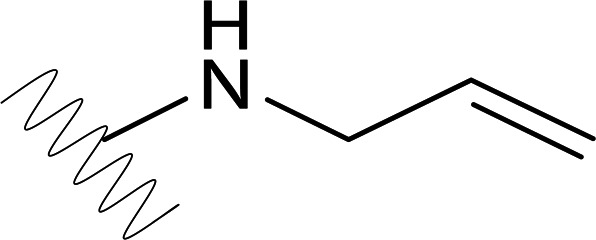 |
1.35 (±0.29) | >50 | 2.21 (±0.04) | >50 |
| DVR-43 | 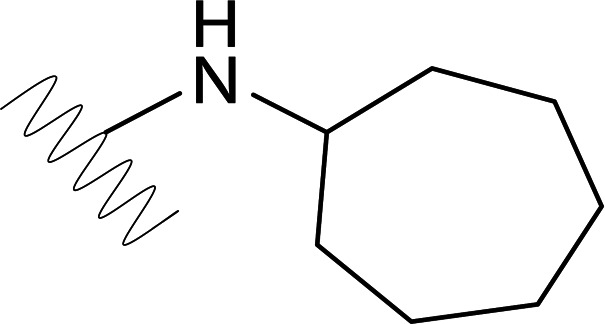 |
1.16 (±0.1) | >50 | 2.32 (±1.22) | >50 |
| DVR-45 | 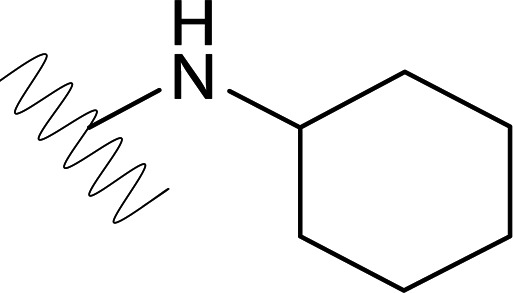 |
1.17 (±0.15) | >50 | 0.46 (±0.38) | >50 |
| DVR-56 | 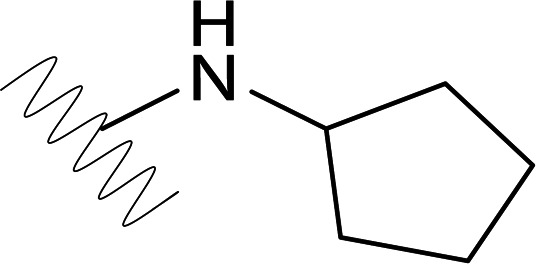 |
0.39 (±0.03) | >50 | 0.14 (±0.09) | >50 |
| DVR-82 | 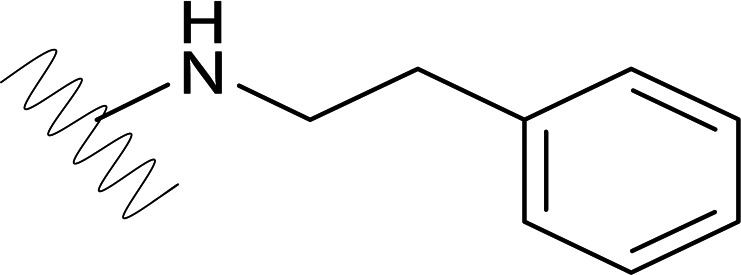 |
0.83 (±0.29) | >50 | 2.22 (±0.64) | >50 |
| DVR-92 | 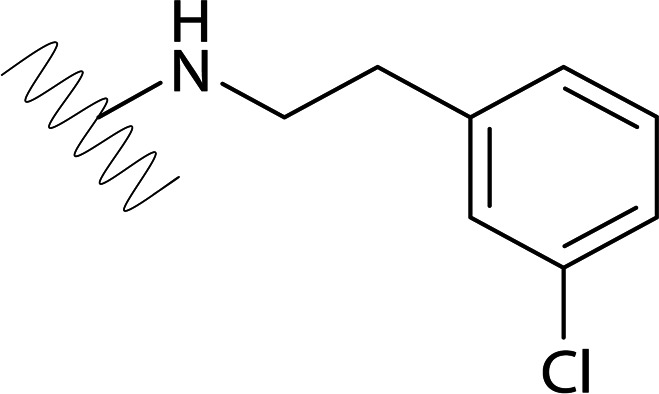 |
2.73 (±0.28) | >50 | 1.64 (±1.48) | >50 |
| DVR-0104 | 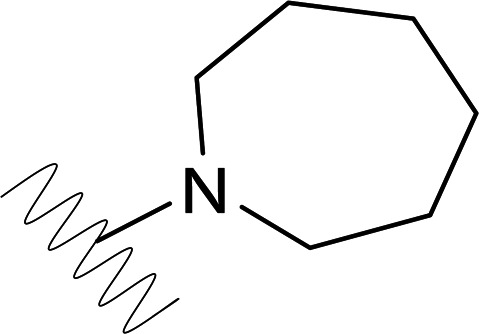 |
4.43 (±1.09) | >50 | 3.80 (±0.29) | >50 |
| DVR-0118 | 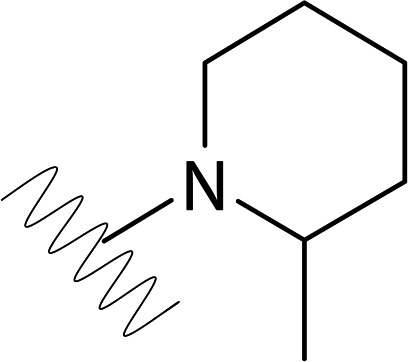 |
>50 | >50 | NDb | ND |
| DVR-0119 | 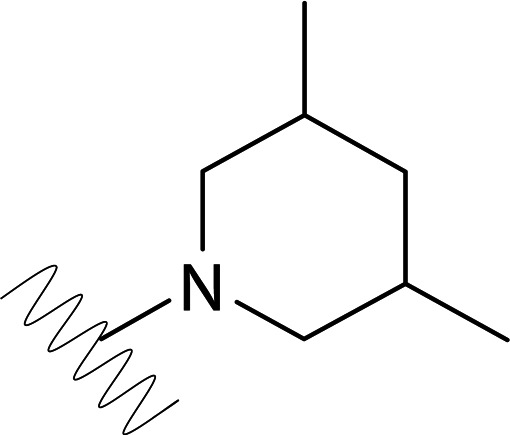 |
10.19 (±2.01) | >50 | 6.99 (±1.83) | >50 |
| DVR-0120 | 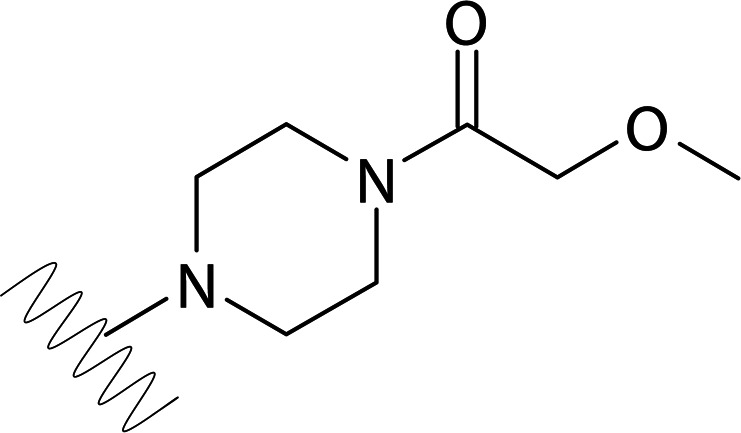 |
>50 | >50 | ND | ND |
| DVR-0121 | 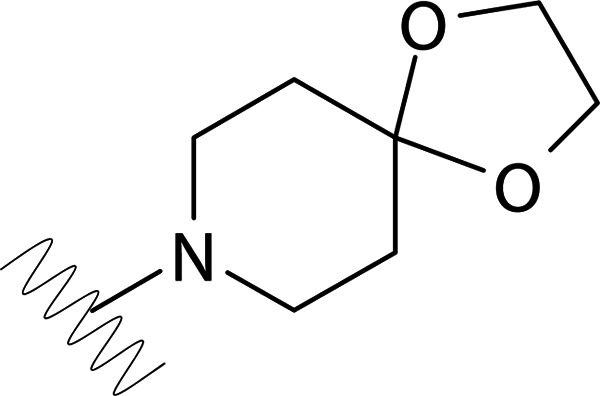 |
>50 | >50 | ND | ND |
| DVR-0122 | 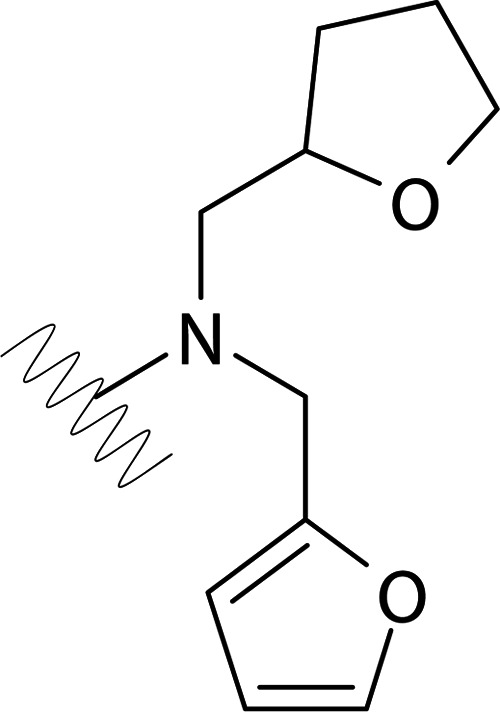 |
>50 | >50 | ND | ND |
| DVR-0123 | 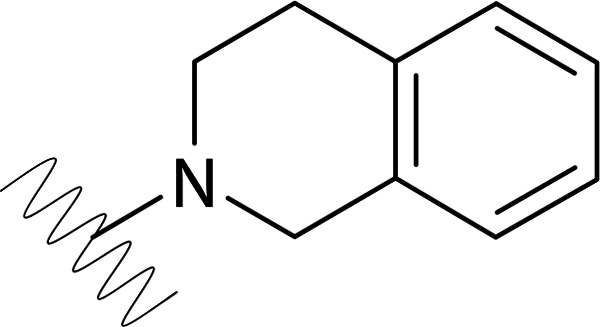 |
>50 | >50 | ND | ND |
| DVR-0124 | 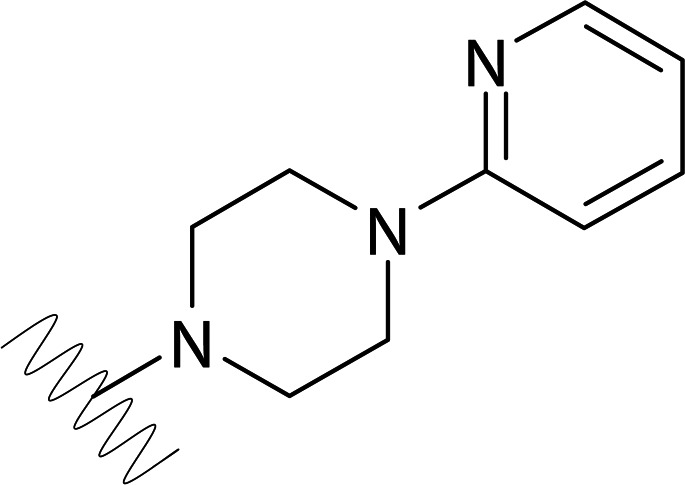 |
>50 | >50 | ND | ND |
| DVR-0125 | 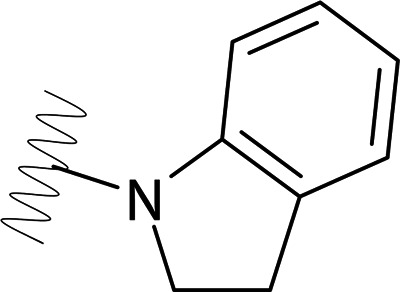 |
>50 | >50 | ND | ND |
Mean values (± standard derivations) are presented from triplicate experiments.
ND, not done.
SBAs inhibit the formation of pgRNA-containing nucleocapsids.
As mentioned above, our screening assay was designed to identify the compounds that inhibited the molecular events between viral protein synthesis and DNA replication. Interestingly, the Southern blot hybridization results presented in Fig. 3A clearly demonstrated that the molecular profiles of HBV DNA replication intermediates in HepDES19 cells treated with DVR-01, DVR-23, or Bay 41-4109 were distinct from those treated with lamivudine. As a viral DNA polymerase inhibitor, lamivudine directly inhibited the elongation of HBV DNA synthesis as a chain terminator and thus prematurely arrested viral DNA synthesis. As a consequence, lamivudine treatment resulted in the accumulation of immature forms of HBV DNA, such as single-stranded DNA, but caused a reduction in the amounts of the mature rcDNA. In marked contrast, Bay 41-4109, DVR-01, or DVR-23 treatment of HepDES19 cells did not apparently alter the ratios of the single-stranded viral DNA to rcDNA (Fig. 3B), suggesting that, like Bay 41-4109, a capsid assembly inhibitor, the two SBAs did not directly affect viral DNA synthesis but most likely inhibited a viral replication step prior to viral DNA synthesis.
To further map the HBV replication step(s) inhibited by the SBAs, AML12HBV10 cells were treated with a serial dilution of three representative SBAs for 48 h. As positive controls, the cells were also treated with Bay 41-4109 or AT-61, the known HBV nucleocapsid assembly effectors that either inhibit capsid formation (Bay 41-4109) (40, 42) or prevent the encapsidation of viral pgRNA into nucleocapsids (AT-61) (39, 43, 44). As shown in Fig. 4, consistent with the proposed mechanism, a particle gel assay revealed that Bay 41-4109 treatment completely abolished capsid formation (Fig. 4B) and thus pgRNA encapsidation and DNA synthesis (Fig. 4C, D, and E). In contrast, AT-61 treatment did not affect capsid formation (Fig. 4B) but reduced the amounts of encapsidated pgRNA (Fig. 4C) and the capsid-associated HBV DNA (Fig. 4D and E). Similar to AT-61, all three SBA compounds did not significantly alter the amounts of total HBV capsids (Fig. 4B) but reduced encapsidated pgRNA and capsid-associated HBV DNA in a dose-dependent manner (Fig. 4C, D, and E). The results thus imply that, like AT-61, the SBAs did not affect the formation of empty capsids but dose dependently inhibited the formation or accelerated the decay of pgRNA-containing nucleocapsids. As a consequence, subsequent HBV DNA replication could not occur.
Fig 4.

Antiviral mechanism of sulfamoylbenzamide derivatives against HBV. AML12HBV10 cells were left untreated (control) or treated with the indicated concentrations of the compounds of DVR-01, DVR-56, and DVR-23 for 2 days. Bay 41-4109 (5 μM) or AT-61 (25 μM) served as a positive control. (A) Intracellular viral RNA was determined by Northern blot hybridization. 28S and 18S rRNA served as loading controls. (B) The total amounts of nucleocapsids were determined by a particle gel assay. (C) Encapsidated pgRNA was extracted and measured by Northern blotting. (D) Nucleocapsid-associated HBV DNA was quantified by alkaline treatment of nucleocapsids on the membrane following the particle gel assay and hybridized with a 32P-labeled HBV-specific riboprobe. (E) HBV DNA replication intermediates were extracted and determined by Southern blot hybridizations. Relative HBV RNA, capsids, or DNA level in each sample is expressed as the percentage of the average level of RNA, capsids, or DNA in the two mock-treated controls and is presented underneath each of the blots. RC, DSL, and SS indicate relaxed circular, double-stranded linear, and single-stranded HBV DNA, respectively.
SBAs do not inhibit WHV and DHBV replication.
Nucleocapsid assembly of hepadnaviruses requires the orchestrated interaction of multiple viral and host components (45). The process begins with the binding of viral DNA polymerase, assisted by the host cellular heat shock protein 90 (HSP90) chaperone complex, to the stem-loop structure (ε) located at the 5′ end of pgRNA (22, 45–48). The protein-pgRNA complex then recruits core protein dimers or oligomers to assemble into a nucleocapsid (49, 50). It is therefore conceivable that the observed inhibition of pgRNA-containing nucleocapsid formation by SBAs could result from a direct or indirect alteration of the structure and/or function of any host and/or viral components involved in nucleocapsid assembly. In order to identify the host or viral component(s) that mediate the antiviral activity of SBAs, we first tested the antiviral activity of the compounds against other animal hepadnaviruses, including WHV and DHBV, in human hepatoma cells. This simple assay not only determines the antiviral spectrum of the compounds, which is important for selection of proper animal models for in vivo antiviral efficacy studies in the future, but also helps to determine if any of the common host components required for the folding and subsequent binding of viral DNA polymerase to pgRNA are targeted by the SBA compounds.
The results presented in Fig. 5 and Table 2 show that, as expected, the viral DNA polymerase inhibitor lamivudine inhibited the replication of all three hepadnaviruses. Bay 41-4109 efficiently inhibited HBV and WHV but not DHBV replication, suggesting that the compound is able to bind and disrupt the interaction of the capsid proteins of only HBV and its closely related WHV but not distantly related DHBV. Interestingly, like AT-61, the two representative SBA compounds efficiently inhibited the replication of HBV but did not significantly inhibit WHV and DHBV. These results indicate that the HSP90 chaperone complex required for the folding and pgRNA binding of both mammalian and avian hepadnavirus DNA polymerases is most likely not the target of the two classes of HBV replication inhibitors (51–54). Instead, the compounds may directly or indirectly target one of three HBV components (pgRNA, DNA polymerase, and capsid protein) that are essential for nucleocapsid assembly.
Table 2.
Antiviral spectrum of the representative SBA compounds against hepadnaviruses in human hepatoma cells
| Compound | EC50 (μM) against:a |
||
|---|---|---|---|
| HBV | WHV | DHBV | |
| Lamivudine | 0.354 (±0.006) | ND | 0.433 (±0.057) |
| Bay 41-4109 | 0.343 (±0.012) | 0.193 (±0.029) | >2 |
| AT-61 | 8.5 | >20 | >20 |
| DVR-01 | 2.899 (±0.140) | >10 | >25 |
| DVR-23 | 0.779 (±0.008) | 3.867 (±0.856) | >10 |
Mean values (± standard derivations) are presented from duplicate experiments. ND, not done.
HBV core protein confers the sensitivity of the virus to SBAs.
It was demonstrated previously that HBV and WHV polymerases were each competent to recognize both HBV and WHV pgRNA packaging signals to form heterologous DNA polymerase-pgRNA complexes and to be encapsidated by either HBV or WHV capsid protein (55, 56). The differential sensitivity of HBV and WHV to the SBAs provided us a unique opportunity to identify the viral target of the compounds through a heterologous genetic complementation study of the capsid protein, DNA polymerase, and pgRNA between HBV and WHV.
We first intended to determine whether SBAs were able to inhibit the encapsidation of the HBV DNA polymerase-pgRNA complex by WHV capsid protein and subsequent HBV DNA synthesis. To this end, a plasmid encoding a capsid protein-deficient HBV pgRNA was cotransfected with a plasmid expressing either HBV or WHV capsid protein into HepG2 cells. The transfected cells were mock treated or treated with the indicated concentrations of lamivudine, Bay 41-4109, AT-61, or DVR-01, as specified in the legend to Fig. 6, for 3 days. As shown in Fig. 6A and B, analysis of cytoplasmic HBV core DNA by Southern blot hybridization demonstrated that, as expected, all the five drugs efficiently inhibited HBV DNA replication in the cells cotransfected with the plasmid expressing HBV capsid protein. In contrast, only lamivudine and Bay 41-4109 efficiently inhibited HBV DNA replication in the cells cotransfected with the plasmid expressing WHV capsid protein. The results thus indicate that AT-61 and DVR-01 selectively disrupt the pgRNA encapsidation by HBV but not WHV capsid protein. In other words, it is the HBV capsid protein that confers the sensitivity of the virus to SBAs and AT-61.
To further validate this notion, the plasmid encoding core-deficient HBV pgRNA was cotransfected into HepG2 cells with a plasmid expressing a polymerase-deficient WHV pgRNA. In the transfected cells, core protein was provided by WHV, but DNA polymerase was provided by HBV. However, the pgRNAs from both HBV and WHV were competent for interaction with the HBV polymerase and thus were encapsidated by WHV capsid protein to form two distinct populations of nucleocapsids that synthesized HBV and WHV DNA, respectively. Indeed, Southern blot analysis of cytoplasmic core DNA revealed the synthesis of both HBV and WHV DNA in the transfected cells was efficiently inhibited by lamivudine and Bay 41-4109 but not AT-61 and DVR-01 (Fig. 6C and D). The results thus reinforce the notion that HBV capsid protein, but not DNA polymerase and pgRNA, is directly or indirectly targeted by the two antiviral compounds, and thus they selectively inhibit HBV but not WHV replication.
SBAs efficiently inhibit the replication of nucleos(t)ide analogue-resistant HBV variants.
Long-term treatment of chronic hepatitis B with nucleos(t)ide analogs can lead to the emergence of drug-resistant HBV variants that bear mutations in the polymerase gene of the virus and that ultimately result in treatment failure (5). Antivirals effective against HBV variants resistant to the currently available HBV DNA polymerase inhibitors would have enormous clinical value. Based on their distinct antiviral mechanism, we expected that SBA compounds should be effective against nucleos(t)ide analogue-resistant HBV. To confirm this hypothesis, HepG2 cells were transiently transfected with a plasmid expressing wild-type (wt) HBV or one of the five main HBV mutants that are resistant to lamivudine, adefovir, and/or entecavir and treated with one of the three nucleoside analogues or the two representative SBAs for 2 days. As expected, all five compounds efficiently inhibited wt HBV DNA replication (Table 3). Their antiviral potency against the wt HBV in this transient-transfection assay system is generally consistent with that reported by other investigators or obtained with the stable cell line-based assays reported herein (57). Also in agreement with the previous reports, HBV variants bearing the mutation(s) rtM204I, rtL180M/204V, or rtL180M/rtM204V/rtT184G/rtS202I were resistant to lamivudine and entecavir but sensitive to adefovir (58, 59). As expected, the two known adefovir-resistant HBV mutants were sensitive to lamivudine and entecavir. However, they were also sensitive (rtA181V) or only slightly resistant (rtN236T) to adefovir under this experimental condition. Interestingly, as predicted, all nucleoside analogue-resistant viruses were as sensitive to DVR-01 and DVR-23 as the wt HBV. The antiviral efficacy of the two SBAs in this assay system is consistent with that obtained in HepDES19 and AML12HBV10 cells (Tables 1 and 3). Hence, these results suggest that targeting nucleocapsid functions may represent an interesting approach to the development of novel HBV inhibitors to prevent and combat nucleos(t)ide analogue drug resistance.
DISCUSSION
The therapeutic goals of chronic hepatitis B treatment are to restore normal liver function and prevent the development of cirrhosis, HCC, and liver failure. It has been convincingly demonstrated thus far that inhibition of HBV replication by the currently available antiviral drugs results in significant histological, biochemical, and serological improvement and reduction of HCC mortality and morbidity (60). However, the current antiviral therapeutics fail to resolve HBV infection in the vast majority of treated patients, and the achievement of therapeutic goals in these patients requires long-term, possibly life-long, treatment, which may ultimately fail due to the emergence of drug resistance and/or intolerability from the long-term use of these drugs (10). Hence, there is a pressing need for therapeutics that are able to either cure HBV infection or induce a durable off-drug suppression of the virus, which requires elimination and/or transcriptional silencing of cccDNA, the most stable HBV replication intermediate. We believe that therapeutic regimes with such pharmacological properties are most likely to come in the form of combination therapies with multiple drugs that target different steps of HBV replication. Thus, cccDNA formation can be prevented through more efficient inhibition of nucleocapsid assembly or rcDNA production and conversion to cccDNA, as well as by suppressing the emergence of drug-resistant mutants and accelerating the loss of preformed cccDNA.
To fulfill this goal, we have been developing cell-based assays suitable for the discovery of antiviral drugs that inhibit HBV replication in a high-throughput manner (33, 61, 62). The AML12HBV10 cell-based assay reported herein is a reliable system for discovering antivirals inhibiting key steps of viral replication, such as polymerase folding and interaction with pgRNA, priming of minus-strand DNA synthesis, nucleocapsid assembly, and DNA synthesis. Although the necessity to quantify HBV DNA via dot blot hybridization significantly reduces the throughput of the assay, to our knowledge, this is still the most efficient cell-based assay for screening HBV replication inhibitors due to easier handling of the cells, robust HBV replication, and the short time of drug treatment. Except for certain nucleoside analogues, the antiviral activity of compounds in AML12HBV10 cells is generally consistent with that obtained in human hepatoma-derived cell lines. It is expected that automation of HBV DNA quantification technology in the future will transform this assay into a true high-throughput system.
Discovery of the SBA derivatives as antiviral agents against HBV provides a novel chemical entity for the development of therapeutics to treat chronic hepatitis B. In addition to their superior antiviral activity, the SBAs are attractive drug-like molecules that meet the classic Lipinsky rules of five, including calculated logP (cLogP) values of <5.0, molecular weight of <500, and acidic pKa values that suggest good oral bioavailability (63). Indeed, the lead SBAs, DVR-23 and DVR-56, demonstrated favorable in vitro absorption, distribution, metabolism, and excretion (ADME) characteristics and pharmacokinetic profiles in mice (data not shown). The sulfonamide moiety has also been the basis of pharmacophores for drugs that are currently on the market. Our SAR studies have thus far focused on the Rx moiety (selected analogs are shown in Table 1), and additional modifications on the amide and phenyl rings are being explored to further improve the antiviral potency and pharmacological properties of these compounds.
In all the mechanistic studies performed so far, the SBAs demonstrated the same phenotype as AT-61. Specifically, both AT-61 and SBAs do not apparently affect the formation of empty capsids but dose dependently inhibit the formation of pregenomic RNA-containing nucleocapsids of HBV but not other hepadnaviruses, such as WHV and DHBV (Fig. 4 and 5). Like AT-61 (16, 64), the SBAs inhibited wild-type and nucleoside-resistant HBV with similar potencies (Table 3). Furthermore, it is the HBV capsid protein, but not polymerase or pgRNA, that confers the sensitivity of HBV to both AT-61 and SBAs. In fact, this is the first biological evidence suggesting that HBV capsid protein is the molecular target of AT-61 (Fig. 6).
Interestingly, AT-61 and its analogue AT-130, two representative phenylpropenamide derivatives (65), were reported recently by Katen et al. as HBV capsid assembly effectors (44). Unlike heteroaryldihydropyrimidines (HAPs; Bay 41-4109 as an example), which interact with HBV capsid assembly intermediates to misdirect the assembly reaction and induce the formation of noncapsid polymers (42, 66), the phenylpropenamides increase both the rate and extent of capsid assembly in vitro (44). Specifically, the phenylpropenamides bind to the core protein with weak affinity and induce the protein into a more assembly-active state. This increase in the concentration of dimer in the active conformation results in an increased nucleation rate, driving capsid assembly forward. Based on this in vitro observation, it was proposed that the phenylpropenamides inhibit HBV replication by accelerating the nucleation of capsid proteins and driving capsid assembly at a rate high enough to exclude the polymerase-pgRNA complex (44).
However, although these in vitro observations are consistent with the findings made in our cell-based biological study indicating that HBV capsid protein is the molecular target of AT-61, it remains to be determined whether the compound inhibits HBV nucleocapsid assembly in virally infected cells through such a mechanism. In fact, it is well documented that the carboxyl-terminal domain (CTD) of capsid protein is essential for encapsidation of pgRNA (67–69). Although the dynamic phosphorylation/dephosphorylation of core protein has been biochemically demonstrated to occur only in DHBV infection and associates with nucleocapsid maturation and virion assembly/secretion (70, 71), accumulating genetic evidence suggests that the dynamic phosphorylation/dephosphorylation of three serine residues in the CTD of the HBV capsid protein is required for the assembly of pgRNA-containing nucleocapsids (72–74). However, the in vitro capsid assembly studies typically utilized C-terminally truncated capsid protein devoid of the CTD (44). Hence, it is possible that AT-61 disrupts pgRNA encapsidation through either direct interaction with the CTD or indirect interruption of the phosphorylation/dephosphorylation of HBV capsid protein. Furthermore, a genetic study has also identified mutations in the N-terminal assembly domain (NTD) of HBV capsid protein that do not interfere with empty capsid assembly but prevent pgRNA packaging (75). These observations imply that the assembly of empty capsids and pgRNA-containing nucleocapsids has a distinct structural requirement for capsid protein. It is thus also possible that AT-61 targets a distinct capsid protein interaction that is unique for the assembly of nucleocapsids but not empty capsids.
Due to the complexity of nucleocapsid assembly, SBAs and AT-61, despite having the same phenotypes displayed in our mechanistic studies, do not necessarily inhibit HBV nucleocapsid assembly via the same mechanism. Furthermore, the observation that HBV capsid protein, but not polymerase and pgRNA, confers the sensitivity of HBV to both AT-61 and SBAs does not necessarily indicate that the two classes of antiviral compounds directly target the capsid protein to disrupt its interaction with other capsid proteins. It is conceivable that the compounds may alternatively target the interface between the HBV capsid protein and DNA polymerase and/or pgRNA and consequentially inhibit pgRNA-containing nucleocapsid assembly. In addition, the compounds could also indirectly alter the posttranslational modifications of HBV capsid proteins to prevent pgRNA encapsidation. Nonetheless, deeper mechanistic insight into the two classes of anti-HBV compounds should be obtained through structural analysis of the interaction between the antiviral compounds and HBV capsid protein and selection of drug-resistant HBV mutants during in vivo antiviral efficacy studies in the future. Obviously, these studies should help not only in elucidating the antiviral mechanism of the compounds but also in understanding the molecular pathway of HBV nucleocapsid assembly.
In conclusion, a convenient cell-based assay has been designed and implemented for the discovery of anti-HBV compounds. SBAs represent a novel chemical entity with superior antiviral activity against both wild-type and nucleoside analogue-resistant HBVs, having a unique antiviral mechanism and favorable pharmacological profiles. The compounds are thus warranted for further development as antiviral therapeutics for the management of chronic hepatitis B.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health (R43AI1098200-01A1) and by the Hepatitis B Foundation through an appropriation of the Commonwealth of Pennsylvania. Ju-Tao Guo is the Bruce Witte Scholar of the Hepatitis B Foundation.
Footnotes
Published ahead of print 10 April 2013
REFERENCES
- 1. McMahon BJ. 2005. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 25(Suppl 1):3–8 [DOI] [PubMed] [Google Scholar]
- 2. Lee WM. 1997. Hepatitis B virus infection. N. Engl. J. Med. 337:1733–1745 [DOI] [PubMed] [Google Scholar]
- 3. Lok AS. 2004. Prevention of hepatitis B virus-related hepatocellular carcinoma. Gastroenterology 127:S303–309 [DOI] [PubMed] [Google Scholar]
- 4. Keeffe EB, Dieterich DT, Han SH, Jacobson IM, Martin P, Schiff ER, Tobias H. 2008. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 update. Clin. Gastroenterol. Hepatol. 6:1315–1341 [DOI] [PubMed] [Google Scholar]
- 5. Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608.e2 doi: 10.1053/j.gastro.2009.08.063 [DOI] [PubMed] [Google Scholar]
- 6. Gish R, Jia JD, Locarnini S, Zoulim F. 2012. Selection of chronic hepatitis B therapy with high barrier to resistance. Lancet Infect. Dis. 12:341–353 [DOI] [PubMed] [Google Scholar]
- 7. Perrillo R. 2009. Benefits and risks of interferon therapy for hepatitis B. Hepatology 49:S103–111 [DOI] [PubMed] [Google Scholar]
- 8. Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, Simon C, So TM, Gerken G, de Man RA, Niesters HG, Zondervan P, Hansen B, Schalm SW. 2005. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 365:123–129 [DOI] [PubMed] [Google Scholar]
- 9. Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, Gane E, Fried MW, Chow WC, Paik SW, Chang WY, Berg T, Flisiak R, McCloud P, Pluck N. 2005. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 352:2682–2695 [DOI] [PubMed] [Google Scholar]
- 10. Dienstag JL. 2009. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology 49:S112–121 [DOI] [PubMed] [Google Scholar]
- 11. Wiegand J, van Bommel F, Berg T. 2010. Management of chronic hepatitis B: status and challenges beyond treatment guidelines. Semin. Liver Dis. 30:361–377 [DOI] [PubMed] [Google Scholar]
- 12. Akbar SM, Hiasa Y, Mishiro S, Onji M. 2009. Treatment of hepatitis B virus-infected patients: utility of therapeutic recommendations in developing countries. Expert Opin. Pharmacother. 10:1605–1614 [DOI] [PubMed] [Google Scholar]
- 13. Liaw YF. 2009. Antiviral therapy of chronic hepatitis B: opportunities and challenges in Asia. J. Hepatol. 51:403–410 [DOI] [PubMed] [Google Scholar]
- 14. Peters MG. 2009. Special populations with hepatitis B virus infection. Hepatology 49:S146–S155 [DOI] [PubMed] [Google Scholar]
- 15. Este JA, Cihlar T. 2010. Current status and challenges of antiretroviral research and therapy. Antiviral Res. 85:25–33 [DOI] [PubMed] [Google Scholar]
- 16. Billioud G, Pichoud C, Puerstinger G, Neyts J, Zoulim F. 2011. The main Hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antiviral Res. 92:271–276 [DOI] [PubMed] [Google Scholar]
- 17. Summers J, O'Connell A, Millman I. 1975. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc. Natl. Acad. Sci. U. S. A. 72:4597–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerlich WH, Feitelson MA, Marion PL, Robinson WS. 1980. Structural relationships between the surface antigens of ground squirrel hepatitis virus and human hepatitis B virus. J. Virol. 36:787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Molnar-Kimber KL, Summers J, Taylor JM, Mason WS. 1983. Protein covalently bound to minus-strand DNA intermediates of duck hepatitis B virus. J. Virol. 45:165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. 1995. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 69:3350–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415 [DOI] [PubMed] [Google Scholar]
- 22. Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670 [DOI] [PubMed] [Google Scholar]
- 24. Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ganem D, Varmus HE. 1987. The molecular biology of the hepatitis B viruses. Annu. Rev. Biochem. 56:651–693 [DOI] [PubMed] [Google Scholar]
- 26. Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255–261 [DOI] [PubMed] [Google Scholar]
- 27. Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J. Virol. 84:9332–9340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J. Virol. 81:12472–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mao R, Zhang J, Jiang D, Cai D, Levy JM, Cuconati A, Block TM, Guo JT, Guo H. 2011. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J. Virol. 85:1048–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 64:2819–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zoulim F, Saputelli J, Seeger C. 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 68:2026–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu W, Goddard C, Clearfield E, Mills C, Xiao T, Guo H, Morrey JD, Motter NE, Zhao K, Block TM, Cuconati A, Xu X. 2011. Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. J. Med. Chem. 54:5660–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dougherty AM, Guo H, Westby G, Liu Y, Simsek E, Guo JT, Mehta A, Norton P, Gu B, Block T, Cuconati A. 2007. A substituted tetrahydro-tetrazolo-pyrimidine is a specific and novel inhibitor of hepatitis B virus surface antigen secretion. Antimicrob. Agents Chemother. 51:4427–4437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 77:1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korba BE, Gerin JL. 1992. Use of a standardized cell culture assay to assess activities of nucleoside analogs against hepatitis B virus replication. Antiviral Res. 19:55–70 [DOI] [PubMed] [Google Scholar]
- 36. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korba BE, Milman G. 1991. A cell culture assay for compounds which inhibit hepatitis B virus replication. Antiviral Res. 15:217–228 [DOI] [PubMed] [Google Scholar]
- 38. Stuyver LJ, Lostia S, Adams M, Mathew JS, Pai BS, Grier J, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. 2002. Antiviral activities and cellular toxicities of modified 2′,3′-dideoxy-2′,3′-didehydrocytidine analogues. Antimicrob. Agents Chemother. 46:3854–3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-l-2′,3′-dideoxy-3′-thiacytidine. Antimicrob. Agents Chemother. 42:3179–3186 (Erratum, 43:726, 1999.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299:893–896 [DOI] [PubMed] [Google Scholar]
- 41. Zhou T, Guo JT, Nunes FA, Molnar-Kimber KL, Wilson JM, Aldrich CE, Saputelli J, Litwin S, Condreay LD, Seeger C, Mason WS. 2000. Combination therapy with lamivudine and adenovirus causes transient suppression of chronic woodchuck hepatitis virus infections. J. Virol. 74:11754–11763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stray SJ, Zlotnick A. 2006. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. 19:542–548 [DOI] [PubMed] [Google Scholar]
- 43. Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. 2007. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 76:168–177 [DOI] [PubMed] [Google Scholar]
- 44. Katen SP, Chirapu SR, Finn MG, Zlotnick A. 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem. Biol. 5:1125–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249 [DOI] [PubMed] [Google Scholar]
- 46. Wang GH, Zoulim F, Leber EH, Kitson J, Seeger C. 1994. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol. 68:8437–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 93:1060–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen Y, Robinson WS, Marion PL. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol. 68:5232–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu J, Boyer M. 2006. Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J. Virol. 80:2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu J, Toft D, Anselmo D, Wang X. 2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol. 76:269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 86:5134–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ziermann R, Ganem D. 1996. Homologous and heterologous complementation of HBV and WHV capsid and polymerase functions in RNA encapsidation. Virology 219:350–356 [DOI] [PubMed] [Google Scholar]
- 56. Okamoto H, Omi S, Wang Y, Imai M, Mayumi M. 1990. Trans-complementation of the C gene of human and the P gene of woodchuck hepadnaviruses. J. Gen. Virol. 71:959–963 [DOI] [PubMed] [Google Scholar]
- 57. Delaney WE, IV, Yang H, Miller MD, Gibbs CS, Xiong S. 2004. Combinations of adefovir with nucleoside analogs produce additive antiviral effects against hepatitis B virus in vitro. Antimicrob. Agents Chemother. 48:3702–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, Plym M, Pokornowski K, Yu CF, Angus P, Ayres A, Bartholomeusz A, Sievert W, Thompson G, Warner N, Locarnini S, Colonno RJ. 2004. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob. Agents Chemother. 48:3498–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Levine S, Hernandez D, Yamanaka G, Zhang S, Rose R, Weinheimer S, Colonno RJ. 2002. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob. Agents Chemother. 46:2525–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lampertico P, Liaw YF. 2012. New perspectives in the therapy of chronic hepatitis B. Gut 61(Suppl 1):i18–i24 [DOI] [PubMed] [Google Scholar]
- 61. Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo JT, Block TM, Cuconati A, Guo H. 2012. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob. Agents Chemother. 56:4277–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou T, Guo H, Guo JT, Cuconati A, Mehta A, Block TM. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res. 72:116–124 [DOI] [PubMed] [Google Scholar]
- 63. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46:3–26 [DOI] [PubMed] [Google Scholar]
- 64. Delaney WEt., Edwards R, Colledge D, Shaw T, Furman P, Painter G, Locarnini S. 2002. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 46:3057–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang P, Naduthambi D, Mosley RT, Niu C, Furman PA, Otto MJ, Sofia MJ. 2011. Phenylpropenamide derivatives: anti-hepatitis B virus activity of the Z isomer, SAR and the search for novel analogs. Bioorg. Med. Chem. Lett. 21:4642–4647 [DOI] [PubMed] [Google Scholar]
- 66. Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. U. S. A. 102:8138–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nassal M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 66:4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kock J, Nassal M, Deres K, Blum HE, von Weizsacker F. 2004. Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 78:13812–13818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lewellyn EB, Loeb DD. 2011. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 85:1298–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pugh J, Zweidler A, Summers J. 1989. Characterization of the major duck hepatitis B virus core particle protein. J. Virol. 63:1371–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Perlman DH, Berg EA, O'Connor P, Costello BCE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. U. S. A. 102:9020–9025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348 [DOI] [PubMed] [Google Scholar]
- 73. Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J. Virol. 69:1025–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee SM, Park SG, Park E, Lee JY, Jung G. 2003. The 113th and 117th charged amino acids in the 5th alpha-helix of the HBV core protein are necessary for pgRNA encapsidation. Virus Genes 27:227–235 [DOI] [PubMed] [Google Scholar]