

Abstract
Primary Tupaia hepatocytes (PTHs) are susceptible to woolly monkey hepatitis B virus (WMHBV) infection, but the identity of the cellular receptor(s) mediating WMHBV infection of PTHs remains unclear. Recently, sodium taurocholate cotransporting polypeptide (NTCP) was identified as a functional receptor for human hepatitis B virus (HBV) infection of primary human and Tupaia hepatocytes. In this study, a synthetic pre-S1 peptide from WMHBV was found to bind specifically to cells expressing Tupaia NTCP (tsNTCP) and it efficiently blocked WMHBV entry into PTHs; silencing of tsNTCP in PTHs significantly inhibited WMHBV infection. Ectopic expression of tsNTCP rendered HepG2 cells susceptible to WMHBV infection. These data demonstrate that tsNTCP is a functional receptor for WMHBV infection of PTHs. The result also indicates that NTCP's orthologs likely act as a common cellular receptor for all known primate hepadnaviruses.
TEXT
Human hepatitis B virus (HBV) is a leading cause of liver diseases ranging from acute hepatitis to chronic hepatitis, cirrhosis, and hepatocellular carcinoma. With approximately 240 million cases of chronic infection worldwide, HBV is responsible for about 600,000 deaths annually (1). Despite its enormous medical and social relevance, progress in HBV research has been impeded by the lack of understanding of HBV entry by which the virus specifically infects human liver cells. Recently we found that sodium taurocholate cotransporting polypeptide (NTCP; also known as SLC10A1 [solute carrier family 10 member 1]), a hepatic sodium/bile acid symporter presumed to span the cellular membrane up to 10 times with small extracellular loops (2–5), is a functional receptor for human HBV and hepatitis D virus (HDV) infections of human and Tupaia hepatocytes (6). The pre-S1 domain of the HBV large envelope protein (L protein) is the key determinant of interaction with the cellular receptor NTCP (6). Furthermore, the critical receptor-binding motif in the pre-S1 domain of human HBV (7–10) is conserved among all hepadnaviruses from humans and nonhuman primates, including chimpanzees, gorillas, orangutans, gibbons, and woolly monkeys (Fig. 1A). Therefore, it is tempting to speculate that NTCP may play an important role in the entry of all known primate hepadnaviruses into host cells.
Fig 1.

Sequence alignment of the pre-S1 N-terminal domain of primate hepadnaviruses and phylogenetic analysis of the L proteins. (A) Amino acid sequences of the receptor-binding region (aa −10 to 48 or 2 to 48) in the pre-S1 domain of human HBVs were aligned with the corresponding regions of other primate HBVs. The residue numbering was based on HBV genotype D. The GenBank accession numbers of the hepadnaviruses compared herein are as follows: HBV genotype A, JQ687533; genotype B, JX978431; genotype C, AY167095; genotype D, X02496; genotype E, GQ161835; genotype F, DQ899142; genotype G, AP007264; genotype H, AB205010; genotype J, AB486012; chimpanzee HBV, AF222322; gorilla HBV, AJ131567; orangutan HBV, EU155825; gibbon HBV, U46935; WMHBV, AY226578. For nonhuman primate HBVs, the values in parentheses are the numbers of recorded complete genomes of the corresponding viruses. The receptor-binding motif of human HBV (aa 9 to 15) and the corresponding regions of other primate HBVs are shaded in gray. The phylogenetic tree was based on the amino acid sequences of the L proteins of the compared primate HBVs and woodchuck hepatitis virus (GenBank accession number J02442), a rodent hepadnavirus. The distance along the horizontal axis among isolates is proportional to the amino acid divergence. The scale bar (upper right) represents 5% divergence. (B) Amino acid sequences of four synthetic peptides used in this study. The peptide WM-Myr47b corresponds to aa 2 to 47 of the L protein of WMHBV (GenBank accession number AY226578). The amino acid sequences of the three HBV peptides are derived from aa 2 to 47 of the L protein of the genotype C isolate (GenBank accession number AY167095). Peptides of this region were synthesized with or without a lysine residue at the C terminus for biotinylation and with or without myristoylation modification at the N terminus. Myr, myristoyl group.
Woolly monkey HBV (WMHBV) is the only hepadnavirus of a nonhuman primate with an established infectious clone and has been studied in some detail (10–18). It was originally isolated from a woolly monkey (Lagothrix lagotricha) with fulminant hepatitis. It has the same genetic organization as and shares 78% sequence identity with human HBV (12). Both the woolly monkey and a closely related New World monkey, the spider monkey (Ateles geoffroyi), are susceptible to WMHBV infection, while chimpanzees are minimally susceptible (11, 12). In cell cultures, WMHBV or WMHBV envelope pseudotyped HDV could consistently infect spider monkey and, with much lower efficiency, chimpanzee and human hepatocytes (10, 14, 15). Similar to HBV, WMHBV infects hepatocytes isolated from Tupaia belangeri, a nonrodent small animal phylogenetically related to primates (17–20). Because of the better availability and higher repopulation rates of primary Tupaia hepatocytes (PTHs) than primary human hepatocytes (PHHs) (18, 21) and higher replication rates of WMHBV than HBV in PTHs (17, 18), WMHBV infection of immunodeficient urokinase-type plasminogen activator transgenic (uPA) mice transplanted with PTHs has been used as a surrogate for studying HBV in vivo infection of PHH-transplanted uPA mice (16). However, as shown in Fig. 1A, phylogenetic analysis of all of the primate HBV family members on the basis of their L proteins showed that WMHBV is the most distant from the human HBV group, forming a separate branch distinct from the monophyletic group of human and ape HBVs. Therefore, studies to demonstrate the same receptor engagement of WMHBV and HBV are of great importance to consolidate the use of WMHBV as a surrogate for HBV. On the other hand, investigation into whether WMHBV utilizes NTCP as a receptor will shed light on the likelihood of NTCP's orthologs serving as a common receptor for all known HBVs from nonhuman primates.
The pre-S1 domain of the L protein is a key determinant of HBV entry (22–24). Synthetic myristoylated peptides corresponding to the pre-S1 N-terminal domain of HBV are sufficient to bind to the human or Tupaia receptor NTCP (6). Considering that the region (amino acids [aa] 9 to 15) essential for receptor binding in the pre-S1 N-terminal domain of HBV is conserved in the corresponding region of WMHBV (Fig. 1A), we first tested whether synthetic pre-S1 N-terminal peptide of WMHBV could directly bind to Tupaia NTCP (tsNTCP) on the cell surface. Four peptides (Fig. 1B), each comprising aa 2 to 47 of the pre-S1 domain of the WMHBV or HBV L protein, were synthesized with or without a lysine residue at the C terminus for biotinylation and with or without myristoylation modification at the N terminus as indicated. 293T cells transfected with plasmid tsNTCP-green fluorescent protein (GFP) or hSDC2-GFP, a control plasmid encoding human syndecan 2 (also known as heparan sulfate proteoglycan core protein, HSPG1) fused with a GFP tag at the C terminus, were incubated with the test peptides at 400 nM. The cells were then stained with phycoerythrin (PE)-conjugated streptavidin (eBioscience, San Diego, CA) to detect the biotin tag of the peptides. As shown in Fig. 2A, similar to myristoylated HBV pre-S1 peptide H-Myr47b, the WMHBV pre-S1 peptide WM-Myr47b specifically bound to cell surface tsNTCP-GFP but not to control protein hSDC2-GFP. As expected, the control peptide H-47b, which is the HBV pre-S1 peptide without N-terminal myristoylation, failed to bind to tsNTCP-GFP (bottom panel). Furthermore, the binding of WM-Myr47b to tsNTCP-GFP was inhibited by the pretreatment of cells with nonbiotinylated HBV pre-S1 peptide H-Myr47 (Fig. 2B). These results demonstrated that tsNTCP is a specific binding partner for the WMHBV pre-S1 peptide.
Fig 2.
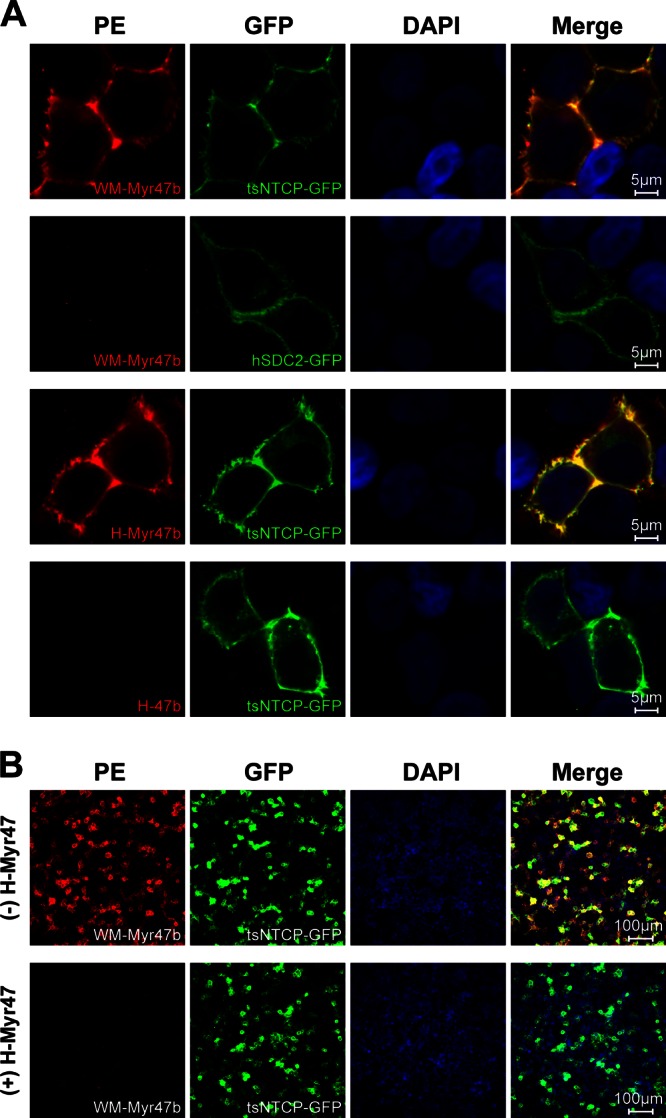
Specific binding of WM-Myr47b peptide to cell surface tsNTCP. (A) 293T cells were transiently transfected with plasmids encoding tsNTCP-GFP or hSDC2-GFP as a control. Transfected cells were cultured in PMM for 36 to 48 h, blocked with 3% bovine serum albumin–phosphate-buffered saline for 1 h, and then incubated with the indicated peptides at 400 nM at 37°C for 2 h. Subsequently, the cells were fixed with 4% paraformaldehyde, stained with 0.6 μg/ml PE-streptavidin, and visualized with a Zeiss LSM 510 Meta confocal microscope. (B) 293T cells transfected with tsNTCP-GFP were blocked with 3% bovine serum albumin–phosphate-buffered saline in the absence (top) or presence (bottom) of 800 nM nonbiotinylated HBV pre-S1 peptide, H-Myr47, followed by washing and incubation with the biotinylated WMHBV pre-S1 peptide, WM-Myr47b, at 400 nM at 37°C for 2 h. Cells were then stained and visualized as described for panel A. DAPI, 4′,6-diamidino-2-phenylindole.
To determine if the endogenous tsNTCP protein is required for WMHBV infection of PTHs, we then examined whether the WM-Myr47b peptide is capable of blocking WMHBV infection on PTHs. To detect WMHBV infection, we used a commercial human hepatitis B e antigen (HBeAg) enzyme-linked immunosorbent assay (ELISA) kit (Wantai Pharmacy, Beijing, China) to measure HBeAg secreted from WMHBV-infected cells. The kit was first assessed by determination of the dose-response curves of inoculated virus versus the level of secreted HBeAg in WMHBV-infected PTHs and PHHs. To ensure detection of the de novo synthesized and secreted HBeAg, cell culture supernatants from different days postinoculation were measured by ELISA. As shown in Fig. 3A and B, HBeAg levels increased with the culturing time and correlated well with the virus inoculation doses. Furthermore, the signals from infected PTHs are around 50-fold higher than that from PHHs, which is consistent with previous reports that WMHBV or WMHBV envelope pseudotyped HDV infection of PHHs is inefficient (14, 15). These data demonstrated that the ELISA kit detects WMHBV HBeAg with good specificity and sensitivity. The two myristoylated pre-S1 peptides from WMHBV and human HBV, WM-Myr47b and H-Myr47b, respectively, were then tested and compared for the efficiency with which they block HBV and WMHBV infections of PTHs. WMHBV was produced by transfection of Huh7 cells with a plasmid expressing the viral genome as previously described by Lanford et al. (11). PTHs were isolated and cultured in primary hepatocyte maintenance medium (PMM) as previously reported (6). Within 3 days after initial plating, freshly isolated PTHs were inoculated in the presence of 4% polyethylene glycol (PEG) 8000 for 16 h with WMHBV at a multiplicity of genome equivalents (MGE) of 100 or with HBV (genotype D) at an MGE of 200. The test peptides were added at different concentrations immediately before viral inoculation and were present in the culture during the inoculation process. Cell culture supernatants were collected every 2 days for the detection of secreted HBeAg (Fig. 4A). HBeAg secretion from both HBV- and WMHBV-infected PTHs in the absence of pre-S1 peptide increased rapidly from 3 to 7 days postinoculation (dpi), demonstrating the successful infection of PTHs with both viruses. We found that WMHBV infection is, in general, more robust than HBV infection of PTHs, which is consistent with previous reports by Köck et al. (17) and Dandri et al. (18). Nonetheless, WM-Myr47b potently inhibited both WMHBV and HBV infections by 95% at a concentration of 40 nM; the efficiency was comparable to that of H-Myr47b, its counterpart peptide derived from the pre-S1 domain of human HBV. To further validate HBeAg ELISA results for WMHBV infection, WMHBV-infected PTHs were analyzed for de novo viral RNA synthesis by Northern blotting with a WMHBV-specific probe covering the whole genome. Three species of viral RNAs, 3.5-, 2.4-, and 2.1-kb RNAs, were all detectable at 5 dpi and increased in the following 2 days, while the viral RNAs were not detected in the PTHs treated with WM-Myr47b during inoculation (Fig. 4B). To demonstrate that WM-Myr47b peptide blocks viral infection at the entry level, the kinetics of peptide-mediated inhibition of WMHBV infection was determined by adding the peptide at 200 nM to PTHs at different times. In addition to the measurement of secreted HBeAg in the supernatants by ELISA (Fig. 4C), viral RNA synthesis was also examined by Northern blotting at 7 days after infection (Fig. 4D). Furthermore, the infected PTHs were stained for intracellular hepatitis B core antigen (HBcAg) of WMHBV at 14 days after infection with mouse monoclonal antibody (MAb) 1C10 (Fig. 4E), which recognizes WMHBV HBcAg with high sensitivity and specificity (Fig. 3C to E). WMHBV infection was effectively blocked by the addition of the WM-Myr47b peptide and virus simultaneously to cells, followed by incubation for 16 h; the blocking efficiency was slightly reduced when the peptide was incubated with the cells for only 1 h before inoculation, while the addition of the WM-Myr47b peptide to cells after removal of the viral inoculum had no detectable inhibitory effect. It has been reported that HBV infection is initiated with virus attachment to cell surface heparan sulfate proteoglycans (HSPGs) via the antigenic loop of the small envelope proteins (25). To examine whether the WM-Myr47b peptide blocks entry steps after primary attachment, we measured the kinetics of peptide-mediated inhibition of WMHBV infection with a much shorter inoculation window (4 h inoculation) in either the presence or the absence of PEG 8000 (Fig. 4F). Under both conditions, the WM-Myr47b peptide exhibited high efficiency in blocking the infection when it was added before or at viral inoculation, while it still partially reduced the infection if it was added immediately after the 4-h inoculation process. These data demonstrated that WM-Myr47b blocks WMHBV infection at the entry level but after initial viral attachment. Together with the cross inhibitory and comparable activities of WM-Myr47b and H-Myr47b against both HBV and WMHBV infections, it suggests that WM-Myr47b and H-Myr47b engage the same cellular receptor on PTHs, most likely tsNTCP.
Fig 3.

Validation of the ELISA kit for WMHBV HBeAg detection and MAb 1C10 for WMHBV HBcAg detection. (A, B) PTHs or PHHs were inoculated for 16 h with the indicated MGE of WMHBV in the presence of 4% PEG 8000 with or without 200 nM H-Myr47 peptide. Cell culture supernatants were collected every 2 days. Supernatants from different days postinoculation were measured by ELISA. The relative quantity of HBeAg is presented as arbitrary units per 50 μl of culture supernatant, which was calculated by multiplying the optical density at 450 nm by the dilution factors. (C) 1C10 is a mouse MAb developed by the hybridoma technique with recombinant WMHBV core protein and recognizes the core proteins of both WMHBV and HBV. Protein G-purified MAb 1C10 and control MAb 4G5 targeting HDV delta antigen were serially diluted in 3% bovine serum albumin–phosphate-buffered saline, incubated in an ELISA plate precoated with 5 μg/ml recombinant WMHBV core protein or HDV delta antigen, and detected by goat anti-mouse IgG-horseradish peroxidase conjugate. 1C10 recognized WMHBV core protein (yellow wells, top) but not HDV delta antigen. (D) Huh7 cells in a 48-well plate were transfected with 0, 30, or 150 ng of WMHBV core expression plasmid. At 24 h after transfection, cells were fixed, permeabilized, and stained with 5 μg/ml MAb 1C10. (E) PTHs or PHHs infected with WMHBV at an MGE of 0 or 100 in panels A and B were fixed at 14 dpi, and intracellular HBcAg of WMHBV was stained with 5 μg/ml of MAb 1C10. Red, WMHBV HBcAg; blue, cell nucleus; white, autofluorescence.
Fig 4.

Inhibition of WMHBV infection in PTHs by tsNTCP-binding peptide or tsNTCP-specific siRNAs. (A) Inhibition of HBV and WMHBV infection by peptide WM-Myr47b or H-Myr47b. PTHs were inoculated with Huh7-produced WMHBV at an MGE of 100 or HBV (genotype D) at an MGE of 200 in the presence of 4% PEG 8000 for 16 h. The indicated peptide was added immediately before inoculation and was present during the inoculation process. Cell culture supernatants were collected every 2 days, and secreted HBeAg was measured by ELISA. The relative quantity of HBeAg is presented as arbitrary units per 50 μl of culture supernatant, which was calculated by multiplying the optical density at 450 nm by the dilution factors. (B) PTHs in six-well plates were inoculated with WMHBV as in panel A. At 0, 1, 5, and 7 dpi, cells were homogenized for total RNA extraction. A 10-μg sample of total RNA was separated on a 1.5% agarose gel and then blotted to Hybond-N+ positively charged nylon membrane (GE Healthcare Life Sciences) by overnight capillary transfer in 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Hybridization was performed at 52°C for 6 h with a random primed digoxigenin-dUTP (Roche Applied Science)-labeled probe covering the whole genome of WMHBV and a probe spanning human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript variant 1 mRNA nucleotides 181 to 690. Hybridized probes were detected with anti-digoxigenin antibody–alkaline phosphatase conjugate and CSPD chemiluminescence substrate (Roche Applied Science). The chemiluminescence signal was detected with BioMax Light chemiluminescence film (Sigma). Samples (100 pg) of denatured 3,032- and 1,554-bp PCR fragments of the WMHBV genome were included as markers. (C to E) Blocking of WMHBV infection by adding WM-Myr47b at different time points. PTHs were inoculated with WMHBV at an MGE of 100 in the presence of 4% PEG 8000 for 16 h. The WM-Myr47b peptide was added to PTHs at 200 nM and incubated for different times as indicated. The time of virus addition to cells is designated 0 h. N.P., no peptide treatment. (C) Cell culture supernatants were collected every 2 days, and secreted HBeAg was measured on the indicated days by ELISA. (D) At 7 dpi, infected PTHs in a six-well plate were homogenized for total RNA extraction and 10 μg of RNA was subjected to Northern blot analysis as in panel B. Lane M corresponds to 10 pg of denatured 3,032- and 1,554-bp PCR fragments from the WMHBV genome. (E) At 14 dpi, infected PTHs were fixed with 4% paraformaldehyde and intracellular HBcAg of WMHBV was stained with 5 μg/ml MAb 1C10 and then with Alexa 488-conjugated goat anti-mouse IgG (Life Technologies). Fluorescence images were captured with a Zeiss LSM 510 Meta confocal microscope. Red, WMHBV HBcAg; blue, cell nucleus. (F) WM-Myr47b-mediated inhibition of WMHBV infection at 4 h after inoculation. PTHs were inoculated with WMHBV at an MGE of 100 in the presence or absence of 4% PEG 8000 for 4 h. The WM-Myr47b peptide was added at 200 nM to PTHs and incubated for the times indicated. The time at which the virus was added to the cells was marked as 0 h. Secreted HBeAg was measured by ELISA at 5 dpi. N.P., no peptide treatment. (G) Validation of the knockdown efficiencies of siRNAs at the mRNA level. Freshly isolated PTHs were transfected with 20 nM siRNA targeting tsNTCP (SLC10A1: siR1, siR2, siR3, and siR4), or tsSLC44A1 (SLC44A1-siRs) or with a nontargeting control siRNA (NC-siR). Three days after transfection, PTHs were lysed for total RNA isolation and cDNA preparation. Tupaia NTCP (SLC10A1) and SLC44A1 mRNA levels were then determined by qRT-PCR with tsNTCP- and tsSLC44A1-specific primers, respectively. qRT-PCR was performed with the SYBR Premix Ex Taq kit (TaKaRa, Tokyo, Japan) on an ABI Fast 7500 RT-PCR system (Applied Biosystems). Data are presented as the relative mRNA levels of NTCP and SLC44A1 compared to that in cells transfected with the negative-control siRNA (NC-siR). (H) WMHBV infection of PTHs is reduced by tsNTCP knockdown. Freshly isolated PTHs were transfected as described for panel G. Three days after transfection, cells were inoculated with WMHBV at an MGE of 100 for 16 h. The culture medium was refreshed every 2 days, and secreted HBeAg was measured at 5 dpi. Data are presented as the relative optical density (OD) value compared to the optical density at 450 nm for HBeAg from cells transfected with the negative-control siRNA (NC-siR). (I) Infection with control virus lenti-VSV-G was not affected by tsNTCP knockdown. Freshly isolated PTHs were transfected as described for panel G. Three days after transfection, cells were inoculated with lenti-VSV-G for 16 h. The culture medium was refreshed every 2 days, and cells were lysed for luciferase activity detection at 5 dpi. RLU, relative luminescence units.
Then we directly tested the requirement of endogenous tsNTCP protein for WMHBV infection by silencing tsNTCP expression on PTHs prior to viral infection. Freshly isolated PTHs were transfected with 20 nM small interfering RNA (siRNAs) targeting tsNTCP (siR1, 5′-CUAUGUAGGCAUUGUGAUAdTdT-3′; siR2, 5′-GGGCAAGAGCAUCAUGUUUdTdT-3′; siR3, 5′-GUGUUAUCCUGGUGGUUAUdTdT-3′; siR4, 5′-GGACAUGAAUCUCAGCAUUdTdT-3′) or a set of control siRNAs, including siRNAs targeting Tupaia SLC44A1 (tsSLC44A1, a multipass membrane transporter responsible for choline transportation) (SLC44A1-siRs, 5′-AGAAGUUUGCAGAGAUAAAdTdT-3′ and 5′-CGAAUGAUCCUUAUGUAUAdTdT-3′) and a nontargeting control siRNA (NC-siR, 5′-UUCUCCGAACGUGUCACGUdTdT-3′). Gene knockdown efficiency was examined 3 days after transfection; Tupaia NTCP (SLC10A1) and SLC44A1 mRNA levels were determined by quantitative real-time PCR (qRT-PCR) with tsNTCP-specific primers (NTCP-F, 5′-TTCAGCAAGATCAAGGC-3′; NTCP-R, 5′-TGGAGCAGGTGGTCATC-3′) and tsSLC44A1-specific primers (SLC44A1-F, 5′-CATTTGTTGCCTTTGGTGTC-3′; SLC44A1-R,5′-TTTGCTGAGGTGCAGAAGTT-3′), respectively. Cells were then incubated for 16 h after inoculation with WMHBV or a control virus, lenti-VSV-G, which is an HIV-1 genome-based lentivirus pseudotyped with the glycoprotein of vesicular stomatitis virus (VSV-G) and carries a firefly luciferase reporter gene. HBeAg secretion from WMHBV-infected cells and luciferase activity of the cell lysates from lenti-VSV-G-infected cells were examined at 5 dpi. Compared to the nontargeting control siRNA, the four tsNTCP-specific siRNAs led to a 60 to 85% reduction of the tsNTCP mRNA level (Fig. 4G). Consequently, HBeAg secretion by WMHBV-infected cells was reduced by 35 to 62% and the reduction of viral infection correlated with the knockdown efficiency of the corresponding siRNAs (Fig. 4H). As a control, when siRNAs targeting tsSLC44A1, a solute carrier family member responsible for choline transport, reduced the tsSLC44A1 mRNA level by 80% (Fig. 4G), WMHBV infection was not reduced (Fig. 4H). Furthermore, in contrast to WMHBV, the lenti-VSV-G infection was not affected by silencing of tsNTCP (Fig. 4I). Collectively, these data indicate that tsNTCP is critical for the efficient infection of PTHs by WMHBV.
We then examined whether ectopic expression of tsNTCP is sufficient to reconstitute susceptibility to WMHBV infection in HepG2 cells, a human hepatoma cell line in which the NTCP mRNA level is negligible compared to that in PTHs (6). HepG2 cells were transiently transfected with a plasmid encoding tsNTCP or a vector control plasmid. The total and surface expression of tsNTCP was confirmed (Fig. 5A), and the transfected cells were infected with WMHBV at an MGE of 100 in the absence or presence of 4% PEG 8000 in the viral inoculum. Culture medium was replenished every 2 to 3 days thereafter. At 11 dpi, culture supernatants were collected for HBeAg level measurement by ELISA and the cells were fixed for intracellular HBcAg staining or lysed with TRIzol reagent for total RNA preparation and detection of WMHBV-specific RNAs by Northern blotting or the same qRT-PCR procedure as previously described for the quantification of human HBV-specific RNAs (6). Primers (WM-402F, 5′-ACCCAATGCCCCTATCTTATC-3′, WM-555R, 5′-CAGGAAGATGCTGGAGATTG-3′) targeting the viral precore/core gene (positions 2304 to 2457; GenBank accession number AY226578) were used for qRT-PCR to quantify the 3.5-kb WMHBV transcripts, which include precore RNA and pregenomic RNA (pgRNA). Both secreted viral HBeAg and intracellular 3.5-kb viral transcripts were readily detected from cells transfected with tsNTCP but not from cells transfected with the vector (Fig. 5B and C). In previous studies, PEG 8000 in viral inoculum has been shown to enhance HBV and HDV infection of primary human hepatocytes and HepaRG cells without compromising the infection specificity of the viruses (9, 15, 26, 27). Similarly, in the present study we also found that the presence of 4% PEG 8000 led to a significantly enhanced WMHBV infection: up to 10-fold increases in the secretory HBeAg level, as well as the intracellular 3.5-kb viral RNA level, were observed in cells transfected with tsNTCP but not in cells transfected with the vector control (Fig. 5B and C). Northern blot analysis with a WMHBV-specific probe detected the 3.5-, 2.4-, and 2.1-kb species of de novo-synthesized viral RNAs in the HepG2 cells transfected with tsNTCP but not in those transfected with the vector control (Fig. 5D). In addition, with an ∼50% transfection efficiency on HepG2 (data not shown), intracellular HBcAg staining of cells inoculated in the presence of PEG 8000 revealed that 5 to 10% of the cells transfected with tsNTCP were stained positively at 11 dpi, but no HBcAg staining was observed in cells transfected with the vector control (Fig. 5E). Importantly, tsNTCP-mediated WMHBV infections were blocked by the entry inhibitory peptide WM-Myr47b under all of the conditions tested, regardless of the presence or absence of PEG 8000 (Fig. 5B to E). To further confirm the completion of authentic infection of tsNTCP-complemented HepG2 cells, the kinetics of secreted HBeAg, intracellular WMHBV viral RNAs, and HBcAg were also analyzed (Fig. 5F to H). The secreted HBeAg and viral RNAs were detectable at 3 dpi and increased greatly thereafter (Fig. 5F and G). Meanwhile, de novo-synthesized HBcAg became detectable at 5 dpi and increased rapidly in the following days tested (Fig. 5H). Again, addition of entry inhibitory peptide WM-Myr47b to the viral inoculum blocked infection, as shown by inhibition of the accumulation of all three infection markers. Taken together, these results demonstrated that exogenous expression of tsNTCP could support WMHBV infection in naturally nonsusceptible hepatoma HepG2 cells.
Fig 5.

Tupaia NTCP expression supports WMHBV infection of HepG2 cells. (A) Western blot analysis of tsNTCP expression in HepG2 cells. HepG2 cells were transfected with a plasmid encoding C9-tagged tsNTCP or a vector control plasmid. Six hours after transfection, the culture medium was changed to PMM and the cells were cultured for an additional 24 h. The cells were subjected to Western blot analysis for the levels of total and surface expression of tsNTCP protein as previously described (6). (B, C) HepG2 cells were transfected with a plasmid encoding tsNTCP or a vector control plasmid. Six hours after transfection, the culture medium was changed to PMM and the cells were cultured for an additional 24 h. Subsequently, the cells were inoculated with WMHBV at an MGE of 100 in the absence or presence of 4% PEG 8000 and with or without 200 nM peptide WM-Myr47b. (B) The culture medium was refreshed every 2 to 3 days, and secreted HBeAg was measured at 11 dpi. (C) The cells were then lysed for total RNA preparation and quantification of the 3.5-kb WMHBV precore RNA/pgRNA by qRT-PCR with the SYBR Premix Ex Taq kit and primers targeting the viral core gene. The levels of 3.5-kb viral RNA are presented as copy numbers of viral RNA per nanogram of total cellular RNA. (D, E) HepG2 cells were transfected as described for panel B and inoculated with WMHBV in the presence of 4% PEG 8000 with or without 200 nM peptide WM-Myr47b. At 11 dpi, the cells were either examined by Northern blot analysis with 15 μg of total RNA (D) or immunostained for intracellular HBcAg (E). (F to H) Kinetics of WMHBV infection of tsNTCP-complemented HepG2 cells. HepG2 cells were transfected as described for panel A and inoculated with WMHBV in the presence of 4% PEG 8000 with or without 200 nM peptide WM-Myr47b. Levels of secreted HBeAg (F), viral RNAs (F and G), and intracellular HBcAg (H) were determined by ELISA, qRT-PCR, Northern blot analysis, and intracellular immunostaining, respectively, on the days indicated. Lane M in panels D and G corresponds to 100 pg of denatured 3,032- and 1,554-bp PCR fragments from the WMHBV genome. In panels E and H, red is WMHBV HBcAg and blue is the cell nucleus.
We further tested human, mouse, rat, and crab-eating macaque (Macaca fascicularis) NTCPs for the ability to support WMHBV infection in HepG2 cells. Besides tsNTCP, human NTCP (hNTCP) also supported the WMHBV infection of HepG2 cells, but the efficiency was about 2-fold lower than that mediated by tsNTCP (Fig. 6A). A minimal increase in the HBeAg level during culture was detected in HepG2 cells transfected with mouse NTCP (mNTCP) and rat NTCP (rNTCP), indicating that mNTCP and rNTCP are very inefficient in supporting infection. NTCP from an Old World monkey, the crab-eating macaque, showed no ability to support WMHBV infection. We then detected the response curves for the amount of tsNTCP and virus dose versus infection, respectively. The infection efficiency of WMHBV on tsNTCP-transfected HepG2 cells correlated well with both the amount of tsNTCP and the viral inoculation dose (Fig. 6B and C).
Fig 6.

Species specificity of NTCP-mediated WMHBV infection and correlation of WMHBV infection with the amount of tsNTCP and the virus inoculation dose. (A) Species specificity of NTCP-mediated WMHBV infection of HepG2 cells. HepG2 cells were transfected with plasmids encoding NTCPs of different species or with a vector control plasmid. The cells were then cultured in PMM for 24 h. Subsequently, the cells were inoculated with WMHBV in the presence of 4% PEG 8000 with or without 200 nM WM-Myr47b peptide. Secreted HBeAg was determined by ELISA on the days indicated. (B) Correlation of the amount of transfected tsNTCP plasmid and the WMHBV infection level. tsNTCP plasmid was serially diluted as indicated, and the total amount of DNA was compensated with the vector plasmid to ensure that the same amount of DNA was transfected into HepG2 cells in 48-well plates. The transfected cells were then cultured and inoculated as described for panel A. Secreted HBeAg was determined by ELISA on the days indicated. (C) Correlation of the virus inoculation dose and the WMHBV infection level. HepG2 cells were transfected with a plasmid encoding tsNTCP or hNTCP or with a vector control plasmid. The cells were then cultured in PMM for 24 h. Subsequently, the cells were inoculated for 16 h with WMHBV at the indicated MGE in the presence of 4% PEG 8000 with or without 200 nM WM-Myr47b peptide. Secreted HBeAg in the culture supernatant was measured by ELISA at 11 dpi.
In summary, in the present study, we showed that synthetic pre-S1 peptide (aa 2 to 47) from WMHBV specifically bound to cells expressing tsNTCP but not to cells expressing the control protein SDC2 (HSPG1), which is a transmembrane heparan sulfate proteoglycan. This tsNTCP-binding peptide efficiently blocked WMHBV entry into PTHs; meanwhile, silencing of tsNTCP expression in PTHs significantly reduced WMHBV infection. Moreover, ectopic expression of tsNTCP rendered HepG2 cells susceptible to WMHBV infection. Collectively, these results demonstrate that tsNTCP is also a functional receptor for WMHBV infection of PTHs. Petersen et al. (16) have reported that the synthetic pre-S1 peptide from human HBV blocked WMHBV infection in the uPA mouse model transplanted with PTHs. Our data reported here are consistent with those in vivo data, indicating that tsNTCP likely serves as a functional receptor in vivo for WMHBV infection in the mouse model transplanted with PTHs. Our finding that NTCP functions as a common cellular receptor for WMHBV and HBV infection of PTHs provides direct evidence that WMHBV could serve as a surrogate for HBV studies.
As shown in our previous study, the efficiency of HBV infection in hNTCP-complemented HepG2 cells is comparable to that in PHHs (6). HBV infection of PTHs achieves comparable efficiency (data not shown). In contrast, we found in this study that the efficiencies of WMHBV infection in PTHs and tsNTCP-complemented HepG2 cells were considerably different: with the same inoculation dose, about 60% of PTHs were positively infected, as indicated by intracellular WMHBV HBcAg staining (Fig. 4E), while only up to 10% of the transfected HepG2 cells were infected (Fig. 5E and H). It has been reported that WMHBV or WMHBV envelope pseudotyped HDV infection of human hepatocytes in primary culture is inefficient (14, 15). We consistently observed that WMHBV infection of PHHs was around 50-fold less efficient than that on PTHs (Fig. 3A, B, and E); nevertheless, the infection of PHHs was blocked by the HBV pre-S1 peptide H-Myr47 (Fig. 3B), whereas WMHBV infection of hNTCP-complemented HepG2 cells is only about 2-fold lower than that of tsNTCP-complemented ones (Fig. 6A and C). These observations indicate that hNTCP is unlikely to be the key determinant responsible for the low efficiency of WMHBV infection of human hepatocytes, and there may exist an additional factor(s) that could contribute to the high infection efficiency of WMHBV in Tupaia and its low infection efficiency in human hepatocytes at the entry or postentry level or even both.
The natural host of WMHBV is the New World primate the woolly monkey; however, it is an endangered species not available for experimental purposes (12). The spider monkey, a close relative of the woolly monkey, has been used instead for WMHBV studies (10–12, 15). It is very difficult to obtain liver samples from these monkeys for NTCP cloning or primary cultivation for viral infection. We therefore could not directly demonstrate that WMHBV infects spider monkey or woolly monkey hepatocytes via their NTCPs. Nonetheless, previous studies using spider monkey hepatocytes have suggested that WMHBV and HBV have a critical receptor in common (10). In this study, we demonstrated that WMHBV, like human HBV, uses Tupaia NTCP as a cellular receptor for entry into PTHs. Given that among all of the primate HBV family members, WMHBV is phylogenetically the most distant from human HBV (12, 28), and both WMHBV and HBV utilize Tupaia NTCP to infect PTHs, it is conceivable that NTCP likely serves as a common cellular receptor for the entry of all known primate hepadnaviruses into host cells.
Previous studies have reported that carboxypeptidase D (CPD) is a receptor for one of the avian hepadnaviruses, duck HBV (DHBV) (29–32). However, transfection with CPD failed to confer susceptibility to DHBV infection on a nonpermissive chicken hepatoma cell line (30, 32). Furthermore, unlike the multipass membrane transporter NTCP, which is enriched in the liver (3), CPD is a ubiquitously distributed type I transmembrane protease (29–31), which alone is difficult to reconcile with the narrow liver tropism of avian hepadnaviruses. The core element in the DHBV L protein for binding to CPD is not the very N-terminal domain, and their interaction is independent of the N-terminal myristoylation of pre-S (32–34). Nevertheless, a synthetic peptide corresponding to the N-terminal 41 aa of DHBV pre-S, which is incapable of binding to CPD, was demonstrated to bind duck hepatocytes and inhibit DHBV infection more efficiently than the peptide corresponding to the CPD-binding region (35). These results suggest the presence of another entry factor(s) that acts independently or cooperates with CPD to facilitate avian HBV infection. It will be interesting to study whether NTCP or an NTCP-like protein of avian or rodent origin could also mediate or facilitate infections with avian or rodent hepadnaviruses. Such investigations will determine if the viral entry step(s) in which NTCP participates is common to all hepadnaviruses.
ACKNOWLEDGMENTS
We thank Robert E. Lanford (Texas Biomedical Research Institute) for providing the WMHBV-producing plasmid. We are also grateful to Cheng Zhan at the imaging center and Zhihua Qiu and Ping Qu at the animal facility of the National Institute of Biological Science, Beijing (NIBS), for their excellent technical assistance.
This work was supported by the Major State Basic Research Development Program of China (2010CB530101 and 2011CB812501) and by the Science and Technology Bureau of the Beijing Municipal Government.
Footnotes
Published ahead of print 17 April 2013
REFERENCES
- 1. Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219 [DOI] [PubMed] [Google Scholar]
- 2. Hagenbuch B, Meier PJ. 1994. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J. Clin. Invest. 93:1326–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stieger B. 2011. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb. Exp. Pharmacol. 201:205–259 [DOI] [PubMed] [Google Scholar]
- 4. Hu NJ, Iwata S, Cameron AD, Drew D. 2011. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 478:408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mareninova O, Shin JM, Vagin O, Turdikulova S, Hallen S, Sachs G. 2005. Topography of the membrane domain of the liver Na+-dependent bile acid transporter. Biochemistry 44:13702–13712 [DOI] [PubMed] [Google Scholar]
- 6. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049 doi: 10.7554/eLife.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schulze A, Schieck A, Ni Y, Mier W, Urban S. 2010. Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol. 84:1989–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glebe D, Urban S, Knoop EV, Cag N, Krass P, Grun S, Bulavaite A, Sasnauskas K, Gerlich WH. 2005. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 129:234–245 [DOI] [PubMed] [Google Scholar]
- 9. Gripon P, Cannie I, Urban S. 2005. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 79:1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barrera A, Guerra B, Notvall L, Lanford RE. 2005. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J. Virol. 79:9786–9798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lanford RE, Chavez D, Barrera A, Brasky KM. 2003. An infectious clone of woolly monkey hepatitis B virus. J. Virol. 77:7814–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lanford RE, Chavez D, Brasky KM, Burns RB, III, Rico-Hesse R. 1998. Isolation of a hepadnavirus from the woolly monkey, a New World primate. Proc. Natl. Acad. Sci. U. S. A. 95:5757–5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lott L, Notvall L, Lanford RE. 2003. Transcomplementation of core and polymerase functions of the woolly monkey and human hepatitis B viruses. Virology 308:330–339 [DOI] [PubMed] [Google Scholar]
- 14. Chouteau P, Le Seyec J, Cannie I, Nassal M, Guguen-Guillouzo C, Gripon P. 2001. A short N-proximal region in the large envelope protein harbors a determinant that contributes to the species specificity of human hepatitis B virus. J. Virol. 75:11565–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barrera A, Guerra B, Lee H, Lanford RE. 2004. Analysis of host range phenotypes of primate hepadnaviruses by in vitro infections of hepatitis D virus pseudotypes. J. Virol. 78:5233–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petersen J, Dandri M, Mier W, Lutgehetmann M, Volz T, von Weizsacker F, Haberkorn U, Fischer L, Pollok JM, Erbes B, Seitz S, Urban S. 2008. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 26:335–341 [DOI] [PubMed] [Google Scholar]
- 17. Köck J, Nassal M, MacNelly S, Baumert TF, Blum HE, von Weizsacker F. 2001. Efficient infection of primary tupaia hepatocytes with purified human and woolly monkey hepatitis B virus. J. Virol. 75:5084–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dandri M, Burda MR, Zuckerman DM, Wursthorn K, Matschl U, Pollok JM, Rogiers X, Gocht A, Köck J, Blum HE, von Weizsacker F, Petersen J. 2005. Chronic infection with hepatitis B viruses and antiviral drug evaluation in uPA mice after liver repopulation with tupaia hepatocytes. J. Hepatol. 42:54–60 [DOI] [PubMed] [Google Scholar]
- 19. Cao J, Yang EB, Su JJ, Li Y, Chow P. 2003. The tree shrews: adjuncts and alternatives to primates as models for biomedical research. J. Med. Primatol. 32:123–130 [DOI] [PubMed] [Google Scholar]
- 20. Murphy WJ, Eizirik E, O'Brien SJ, Madsen O, Scally M, Douady CJ, Teeling E, Ryder OA, Stanhope MJ, de Jong WW, Springer MS. 2001. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 294:2348–2351 [DOI] [PubMed] [Google Scholar]
- 21. Dandri M, Burda MR, Torok E, Pollok JM, Iwanska A, Sommer G, Rogiers X, Rogler CE, Gupta S, Will H, Greten H, Petersen J. 2001. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology 33:981–988 [DOI] [PubMed] [Google Scholar]
- 22. Le Seyec J, Chouteau P, Cannie I, Guguen-Guillouzo C, Gripon P. 1999. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J. Virol. 73:2052–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blanchet M, Sureau C. 2007. Infectivity determinants of the hepatitis B virus pre-S domain are confined to the N-terminal 75 amino acid residues. J. Virol. 81:5841–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Le Duff Y, Blanchet M, Sureau C. 2009. The pre-S1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J. Virol. 83:12443–12451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sureau C, Salisse J. 2013. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus A-determinant. Hepatology 57(3):1–10 [DOI] [PubMed] [Google Scholar]
- 26. Gripon P, Diot C, Guguen-Guillouzo C. 1993. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology 192:534–540 [DOI] [PubMed] [Google Scholar]
- 27. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 99:15655–15660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paraskevis D, Magiorkinis G, Magiorkinis E, Ho SY, Belshaw R, Allain JP, Hatzakis A. 2013. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 57:908–916 [DOI] [PubMed] [Google Scholar]
- 29. Kuroki K, Cheung R, Marion PL, Ganem D. 1994. A cell surface protein that binds avian hepatitis B virus particles. J. Virol. 68:2091–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuroki K, Eng Ishikawa FT, Turck C, Harada F, Ganem D. 1995. gp180, a host cell glycoprotein that binds duck hepatitis B virus particles, is encoded by a member of the carboxypeptidase gene family. J. Biol. Chem. 270:15022–15028 [DOI] [PubMed] [Google Scholar]
- 31. Tong S, Li J, Wands JR. 1995. Interaction between duck hepatitis B virus and a 170-kilodalton cellular protein is mediated through a neutralizing epitope of the pre-S region and occurs during viral infection. J. Virol. 69:7106–7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Breiner KM, Urban S, Schaller H. 1998. Carboxypeptidase D (gp180), a Golgi-resident protein, functions in the attachment and entry of avian hepatitis B viruses. J. Virol. 72:8098–8104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Urban S, Breiner KM, Fehler F, Klingmuller U, Schaller H. 1998. Avian hepatitis B virus infection is initiated by the interaction of a distinct pre-S subdomain with the cellular receptor gp180. J. Virol. 72:8089–8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Urban S, Schwarz C, Marx UC, Zentgraf H, Schaller H, Multhaup G. 2000. Receptor recognition by a hepatitis B virus reveals a novel mode of high affinity virus-receptor interaction. EMBO J. 19:1217–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Urban S, Gripon P. 2002. Inhibition of duck hepatitis B virus infection by a myristoylated pre-S peptide of the large viral surface protein. J. Virol. 76:1986–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]