

Abstract
Human herpesvirus 6 is a T lymphotropic herpesvirus, long classified into variants A and B (HHV-6A and HHV-6B) based on differences in sequence and pathogenicity. Recently, however, HHV-6A and HHV-6B were reclassified as different species. Here, we isolated a neutralizing monoclonal antibody (Mab) named AgQ 1-1 that was specific for HHV-6A glycoprotein Q1 (AgQ1), and we showed that amino acid residues 494 to 497 of AgQ1 were critical for its recognition by this Mab. This region was also essential for AgQ1's complex formation with gH, gL, and gQ2, which might be important for viral binding to the cellular receptor, CD46. In addition, amino acid residues 494 to 497 are essential for viral replication. Interestingly, this sequence corresponds to the domain on HHV-6B gQ1 that is critical for recognition by an HHV-6B-specific neutralizing Mab. Within this domain, only Q at position 496 of HHV-6A is distinct from the HHV-6B sequence; however, the mutant AgQ1(Q496E) was still clearly recognized by the Mab AgQ 1-1. Surprisingly, replacement of the adjacent amino acid, in mutant AgQ1(C495A), resulted in poor recognition by Mab AgQ 1-1, and AgQ1(C495A) could not form the gH/gL/gQ1/gQ2 complex. Furthermore, the binding ability of mutant AgQ1(L494A) with CD46 decreased, although it could form the gH/gL/gQ1/gQ2 complex and it showed clear reactivity to Mab AgQ 1-1. These data indicated that amino acid residues 494 to 497 of AgQ1 were critical for the recognition by Mab AgQ 1-1 and essential for AgQ1's functional conformation.
INTRODUCTION
Human herpesvirus 6 (HHV-6) was first isolated in 1986 from patients with lymphoproliferative disorders (1). HHV-6 was originally classified into two variants, HHV-6A and -B, based on differences in their genetic characteristics and cell tropism (2–5). However, quite recently, HHV-6A and HHV-6B were reclassified into different species (according to the Virus Taxonomy List 2011). HHV-6B is the causative agent of exanthem subitum (6) and mainly causes reactivation in immunocompromised hosts (7, 8), while HHV-6A is involved in the etiology of several diseases, including multiple sclerosis (9), encephalitis (10), and Hashimoto's thyroiditis (11). Almost all children have antibodies against HHV-6A or HHV-6B by 2 years of age (12). Although HHV-6B has been reported to be responsible for primary infections in many countries (13–15), primary HHV-6A infection has been also reported in Africa (16). Both variants can infect the brain, but HHV-6A is thought to be more neurotropic than HHV-6B (17). Several reports have shown that the reactivation of HHV-6A and -B may contribute to several diseases in immunosuppressed patients (18–20).
Human CD46, a regulator of the complement activation receptor expressed on all nucleated cells, is a cellular receptor for HHV-6 (21), and HHV-6A's viral ligand is the envelope glycoprotein complex (gH/gL/gQ1/gQ2) (22, 23). Furthermore, we previously found that the HHV-6A gH/gL/gQ1/gQ2 complex formation itself is important for HHV-6A's trafficking and CD46 binding, thus indicating that the correct folding could not be done without either of those (24). Moreover, in experiments using recombinant viruses, we proved that HHV-6A gQ1 and gQ2 genes are essential genes for virus growth (24, 25).
The gQ1 and gQ2 proteins are conserved in roseoloviruses, but the amino acid identities between HHV-6A strain U1102 and HHV-6B strain HST are 78.0% and 68.7%, respectively. Since the average amino acid identity between HHV-6A U1102 and HHV-6B HST was about 94% (26), gQ1 and gQ2 appear to be key genes for determining various biological differences between HHV-6A and -B. Although the function of HHV-6B gQ1 was unknown, we previously obtained an HHV-6B-specific neutralizing antibody that recognizes a conformational epitope for gQ1, indicating that HHV-6B gQ1, like the gQ1 of HHV-6A, plays a key role in virus infection, especially in cell entry (27). Therefore, to elucidate the viral entry mechanism, it was important to analyze the structure and function of gQ1 in more detail.
In this study, we produced a neutralizing antibody that specifically recognizes the gQ1 of HHV-6A and not that of HHV-6B. We found that the critical domain of gQ1 for antibody recognition was conserved between HHV-6A and -B, although the specific neutralizing antibodies for each virus had distinct determinant(s) in HHV-6A and -B. We further found that the identified domain was also essential for the formation of the HHV-6A gQ1, gQ2, gH, and gL complex, its CD46 binding, and viral replication.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic kidney (HEK) 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 8% fetal bovine serum (FBS), 20 μg/ml gentamicin, and l-glutamine (0.584 g/liter). Umbilical cord blood mononuclear cells (CBMCs) were prepared as described previously (28, 29). CBMCs were kindly provided by Kazushige Adachi (Minoh City Hospital) and Hideto Yamada (Department of Obstetrics and Gynecology, Kobe University Graduate School of Medicine) and purchased from Cell Bank of RIKEN Bioresource Center (RIKEN; the Institute of Physical and Chemical Research, Japan). This study was approved by the ethical committees of all the institutions involved.
Plasmid construction.
Plasmids expressing HHV-6A gH, gL, gQ1, and gQ2 were described previously (24). To construct plasmids expressing the HHV-6A gQ1 carboxyl-terminal deletion mutants AgQ1(Δ1266), AgQ1(Δ1440), AgQ1(Δ1476), AgQ1(Δ1512), and AgQ1(Δ1533), each gQ1 fragment was amplified by the appropriate primer pair, digested with EcoRI and XhoI, and ligated into a digested pcDNA 3.1(−) vector. The HHV-6A gQ1 point mutants AgQ1(L494A), AgQ1(C495A), AgQ1(Q496A), AgQ1(Q496E), and AgQ1(G497A) were constructed with a QuikChange Lightning multisite-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. The constructs were all verified by sequencing. The HHV-6A gQ1 deletion mutant AgQ1(Δ494-497) was constructed as follows: two gQ1 fragments were amplified using two primer pairs (underlines indicate the NotI or XhoI site, as specified), 5′-TCGGACAAGCTGGGCACGTATTTTAC-3′ and 5′-ACCAGCGGCCGCTCAAGTCCTCACGACAGTAGG-3′ (NotI) and 5′-ACCCTCGAGCCACCATGGCAACCGCAAGACTG-3′ (XhoI) and 5′-AAATACGTGCCCAGCTTGTCCGAGGTGTTTATATAC-3′. The mixed fragments were used as the template for the next PCR, which used primers 5′-ACCCTCGAGCCACCATGGCAACCGCAAGACTG-3′ (XhoI) and 5′-ACCAGCGGCCGCTCAAGTCCTCACGACAGTAGG-3′ (NotI). The amplified PCR product was digested with XhoI and NotI, separated by agarose gel electrophoresis, purified, and cloned into XhoI/NotI-digested pcDNA 3.1(−). AgQ1(494AAAA497) was constructed similarly with the primer pair 5′-AGCCGCAGCTGCAGCTTGTCCGAGGTGTTTATATAC-3′ and 5′-GACAAGCTGCAGCTGCGGCTGGGCACGTATTTTACAATC-3′. The CD46-expressing vector pM18S was a generous gift from T. Seya, Hokkaido University, Hokkaido, Japan (30).
Transfections.
293T cells were transfected with the indicated plasmids by using Lipofectamine 2000 reagent (Invitrogen). The transfected cells were harvested at 48 h posttransfection and lysed with TNE buffer (10 mM Tris-HCl [pH 7.8], 1% Nonidet P-40, 0.15 M NaCl, 1 mM EDTA) containing a cocktail of protease inhibitors (Roche). The lysates were cleared by centrifugation at 200,000 × g for 1 h at 4°C. The supernatants were resolved with sample buffer (32 mM Tris-HCl [pH 6.8], 1.5% SDS, 5% glycerol, 2.5% 2-mercaptoethanol) for SDS-PAGE or used for immunoprecipitation.
Antibodies.
The monoclonal antibodies (Mabs) designated AgQ 1-1 and gH F5 were obtained by immunizing mouse footpads with UV-inactivated HHV-6A (strain GS) virions (27). Lymphocytes from the popliteal lymph nodes were subsequently retrieved and fused with the nonproducing myeloma cell line SP2. The hybridoma cells secreting Mabs for HHV-6A were selected by immunofluorescence antibody assay (IFA). Next, the selected Mabs were examined with the neutralizing assay described elsewhere (27). The Mabs designated AgQ1-119, AgL4, gH1-1, and AgQ2B were described previously (24).
Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescein isothiocyanate (FITC)-conjugated rabbit anti-mouse immunoglobulin (Dako) was used as a secondary antibody.
Immunofluorescence assay.
The IFA was performed as described previously (22). Specific immunofluorescence was observed with a fluorescence microscope (Nikon) at ×400 magnification.
Virus neutralization assay.
The virus neutralization assay was performed as described previously (27). Virus titers were determined by measuring the 50% tissue culture infective dose (TCID50) (31).
Immunoprecipitation and Western blot assays.
The immunoprecipitation assay and Western blotting were performed as described previously (24).
CD46 binding assay.
The CD46 binding assay was performed as described previously (24). Briefly, an anti-CD46 antibody was incubated with protein G-Sepharose and then cross-linked with protein G-Sepharose, followed by incubation with CD46-expressing 293T cell lysates. Cell extracts were immunoprecipitated with the protein G-Sepharose-bound antibody. The bound proteins were eluted with 0.1 M glycine (pH 2.5) and neutralized with 1 M Tris-HCl (pH 9.0).
Construction of gQ1 mutant and its revertant.
RecA-mediated recombination was performed as described previously (32–35), with some modifications. We used pST76A-SR for the shuttle vector. We first cloned two homology arms (“gQ1 up” arm, bp 146765 to 147808, and “gQ1 down” arm, bp 150281 to 151227, of the U1102 genome) into the shuttle vector. The gQ1 up arm was amplified from the U1102 genome by PCR with primers 5′-ACAGCGGCCGCGAGCTCAGTAATTCGTTACTATGAG-3′ (underlining indicates the NotI site and italics indicate the SacI site) and 5′-ACCGGTACCTGACCGTTATGTGCGCGTG-3′ (underlining indicates the KpnI site). The gQ1 down arm was amplified with 5′-ACCGGTACCATCAGCGCACAAGATCTC-3′ (underlining indicates the KpnI site) and 5′-ACAGGATCCGTCGACTCCGTCTCCCGGAGAAAC-3′ (italics indicate the BamHI site and underlining the SalI site). The PCR product was digested with NotI and KpnI (gQ1 up arm) and with KpnI and SalI (gQ1 down arm). The fragments were then ligated with pBluescript SK(−), which had been digested with NotI and SalI, followed by sequencing to confirm the lack of PCR-generated mutations. The resultant plasmid was digested with SacI and BamHI and ligated with the shuttle vector pST76A-SR, which had been digested with SacI and BamHI. We named the construct pST76A-SR-fullΔgQ1. Next we cloned the gQ1 up arm, wild-type gQ1, and gQ1 down arm (bp 146765 to 151227 of the U1102 genome) into the shuttle vector, to create pST76A-SR-gQ1 wild type.
We generated gQ1 mutant bacterial artificial chromosome (BAC) DNA, HHV-6ABACgQ1(Δ494-497), in two steps. In the first step, pST76A-SR-fullΔgQ1 was electroporated into DH10B electrocompetent cells harboring wild-type HHV-6A BAC and plated on LB plates containing 17 μg/ml chloramphenicol (Cm) and 50 μg/ml ampicillin (Amp). Ten to 30 colonies were picked, diluted with LB medium, plated onto LB plates containing Cm and Amp, and incubated at 43°C overnight. Six to 10 colonies were picked, diluted 10,000 times with LB medium containing 5% sucrose, and plated onto LB plates containing Cm and 5% sucrose. Colonies growing on these plates were selected by plating single clones onto Cm-containing and Cm- and Amp-containing plates. Cm-resistant but Amp-sensitive clones were tested by PCR. The selected clones were confirmed by digesting the BAC DNA with BamHI. We named the resultant BAC HHV-6ABACfullΔgQ1. In the second step, using DH10B electrocompetent cells harboring HHV-6ABACfullΔgQ1 and pST76A-SR-gQ1(Δ494-497), we constructed HHV-6ABACgQ1(Δ494-497) as described for the first step. The mutation of gQ1 was confirmed by sequencing the isolated BAC DNA. HHV-6ABACgQ1(Δ494-497)rev was constructed in the same way.
Reconstitution of infectious virus using HHV-6A BACs.
The detailed method for reconstituting infectious virus using HHV-6A BACs was described previously (24, 25).
RESULTS
Generation of AgQ1 carboxyl truncation mutants and their reactivity with an HHV-6A-neutralizing Mab.
To analyze HHV-6A's entry mechanism in detail, we first sought to produce neutralizing antibodies for HHV-6A, and we obtained several monoclonal antibodies (Mabs) that could block virus infection. One of them was against HHV-6A gQ1 (AgQ1), which we named AgQ 1-1. Therefore, to elucidate the role of gQ1 in HHV-6A infection, we analyzed this neutralizing Mab and its recognition site on gQ1 in detail.
The Mab AgQ 1-1 neutralized HHV-6A but not HHV-6B, and it specifically recognized AgQ1 (data not shown). In a virus neutralization assay, a 4,000-fold dilution of purified Mab AgQ 1-1 (0.89 mg/ml) completely blocked HHV-6A strain GS infection at 4.0 × 104 TCID50/ml, while a control antibody (AgHgL-A2) never blocked the infection.
Recently, we also produced a neutralizing Mab against HHV-6B gQ1 (BgQ1) and determined the critical domain on BgQ1 for that Mab's recognition (27). Here, we similarly sought to identify the recognition site(s) for Mab AgQ 1-1. For this, we first constructed AgQ1 carboxyl-terminal truncation mutants and examined their reactivity with Mab AgQ 1-1 by IFA. As shown in Fig. 1, the mutant with a C-terminal truncation beginning at residue 504 was recognized by the Mab, while the one with its truncation beginning at residue 492 was not. These results suggested that the important site(s) for Mab AgQ 1-1 recognition was between residues 492 and 504. Surprisingly, the critical site for recognition was completely the same as that for KH-1, the neutralizing Mab for BgQ1 (27). In contrast, another Mab, AgQ1-119, which has an N-terminal (between amino acids 3 and 422) linear epitope on AgQ1, reacted with all the truncation mutants depicted in Figure 1.
Fig 1.
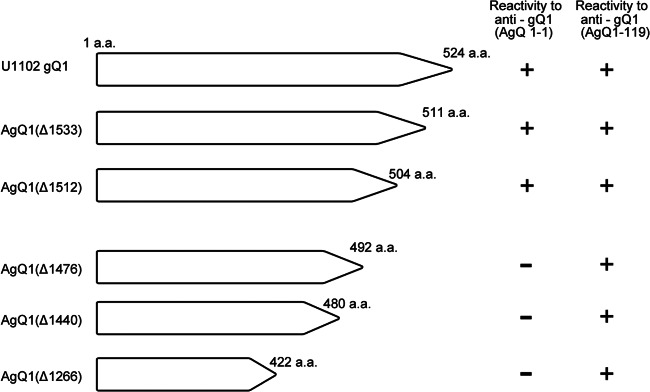
Schematic diagram of HHV-6A gQ1 mutants with various carboxyl-terminal deletions and their reactivity with Mab AgQ 1-1. 293T cells were cotransfected with wild-type AgQ1 or one of its deletion mutants and AgQ2. The transfected cells were harvested 48 h after transfection and subjected to staining with anti-gQ1 Mabs AgQ 1-1 and AgQ1-119. The reactivities to the Mabs are shown at right.
The AgQ1 amino acid residues around 496 are important for its recognition by Mab AgQ 1-1.
The results shown in Figure 1 suggested that the sequence between amino acid residues 492 and 504 of HHV-6A strain U1102 gQ1 might contain an epitope for Mab AgQ 1-1. We next searched for U1102-specific amino acid residue(s) in this region by comparing the gQ1 amino acid sequence of HHV-6A U1102 and HHV-6B strain HST, because the Mab recognized HHV-6A but not HHV-6B. As shown in Figure 2A, only Gln 496 in this region was U1102 specific; this residue in HST was Glu. We therefore constructed a mutant AgQ1 in which Gln496 was replaced with Glu, AgQ1(Q496E) (Fig. 2B) and examined its reactivity with Mab AgQ 1-1. Unexpectedly, this mutant did react with Mab AgQ 1-1 with reactivity similar to that of wild-type AgQ1 (Fig. 3B), which suggested that this region was not the epitope of the Mab AgQ 1-1.
Fig 2.
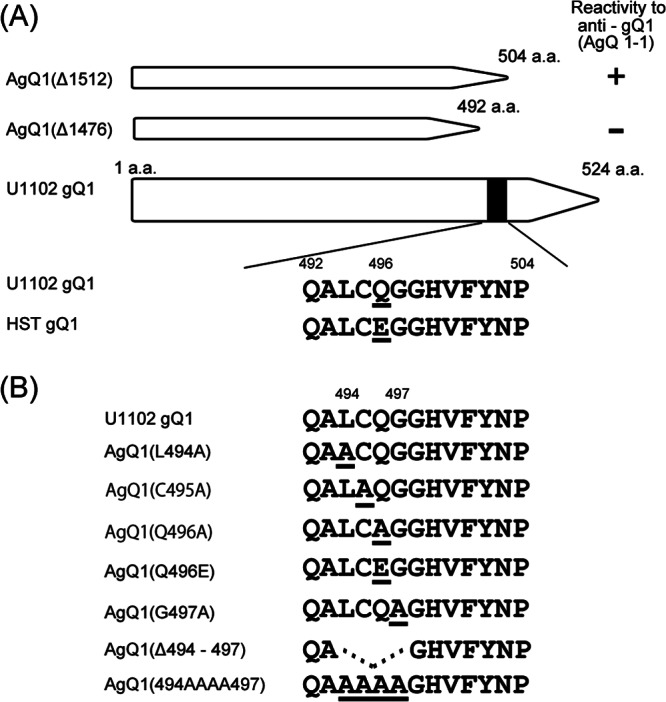
Amino acid sequence comparison between HHV-6A gQ1 and HHV-6B gQ1 and schematic representation of HHV-6A gQ1 mutants. (A) Amino acid sequence comparison of gQ1, from amino acid 492 to 504, between strains U1102 and HST. The differences between these sequences are indicated by underlines. (B) Wild-type AgQ1 and its mutants constructed in this study. Underlines indicate the mutated amino acids.
Fig 3.

AgQ1 region recognized by the neutralizing Mab AgQ 1-1. 293T cells were transfected with wild-type or mutant AgQ1 with AgQ2, AgH, and AgL. The cells were harvested at 48 h posttransfection. (A) The cells were lysed and immunoblotted with the Mab AgQ1-119. WB, Western blotting. (B) The transfected cells were fixed and stained with Hoechst 33258 or with the Mab against AgQ1 (AgQ 1-1 or AgQ1-119) or AgQ2 (AgQ2B).
To analyze this region in more detail, we constructed substitution mutants and a deletion mutant, depicted in Figure 2B. A deletion mutant lacking residues 494 to 497, AgQ1(Δ494-497), and a mutant with serial substitutions, AgQ1(494AAAA497), did not react with Mab AgQ 1-1 at all (Fig. 3B). The expression of all the mutants was confirmed by Western blotting (Fig. 3A) and IFA (Fig. 3B) with Mab AgQ1-119. On the other hand, the single-substitution mutants AgQ1(L494A), AgQ1(C495A), AgQ1(Q496A), and AgQ1(G497A) showed reactivity with Mab AgQ 1-1, although the reactivity of AgQ1(C495A) was severely impaired (Fig. 3B). Since Mab AgQ 1-1 does not react in Western blots (data not shown), we used the AgQ1-119 Mab for Western blotting.
Mab AgQ 1-1 seemed to have a conformational epitope, because the reactivity to AgQ1 clearly increased in the presence of AgQ2, AgH, and AgL (Fig. 3B). In addition, we previously showed that AgQ1 maturation occurs only when all four components, AgH, AgL, AgQ1, and AgQ2, are coexpressed (24). Therefore, the reactivity for Mab AgQ 1-1 was analyzed in the presence of AgH, AgL, and AgQ2. Wild-type or mutant gQ1 showed greater reactivity to Mab AgQ 1-1 in the presence of AgH, AgL, and AgQ2 compared to the reactivity when gQ1 was expressed alone or coexpressed with only gQ2, although AgQ1(Δ494-497) and AgQ1(494AAAA497) showed no reactivity(data not shown). These results indicated that the amino acid residues around positions 494 to 497 were a critical site for Mab AgQ 1-1 recognition, although they were thought not to be the epitope of the Mab.
The amino acid residues around position 496 are important for AgQ1 maturation and AgH/AgL/AgQ1/AgQ2 complex formation.
Next, we performed immunoprecipitation with the Mab AgQ 1-1. As predicted, when AgQ1(Δ494-497) and AgQ1(494AAAA497) were coexpressed with AgH, AgL, and AgQ2, Mab AgQ 1-1 could not immunoprecipitate any component (Fig. 4). In contrast, when wild-type gQ1, AgQ1(L494A), AgQ1(Q496A), AgQ1(Q496E), or AgQ1(G497A) was coexpressed with the other three molecules, Mab AgQ 1-1 could coimmunoprecipitate all of the components. In the case of AgQ1(C495A), although gQ1 and gH could be detected faintly, gL and gQ2 could be scarcely detected.
Fig 4.

Detection of wild-type AgQ1 or its various mutants using the Mab AgQ 1-1. 293T cells were cotransfected with wild-type or mutant HHV-6A gQ1 with AgH, AgL, and AgQ2. Forty-eight hours later, the cells were harvested, lysed, and immunoprecipitated (IP) with the anti-gQ1 Mab, AgQ 1-1, followed by Western blotting (WB) with Mabs for AgQ1 (AgQ1-119), gH (gH1-1), gL (AgL4), and gQ2 (AgQ2B).
We previously showed that with AgQ1's maturation, its size shifted from 74 kDa (gQ1-74K), the immature form, to 80 kDa, the mature form (gQ1-80K); this shift also occurred only when all four components were coexpressed (24). Based on the report, the Western blot assay done here showed that the mature gQ1-80K form of AgQ1(Q494A), AgQ1(C495A), AgQ1(Q496A), AgQ1(Q496E), AgQ1(G497A), or wild-type AgQ1 could be detected, while that of AgQ1(C495A) could be detected but only faintly (Fig. 4, left panels). On the other hand, the gQ1-80K form of AgQ1 (Δ494-497) and AgQ1(494AAAA497) could not be detected at all, even when cotransfected with AgH, AgL, and AgQ2, similar to the results seen in AgH, AgL, and AgQ1 (wild type) transfections, used as a negative control for maturation (Fig. 4). These results may show that this region is also critical for gH/gL/gQ1/gQ2 complex formation, since we have previously shown that gH/gL/gQ1/gQ2 complex formation occurs only when gH, gL, gQ1, and gQ2 are coexpressed (24).
Therefore, to confirm this, we did coimmunoprecipitation experiments by using anti-gH Mab. As expected, AgQ1(Δ494-497) and AgQ1(494AAAA497) could not be immunoprecipitated with anti-gH Mab (Fig. 5). In contrast, AgQ1(Q494A), AgQ1(C495A), AgQ1(Q496A), AgQ1(Q496E), AgQ1(G497A), or wild-type AgQ1 could be immunoprecipitated, although AgQ1(C495A) could barely be detected. These findings collectively show that the amino acid residues around position 496 of AgQ1 are important for both gQ1 maturation and gH/gL/gQ1/gQ2 complex formation, and the region from position 494 to 497 is essential for them.
Fig 5.

Confirmation of complex formation of HHV-6A gH/gL/gQ2 with wild-type or mutant AgQ1. Wild-type HHV-6A gQ1 or each mutant was cotransfected with AgH, AgL, and AgQ2. Forty-eight hours later, the cells were harvested, lysed, and immunoprecipitated (IP) with an anti-gH Mab, gH F5, followed by Western blotting (WB) with Mabs for AgQ1 (AgQ1-119), gH (gH1-1), gL (AgL4), and gQ2 (AgQ2B).
The amino acid residues around 496 are also essential for binding to CD46.
Human CD46 is a cellular receptor for HHV-6A (21), and the HHV-6A gH/gL/gQ1/gQ2 complex is its ligand (22, 23). We previously showed that the binding of AgQ1 to CD46 can be detected only when all three of the other components are coexpressed with AgQ1 (24). Therefore, we examined whether each gQ1 mutant could bind to CD46 when coexpressed with the other three components of the complex. As predicted, the binding of AgQ1(Δ494-497) and AgQ1(494AAAA497) to CD46 could not be detected, although AgQ1(Q496A), AgQ1(Q496E), AgQ1(G497A), or wild-type AgQ1 could bind to CD46 (Fig. 6). Surprisingly, the CD46 binding of AgQ1(L494A) was very weak, although it could form the gH/gL/gQ1/gQ2 complex. As predicted, an interaction of AgQ1(C495A) with CD46 was not seen. This result shows that the amino acid residues around position 496 are also important for binding to CD46 and the region from position 494 to 497 is essential for it.
Fig 6.

Interaction of AgH/AgL/AgQ1 (wild type or each mutant)/AgQ2 with CD46. 293T cells were cotransfected with AgH, AgL, AgQ2, and wild-type or mutant AgQ1, and 48 h later, the cell lysates were prepared and used for a CD46 binding assay. CD46 protein was isolated from 293T cells transfected with a CD46-expressing plasmid using an anti-CD46 Mab cross-linked to protein G-Sepharose. The Sepharose was then incubated with the cell extracts, shown in the left panels, and the eluates from the Sepharose were blotted with the indicated Mabs, shown in the right panels.
Mab AgQ 1-1 does not immunoprecipitate CD46 from HHV-6A-infected cells.
We next analyzed the relationship between the Mab AgQ 1-1 determinant and the CD46 binding domain in HHV-6A-infected cells. For this, we examined whether Mab AgQ 1-1 could coprecipitate CD46 in HHV-6A strain GS-infected HSB-2 cell lysates. CD46 was not coprecipitated by Mab AgQ 1-1, while CD46 was coprecipitated with Mab AgQ1-119 (Fig. 7), suggesting that Mab AgQ 1-1 blocks HHV-6A infection by interfering with CD46 binding.
Fig 7.
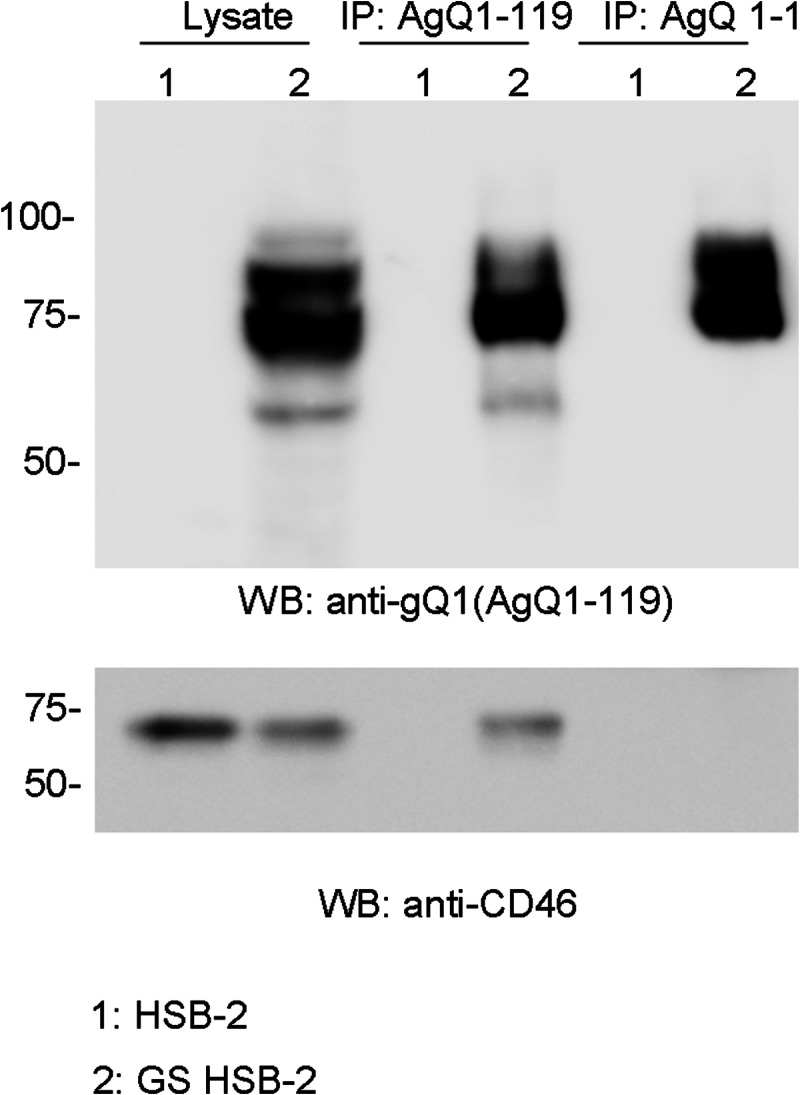
Mab AgQ 1-1 does not immunoprecipitate CD46 from HHV-6A-infected cells. Lysates from HHV-6A strain GS-infected HSB-2 cells were immunoprecipitated with the antibodies indicated above the gels. The immunoprecipitates were blotted with anti-gQ1 (top) and anti-CD46 (bottom) Mabs.
We finally analyzed whether the amino acid residues around position 494 to 497 of AgQ1 are also essential for viral replication, because this region is essential for gH/gL/gQ1/gQ2 complex formation and binding to CD46. In fact, infectious virus could not be reconstituted from the HHV-6ABACgQ1(Δ494-497) genome, while its revertant virus, rHHV-6ABACgQ1(Δ494-497)rev, could be rescued (data not shown).
DISCUSSION
We previously reported that the HHV-6A gH/gL/gQ1/gQ2 complex binds to CD46 (24). In the present study, we established a neutralizing Mab, AgQ 1-1, which specifically reacted with HHV-6A gQ1 but not with HHV-6B gQ1, and analyzed its characteristics.
The Mab AgQ 1-1 was thought to have a conformational epitope, because its reactivity to AgQ1 clearly increased in the presence of AgQ2, AgH, and AgL (Fig. 3) and it did not react in the Western blot assay.
To determine the recognition site(s) of the Mab AgQ 1-1, we constructed several C-terminal deletion mutants of HHV-6A gQ1 and examined whether they could react with Mab AgQ 1-1. The important domain for the Mab recognition appeared to lie between the amino acid residues at positions 496 and 504 (Fig. 1). Interestingly, in HHV-6B gQ1, this region is important for reacting with HHV-6B's neutralizing Mab, KH-1 (27). Although the only amino acid in this region that is different in HHV-6A and -B is Glu 496 of HHV-6B, the replacement of this amino acid with Gln did not affect KH-1's recognition (27). In addition, the replacement of this region in HHV-6A gQ1 with the homologous domain from HHV-6B gQ1 did not abrogate its recognition by Mab AgQ 1-1. This shows that although the epitope for the Mab AgQ 1-1 did not exist within this region, the region appears to be important for AgQ1's recognition by Mab AgQ 1-1, because AgQ1(Δ494-497) and AgQ1(494AAAA497) showed no reactivity with the Mab at all, even in the presence of gH, gL, and gQ2 (Fig. 3). Therefore, we further analyzed the region in detail by constructing other mutants, as depicted in Figure 2.
From the results with AgQ1(Δ494-497) and AgQ1(494AAAA497), we found that this region was also essential for HHV-6A gH/gL/gQ1/gQ2 complex formation (Fig. 5) and for its binding to CD46 (Fig. 6). To investigate whether this region is also essential for virus replication, we constructed an HHV-6A BAC genome with the mutated gQ1, AgQ1(Δ494-497), and tried to reconstitute the infectious virus from the genome. As predicted, infectious virus could not be reconstituted, but its revertant virus could be (data not shown), indicating that this region is also essential for virus infection.
Since this region was thought to be important for AgQ1's proper folding, we next replaced amino acids around position 496 and analyzed the characteristics of the mutants. The characteristics of the wild type and each mutant are summarized in Table 1. The mutants AgQ1(Q496A) and AgQ1(G497A) showed levels of reactivity to Mab AgQ 1-1 in the presence of AgH, AgL, and AgQ2 that were similar to the reactivity of wild-type AgQ1 (Fig. 3). In addition, they could form the gH/gL/gQ1/gQ2 complex and bind to CD46 as well as wild-type AgQ1 or AgQ1(Q496E) (Fig. 4, 5, and 6). However, AgQ1(C495A) showed much less reactivity to Mab AgQ 1-1 even in the presence of AgH, AgL and AgQ2 (Fig. 3). Furthermore, the interaction of AgQ1(C495A) with gH, gL, and gQ2 seemed to be severely impaired (Fig. 4). In addition, the complex containing AgQ1(C495A) could not bind to CD46 (Fig. 6), indicating that gQ1(C495A) with gH, gL, and gQ2 might not be folded properly. Surprisingly, the interaction of the complex containing AgQ1(L494A) with CD46 was very weak (Fig. 6), although AgQ1(L494A) showed reactivity to Mab AgQ 1-1 in the presence of AgH, AgL, and AgQ2 (Fig. 3). Furthermore, unlike AgQ1(C495A), that substitution (AgQ1 494 L → A) did not seem to affect the interaction of AgQ1(L494A) with gH, gL, and gQ2 (Fig. 4). The discrepancy may be explained by two possibilities. First, the CD46 binding domain of AgQ1 might exist within this region. Second, this region might be critical for maintaining the conformation of the CD46 binding domain, although it is not the direct binding site. Consistent with this, the results shown in Figure 7 suggested that the Mab AgQ 1-1 might interfere with the binding of CD46 to its ligand, the gH/gL/gQ1/gQ2 complex. Therefore, since the amino acids around position 496 might not be the epitope of the Mab AgQ 1-1, if this region overlaps the CD46 binding domain, the epitope of the Mab AgQ 1-1 should exist in some other region. If this region does not overlap the CD46 binding domain, the epitope of the Mab AgQ 1-1 may or may not exist in the CD46 binding domain. AgQ2 bands were barely visible in the data obtained here. Since it seemed to be difficult to detect it by Western blotting, this might be explained by low expression of gQ2 or its specific structure.
Table 1.
Summary of characteristics of wild type and mutants of AgQ1
| Protein | Reactivitya | Complex formation | CD46 binding |
|---|---|---|---|
| AgQ1 wild type | + | + | + |
| AgQ1(L494A) | + | + | ±b |
| AgQ1(C495A) | ±b | ±b | − |
| AgQ1(Q496A) | + | + | + |
| AgQ1(Q496E) | + | + | + |
| AgQ1(G497A) | + | + | + |
| AgQ1(Δ494-497) | − | − | − |
| AgQ1(494AAAA497) | − | − | − |
Reactivity to the Mab AgQ 1-1 in IFA (Fig. 3).
±, reactivity to the Mab AgQ 1-1 or interaction between each component seemed to be severely impaired, or binding to CD46 could hardly be detected.
We previously produced a neutralizing Mab to BgQ1, named KH-1, by immunizing mice with UV-inactivated HHV-6B virions (27). Interestingly, we determined that the critical domain for the recognition of Mab AgQ 1-1, which we produced by immunizing mice with UV-inactivated HHV-6A virions, is completely the same as that of Mab KH-1. Furthermore, KH-1 showed reactivity similar to the reactivities of mutants with single amino acid substitutions, like Mab AgQ 1-1. In other words, KH-1 did react to BgQ1(E488Q) to an extent similar to the reactivity of wild-type BgQ1 (BgQ1 488E corresponds to AgQ1 496Q), and the levels of reactivity to mutants with its neighboring amino acid substitutions were significantly decreased (27). However, some point mutants with mutations around BgQ1 488E did not show reactivity to KH-1, although all the point mutants with mutations around AgQ1 496Q showed reactivity. We think this difference mainly comes from the difference of the presence of AgH and AgL; we performed IFA with Mab AgQ 1-1 in the presence of AgH, AgL, and AgQ2. On the other hand, the reactivity to BgQ1 was analyzed only in the presence of BgQ2. We found that the reactivity of AgQ1 point mutants to Mab AgQ 1-1 clearly increased more in the presence of AgQ2, AgH, and AgL than only in the presence of AgQ2 (data not shown). Since both Mabs AgQ 1-1 and KH-1 are neutralizing Mabs, the amino acid residues around position 496 of AgQ1, which corresponds to 488 of BgQ1, are conserved and thought to be critical for virus entry, although they are thought not to be the epitope of the neutralizing Mabs.
Here, we identified a domain of AgQ1 that is critical for maintaining its functional conformation. Although the structure of its receptor, CD46, has been determined (36, 37), the crystal structures of gQ1/gQ2 or gH/gL/gQ1/gQ2 have not been reported. In other herpesviruses, structure-based mutagenesis had been reported to identify functional domains of gH and gB (38–40). The conformation of the HHV-6A gH/gL/gQ1/gQ2 complex needs to be determined to elucidate the functional and conformational changes of the envelope and host membrane, which will help reveal the entry mechanism of HHV-6A.
ACKNOWLEDGMENTS
We thank Ulrich H. Koszinowski (Max von Pettenkofer-Institute, Ludwig-Maximilians-University, Munich, Germany) for the pHA-2 and pST76A-SR plasmids. We thank Kazushige Adachi (Minoh City Hospital) and Hideto Yamada (Department of Obstetrics and Gynecology, Kobe University Graduate School of Medicine) for providing the CBMCs and Emi Hayashi and Eiko Moriishi (National Institute of Biomedical Innovation) for their assistance.
This study was supported in part by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS).
Footnotes
Published ahead of print 17 April 2013
REFERENCES
- 1. Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarsky B, Gallo RC. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596–601 [DOI] [PubMed] [Google Scholar]
- 2. Ablashi DV, Balachandran N, Josephs SF, Hung CL, Krueger GR, Kramarsky B, Salahuddin SZ, Gallo RC. 1991. Genomic polymorphism, growth properties, and immunologic variations in human herpesvirus-6 isolates. Virology 184:545–552 [DOI] [PubMed] [Google Scholar]
- 3. Aubin JT, Collandre H, Candotti D, Ingrand D, Rouzioux C, Burgard M, Richard S, Huraux JM, Agut H. 1991. Several groups among human herpesvirus 6 strains can be distinguished by Southern blotting and polymerase chain reaction. J. Clin. Microbiol. 29:367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Campadelli-Fiume G, Guerrini S, Liu X, Foa-Tomasi L. 1993. Monoclonal antibodies to glycoprotein B differentiate human herpesvirus 6 into two clusters, variants A and B. J. Gen. Virol. 74(Pt 10):2257–2262 [DOI] [PubMed] [Google Scholar]
- 5. Chandran B, Tirawatnapong S, Pfeiffer B, Ablashi DV. 1992. Antigenic relationships among human herpesvirus-6 isolates. J. Med. Virol. 37:247–254 [DOI] [PubMed] [Google Scholar]
- 6. Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, Kurata T. 1988. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet i:1065–1067 [DOI] [PubMed] [Google Scholar]
- 7. Lautenschlager I, Razonable RR. 2012. Human herpesvirus-6 infections in kidney, liver, lung, and heart transplantation: review. Transplant Int. 25:493–502 [DOI] [PubMed] [Google Scholar]
- 8. Scheurer ME, Pritchett JC, Amirian ES, Zemke NR, Lusso P, Ljungman P. 2013. HHV-6 encephalitis in umbilical cord blood transplantation: a systematic review and meta-analysis. Bone Marrow Transplant. 48:574–580 [DOI] [PubMed] [Google Scholar]
- 9. Akhyani N, Berti R, Brennan MB, Soldan SS, Eaton JM, McFarland HF, Jacobson S. 2000. Tissue distribution and variant characterization of human herpesvirus (HHV)-6: increased prevalence of HHV-6A in patients with multiple sclerosis. J. Infect. Dis. 182:1321–1325 [DOI] [PubMed] [Google Scholar]
- 10. Portolani M, Pecorari M, Tamassia MG, Gennari W, Beretti F, Guaraldi G. 2001. Case of fatal encephalitis by HHV-6 variant A. J. Med. Virol. 65:133–137 [PubMed] [Google Scholar]
- 11. Caselli E, Zatelli MC, Rizzo R, Benedetti S, Martorelli D, Trasforini G, Cassai E, degli Uberti EC, Di Luca D, Dolcetti R. 2012. Virologic and immunologic evidence supporting an association between HHV-6 and Hashimoto's thyroiditis. PLoS Pathog. 8:e1002951 doi: 10.1371/journal.ppat.1002951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okuno T, Takahashi K, Balachandra K, Shiraki K, Yamanishi K, Takahashi M, Baba K. 1989. Seroepidemiology of human herpesvirus 6 infection in normal children and adults. J. Clin. Microbiol. 27:651–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hall CB, Caserta MT, Schnabel KC, McDermott MP, Lofthus GK, Carnahan JA, Gilbert LM, Dewhurst S. 2006. Characteristics and acquisition of human herpesvirus (HHV) 7 infections in relation to infection with HHV-6. J. Infect. Dis. 193:1063–1069 [DOI] [PubMed] [Google Scholar]
- 14. Tanaka-Taya K, Kondo T, Mukai T, Miyoshi H, Yamamoto Y, Okada S, Yamanishi K. 1996. Seroepidemiological study of human herpesvirus-6 and -7 in children of different ages and detection of these two viruses in throat swabs by polymerase chain reaction. J. Med. Virol. 48:88–94 [DOI] [PubMed] [Google Scholar]
- 15. Zerr DM, Meier AS, Selke SS, Frenkel LM, Huang ML, Wald A, Rhoads MP, Nguy L, Bornemann R, Morrow RA, Corey L. 2005. A population-based study of primary human herpesvirus 6 infection. N. Engl. J. Med. 352:768–776 [DOI] [PubMed] [Google Scholar]
- 16. Bates M, Monze M, Bima H, Kapambwe M, Clark D, Kasolo FC, Gompels UA. 2009. Predominant human herpesvirus 6 variant A infant infections in an HIV-1 endemic region of sub-Saharan Africa. J. Med. Virol. 81:779–789 [DOI] [PubMed] [Google Scholar]
- 17. Pedersen SM, Hollsberg P. 2006. Complexities in human herpesvirus-6A and -6B binding to host cells. Virology 356:1–3 [DOI] [PubMed] [Google Scholar]
- 18. Clark DA. 2002. Human herpesvirus 6 and human herpesvirus 7: emerging pathogens in transplant patients. Int. J. Hematol. 76(Suppl 2):246–252 [DOI] [PubMed] [Google Scholar]
- 19. Corti M, Villafane MF, Trione N, Mamanna L, Bouzas B. 2011. Human herpesvirus 6: report of emerging pathogen in five patients with HIV/AIDS and review of the literature. Rev. Soc. Bras. Med. Trop. 44:522–525 [DOI] [PubMed] [Google Scholar]
- 20. Fremont M, Metzger K, Rady H, Hulstaert J, De Meirleir K. 2009. Detection of herpesviruses and parvovirus B19 in gastric and intestinal mucosa of chronic fatigue syndrome patients. In Vivo 23:209–213 [PubMed] [Google Scholar]
- 21. Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. 1999. CD46 is a cellular receptor for human herpesvirus 6. Cell 99:817–827 [DOI] [PubMed] [Google Scholar]
- 22. Akkapaiboon P, Mori Y, Sadaoka T, Yonemoto S, Yamanishi K. 2004. Intracellular processing of human herpesvirus 6 glycoproteins Q1 and Q2 into tetrameric complexes expressed on the viral envelope. J. Virol. 78:7969–7983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mori Y, Yang X, Akkapaiboon P, Okuno T, Yamanishi K. 2003. Human herpesvirus 6 variant A glycoprotein H-glycoprotein L-glycoprotein Q complex associates with human CD46. J. Virol. 77:4992–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang H, Hayashi M, Maeki T, Yamanishi K, Mori Y. 2011. Human herpesvirus 6 glycoprotein complex formation is required for folding and trafficking of the gH/gL/gQ1/gQ2 complex and its cellular receptor binding. J. Virol. 85:11121–11130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang H, Kawabata A, Yoshida M, Oyaizu H, Maeki T, Yamanishi K, Mori Y. 2010. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 407:360–367 [DOI] [PubMed] [Google Scholar]
- 26. Isegawa Y, Mukai T, Nakano K, Kagawa M, Chen J, Mori Y, Sunagawa T, Kawanishi K, Sashihara J, Hata A, Zou P, Kosuge H, Yamanishi K. 1999. Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J. Virol. 73:8053–8063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawabata A, Oyaizu H, Maeki T, Tang H, Yamanishi K, Mori Y. 2011. Analysis of a neutralizing antibody for human herpesvirus 6B reveals a role for glycoprotein Q1 in viral entry. J. Virol. 85:12962–12971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mori Y, Akkapaiboon P, Yang X, Yamanishi K. 2003. The human herpesvirus 6 U100 gene product is the third component of the gH-gL glycoprotein complex on the viral envelope. J. Virol. 77:2452–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mori Y, Koike M, Moriishi E, Kawabata A, Tang H, Oyaizu H, Uchiyama Y, Yamanishi K. 2008. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 9:1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iwata K, Seya T, Yanagi Y, Pesando JM, Johnson PM, Okabe M, Ueda S, Ariga H, Nagasawa S. 1995. Diversity of sites for measles virus binding and for inactivation of complement C3b and C4b on membrane cofactor protein CD46. J. Biol. Chem. 270:15148–15152 [DOI] [PubMed] [Google Scholar]
- 31. Asada H, Yalcin S, Balachandra K, Higashi K, Yamanishi K. 1989. Establishment of titration system for human herpesvirus 6 and evaluation of neutralizing antibody response to the virus. J. Clin. Microbiol. 27:2204–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adler H, Messerle M, Koszinowski UH. 2003. Cloning of herpesviral genomes as bacterial artificial chromosomes. Rev. Med. Virol. 13:111–121 [DOI] [PubMed] [Google Scholar]
- 33. Gong S, Yang XW, Li C, Heintz N. 2002. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 12:1992–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Connor M, Peifer M, Bender W. 1989. Construction of large DNA segments in Escherichia coli. Science 244:1307–1312 [DOI] [PubMed] [Google Scholar]
- 35. Somboonthum P, Yoshii H, Okamoto S, Koike M, Gomi Y, Uchiyama Y, Takahashi M, Yamanishi K, Mori Y. 2007. Generation of a recombinant Oka varicella vaccine expressing mumps virus hemagglutinin-neuraminidase protein as a polyvalent live vaccine. Vaccine 25:8741–8755 [DOI] [PubMed] [Google Scholar]
- 36. Cardone J, Le Friec G, Kemper C. 2011. CD46 in innate and adaptive immunity: an update. Clin. Exp. Immunol. 164:301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Persson BD, Schmitz NB, Santiago C, Zocher G, Larvie M, Scheu U, Casasnovas JM, Stehle T. 2010. Structure of the extracellular portion of CD46 provides insights into its interactions with complement proteins and pathogens. PLoS Pathog. 6:e1001122 doi: 10.1371/journal.ppat.1001122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen J, Jardetzky TS, Longnecker R. 2013. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J. Virol. 87:3620–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J, Rowe CL, Jardetzky TS, Longnecker R. 2012. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 3(1):e00290–11 doi: 10.1128/mBio.00290-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oliver SL, Brady JJ, Sommer MH, Reichelt M, Sung P, Blau HM, Arvin AM. 2013. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 110:1911–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]