

Abstract
Neuromyelitis optica (NMO) is an autoimmune ‘aquaporinopathy’ of the central nervous system that causes inflammatory demyelinating lesions primarily in spinal cord and optic nerve, leading to paralysis and blindness. NMO lesions show loss of aquaporin-4 (AQP4), GFAP and myelin, infiltration of granulocytes and macrophages, and perivascular deposition of activated complement. Most patients with NMO are seropositive for immunoglobulin autoantibodies (AQP4-IgG) against AQP4, the principal water channel of astrocytes. There is strong evidence that AQP4-IgG is pathogenic in NMO, probably by a mechanism involving complement-dependent astrocyte cytotoxicity, causing leukocyte infiltration, cytokine release and blood-brain barrier disruption, which leads to oligodendrocyte death, myelin loss and neuron death. Here, we review the evidence for this and alternative proposed NMO pathogenesis mechanisms, such as AQP4-IgG-induced internalization of AQP4 and glutamate transporters, complement-independent cell-mediated cytotoxicity, and AQP4-IgG inhibition of AQP4 water transport function. Based on the initiating pathogenic role of AQP4-IgG binding to astrocyte AQP4 in NMO, selective blocker therapies are under development in which AQP4-targeted monoclonal antibodies or small molecules block binding of AQP4-IgG to astrocytes and consequent downstream pathology.
Keywords: NMO, Devic’s disease, Aquaporin-4, Astrocyte, Complement, Autoimmunity
1. Introduction
The aquaporins (AQPs) are a family of related membrane water channels, most of which, including aquaporin-4 (AQP4), which is the focus of this review, are selective for water transport. Some AQPs, called aquaglyceroporins, also transport glycerol. The water-selective AQPs are involved in a wide range of biological functions including transepithelial fluid transport, cell migration and neuroexcitation (Verkman, 2011, 2012). The aquaglyceroporins are involved in cell proliferation, adipocyte metabolism and epidermal water retention (Hara-Chikuma and Verkman, 2008; Rojek et al., 2008). Loss-of-function mutations in human AQPs cause nephrogenic diabetes insipidus (AQP2) and congenital cataracts (AQP0). The AQPs are assembled in membranes as tetramers in which each ~30kDa monomer contains six transmembrane helical domains and two short helical segments surrounding cytoplasmic and extracellular vestibules connected by a narrow aqueous pore (Fujiyoshi et al., 2002; Walz et al., 2009). Water-selective AQPs are expressed broadly in mammalian tissues involved in fluid transport, such as epithelial and endothelial cells in kidney, lung, eye and the gastrointestinal tract. The aquaglyceroporins are expressed in adipocytes, epidermal keratinocytes, and various cell types involved in tissue regeneration. Many tumors express AQPs as well.
The focus of this review is on cellular pathogenesis mechanisms of the AQP4-based neuroinflammatory disease, neuromyelitis optica (NMO). NMO was originally thought to be a variant of multiple sclerosis in that both diseases are associated with inflammatory, demyelinating lesions of the central nervous system (CNS). However, there are clear differences in the sites and pathology of lesions, and in their clinical course and response to therapies. NMO primarily affects optic nerve and spinal cord, causing blindness and paralysis, with relatively little brain pathology. The prevalence of NMO is ~2–4 per 100,000 individuals, and ~6–8 times more prevalent in women than in men (Wingerchuk et al., 2007). The incidence of NMO is greater in Asians and blacks than in Caucasians. The median age of onset is 39 years, and 80–90% of NMO patients have relapsing optic neuritis and myelitis rather than a monophasic course (Jarius and Wildemann, 2010). Current NMO therapies include immunosuppression, plasmapheresis and B-cell depletion. The clinical aspects of NMO are discussed in more detail in recent reviews (Fazio et al., 2011; Jarius and Wildemann, 2010; Wingerchuk et al., 2007).
A defining feature of NMO is the presence of serum autoantibodies against AQP4 (called AQP4-IgG or NMO-IgG), which is detected in 60–90% of NMO patients (Jarius and Wildemann, 2010; Lennon et al., 2005). AQP4-IgG seropositivity is highly specific for NMO. AQP4 is expressed on the plasma membrane of astrocytes throughout the CNS, as well as in skeletal muscle and in epithelial cells in kidney, stomach and exocrine glands (Frigeri et al., 1995a,b). Though AQP4-IgG was initially thought to be a serum marker of NMO, perhaps related to astrocyte damage, there is now strong evidence that AQP4-IgG is pathogenic in NMO. AQP4-IgG binding to AQP4 on astrocytes is thought to cause complement-dependent cytotoxicity, leading to leukocyte infiltration, cytokine release and blood-brain barrier breakdown. These initial events lead to oligodendrocyte death, myelin loss and neuron death, and consequent clinical neurological deficit.
Familial NMO occurs in approximately 3% of cases and seems to have a complex genetic basis (Matiello et al., 2010). Previous studies failed to establish significant correlation between polymorphisms in AQP4 sequence and NMO susceptibility (Crane et al., 2011b; Matiello et al., 2011). An increased risk of NMO has been associated with particular human leukocyte antigen alleles in specific populations (Wang et al., 2011; Zephir et al., 2009). It remains largely unknown why patients develop antibodies against AQP4. AQP4 autoimmunity frequently coexists with other autoimmune diseases, and it has been suggested in some NMO patients that AQP4 autoimmunity is initiated as an immune response to tumor antigens (Antoine et al., 2004; Ducray et al., 2007; Pittock and Lennon, 2008). In this review we evaluate potential pathogenic effects of AQP4-IgG binding to AQP4 and their relevance in NMO pathogenesis. How AQP4-IgG causes NMO pathology has important consequences for development of new therapies that target the underlying cause(s) of the disease.
2. AQP4 structure and function
AQP4 was originally cloned in 1994 by our lab based on its homology to known AQPs (Hasegawa et al., 1994). X-ray structure analysis showed that each AQP4 monomer consists of six helical, membrane-spanning domains and two short helical segments surrounding a narrow aqueous pore (Ho et al., 2009), similar to other AQPs. AQP4 monomers assemble in membranes as tetramers, which can further associate in the cell plasma membrane as large aggregates called orthogonal arrays of particles (OAPs). OAPs are seen by freeze-fracture electron microscopy as regular square arrays of intramembrane particles (Landis and Reese, 1974; Rash et al., 1974; Wolburg et al., 2011). Our lab identified AQP4 as the major OAP protein from the appearance of OAPs in AQP4-transfected cells (Yang et al., 1996) and the absence of OAPs in AQP4 knockout mice (Verbavatz et al., 1997). AQP4 is present in two major isoforms: a long (M1) isoform with translation initiation at Met-1, and a short (M23) isoform with translation initiation at Met-23 (Fig. 1A) (Lu et al., 1996; Yang et al., 1995). Both isoforms are expressed in astrocytes, forming heterotetramers. OAP formation is stabilized by intermolecular N-terminus interactions involving residues just downstream of Met-23 (Crane and Verkman, 2009a). Our lab has applied biophysical methods, including quantum dot single particle tracking and super-resolution imaging, to study the structure, dynamics and regulation of AQP4 assembly in OAPs (Crane et al., 2008, 2009; Crane and Verkman, 2009a,b; Jin et al., 2011; Rossi et al., in press).
Fig. 1.
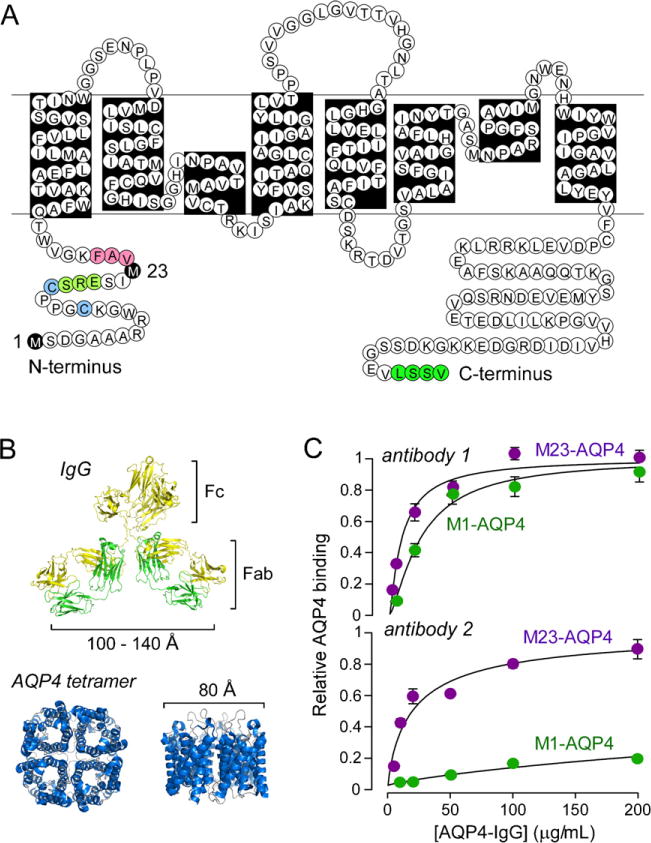
AQP4-IgG binding to its target, AQP4. (A) Amino acid sequence of human AQP4 showing Met-1 and Met-23 translation inhibition sites (black). Pink – residues involved in intermolecular N-terminus associations to form OAPs; light green – residues preventing OAP formation by M1-AQP4; blue – cysteine residues involved in palmitoylation-regulated OAP assembly; and dark green – C-terminus PDZ domain. (B) Crystal structure of human AQP4 (PDB, 3GD8) showing tetrameric association with central aqueous pore, along with structure of a generic IgG antibody shown on the same size scale. (C) Binding of two different monoclonal recombinant NMO autoantibodies to cells expressing M1-AQP4 or M23-AQP4.
Parts of this figure were adapted from work originally published in the Journal of Biological Chemistry (Crane et al., 2011a) © the American Society for Biochemistry and Molecular Biology.
In the central nervous system, AQP4 is by far the most abundant water channel. AQP4 is concentrated in astrocyte membranes facing blood-brain and brain-CSF interfaces and in the basolateral membrane of ependymal cells (Nielsen et al., 1997; Rash et al., 1998). AQP4 is also expressed in astrocyte-like ‘supportive cells’ in sensory organs such as retinal Müller cells (Li et al., 2002). AQP4 facilitates movement of water between the blood and brain and the brain and CSF compartments. Knockout mice lacking AQP4 show reduced cytotoxic (cell swelling) brain edema following water intoxication or ischemic stroke (Manley et al., 2000), increased vasogenic (leaky vessel) brain edema in brain tumor (Papadopoulos et al., 2004) or abscess (Bloch et al., 2005), and increased ventricular enlargement in obstructive hydrocephalus (Bloch et al., 2006). Phenotype analysis of knockout mice has also implicated AQP4 involvement in neuroexcitation (Binder et al., 2006; Li et al., 2002; Li and Verkman, 2001; Lu et al., 2008), astrocyte migration (Auguste et al., 2007; Saadoun et al., 2005) and neuroinflammation (Li et al., 2011). Impaired AQP4 water permeability in astrocytes is likely responsible for each of these phenotypes.
3. Cellular consequences of AQP4-IgG binding to AQP4
When bound to their cellular target, antibodies can cause: (i) altered target function; (ii) target internalization, reducing cell surface expression; (iii) complement activation, causing cell death (complement-dependent cytotoxicity, CDC); and/or (iv) activation of effector cells, including natural-killer cells (NK-cells), causing cell death (antibody-dependent cellular cytotoxicity, ADCC). Available evidence implicates (iii) as the primary mechanism involved in NMO pathogenesis, though (iv) may contribute as well. Though conflicting data have been reported, mechanisms (i) and (ii) are probably not important in NMO pathogenesis.
3.1. AQP4-IgG binds to extracellular epitopes on AQP4
AQP4-IgG is substantially larger in size than AQP4 (Fig. 1B). Ratio-imaging fluorescence microscopy measurements indicate that AQP4-IgG generally binds with higher affinity to M23-AQP4 than to M1-AQP4 (Fig. 1C), though some antibodies bind with similar affinity to both isoforms (Crane et al., 2011a; Nicchia et al., 2009). Because M23-AQP4 forms OAPs and M1-AQP4 does not, this suggests a preference for AQP4-IgG binding to OAPs. Studies with M23-AQP4 mutants that do not form OAPs confirmed preferential binding of AQP4-IgG to OAPs (Crane et al., 2011a). Measurements were done using NMO patient serum, as well as monoclonal recombinant AQP4-IgGs derived from clonally expanded plasma cells from cerebrospinal fluid of NMO patients (Bennett et al., 2009). The dissociation constant of the tightest binding monoclonal AQP4-IgG was 44 nM. Mutagenesis studies suggest that AQP4-IgGs bind to various three-dimensional epitopes on the extracellular surface of AQP4 (Pisani et al., 2011; Yu et al., 2011).
3.2. AQP4-IgG binding to AQP4 causes CDC and ADCC
It has been shown, using different cellular systems and cytotoxicity read-outs, that AQP4-IgG can cause CDC when complement is present, and ADCC when effector cells, such as NK-cells, are present (Hinson et al., 2007, 2009; Kalluri et al., 2010; Vincent et al., 2008). The ability of AQP4-bound AQP4-IgG to cause CDC and ADCC is not unexpected, as the IgG Fc region binds complement protein C1q and effector cell receptor FcR. AQP4-IgG in NMO is predominantly of the IgG1 subtype (Isobe et al., in press), which strongly activates complement and binds all classes of Fc receptors involved in ADCC (Capel et al., 1994). An interesting recent finding was that efficient CDC (but not ADCC) requires AQP4 assembly in OAPs (Phuan et al., 2012). Fig. 2A shows AQP4 immunofluorescence and immunoblot analysis of cells expressing M1-AQP4 or M23-AQP4. M1-AQP4, which does not form OAPs, shows a smooth pattern of cellular fluorescence and a single (tetramer) band by native gel electrophoresis; M23-AQP4, which forms OAPs, shows a punctate pattern of fluorescence and multiple higher-order bands. CDC, as measured by LDH release (Fig. 2B, left) and live/dead cell staining (Fig. 2B, right) assays, was substantial for M23-AQP4-expressing cells, but minimal or absent for M1-AQP4-expressing cells. Measurements of complement protein binding suggested a mechanism for the greatly enhanced CDC involving C1q binding to AQP4-IgG when clustered on AQP4 OAPs (Fig. 2C). C1q is a large, multivalent protein whose binding affinity to IgG is greatly enhanced with increased binding valency. OAP formation by AQP4 thus enhances CDC at two levels: AQP4-IgG binding affinity to AQP4, and C1q binding to clustered AQP4-IgG.
Fig. 2.

Complement-dependent cytotoxicity requires AQP4 assembly in orthogonal arrays. (A) (left) Confocal and TIRFM of M1- and M23-AQP4 transfected CHO cells immunostained for AQP4 using a C-terminus anti-AQP4 antibody. (right) SDS-PAGE (top) and BN-PAGE (bottom) of cell homogenates. (B) Cells expressing M1-AQP4 are resistant to CDC caused by AQP4-IgG in human NMO sera. (left) Cytotoxicity plotted as a function of total IgG concentration (from NMO patient serum) in the presence of human complement. Cells were incubated for 1 h prior to LDH release assay. (right) Fluorescence micrographs showing live/dead (green/red) cell staining. (C) Multivalent binding of C1q to Fc regions of clustered AQP4-IgG bound to OAP-assembled AQP4. Side-view (left) and en-face view (right) shown.
Parts of this figure were adapted from work originally published in the Journal of Biological Chemistry (Phuan et al., 2012).
3.3. AQP4-IgG binding to AQP4 does not inhibit AQP4 water permeability
Because AQP4-IgG is large compared to AQP4, it is sterically not possible to accommodate more than one AQP4-IgG per AQP4 tetramer, which contains four monomers and thus four separate water pores. Significant inhibition of AQP4 water permeability by AQP4-IgG is thus theoretically unlikely. Further, despite considerable search, small-molecule AQP4 inhibitors have not been identified. Two studies of osmotic volume changes in cultured cells have concluded that AQP4-IgG does not inhibit AQP4 water permeability (Melamud et al., 2012; Nicchia et al., 2009). We also found no significant inhibition of AQP4 water permeability by high concentrations of serum from multiple NMO patients, as well as by monoclonal NMO antibodies (Rossi et al., 2012). For these studies AQP4 water permeability was measured by a stopped-flow light scattering method in plasma membrane vesicles from AQP4-expressing cells and in AQP4-reconstitutedproteoliposomes, an approach that can detect differences in water permeability of less than 5% and is insensitive to effects of internalization, unstirred layers, and ion/solute transport. One conflicting study, however, reported inhibition of AQP4 water permeability by AQP4-IgG (Hinson et al., 2012) using a time-to-lysis assay of Xenopus oocytes, which is an inaccurate surrogate of osmotic water permeability. With the exception of the Hinson et al. study, the evidence supports the conclusion that AQP4-IgG does not inhibit AQP4 water permeability.
3.4. AQP4-IgG binding to AQP4 does not cause AQP4 internalization in vivo
An initial study demonstrated that AQP4-IgG addition to cells stably transfected with a GFP-AQP4 chimera caused rapid internalization and degradation of AQP4 (Hinson et al., 2007). Cellular internalization of AQP4 and AQP4-IgG, if it occurs in the CNS in vivo, would have important implications for NMO pathogenesis and therapy, as it would protect astrocytes from CDC and ADCC. Cellular internalization of AQP4 in response to AQP4-IgG has also been shown in cultures of rat and human primary astrocyte cultures (Hinson et al., 2008; Melamud et al., 2012; Vincent et al., 2008). Using a dark quencher to resolve extracellular vs. intracellular localization of a fluorescent AQP4-IgG, we found rapid and selective internalization of AQP4-IgG and AQP4 in transfected cell cultures, in agreement with prior findings; however, there was little or no internalization of AQP4-IgG or AQP4 in primary cultures of mouse astrocytes (Ratelade et al., 2011a). In vivo, after injection of a fluorescent AQP4-IgG in mouse brain, AQP4-IgG was localized to perivascular astrocyte end-feet as long as 24 h after injection, indicating minimal or no AQP4-IgG internalization (Ratelade et al., 2011a). We conclude that AQP4 internalization, which occurs in some cell culture models, is probably not relevant to NMO disease in vivo.
4. Organ culture and rodent models of NMO
Cell culture studies show that AQP4-IgG can produce CDC when complement is present and ADCC when effector cells are present. An animal model of NMO should recapitulate the key pathological features of human NMO lesions, including loss of AQP4 and GFAP immunoreactivity, demyelination, perivascular deposition of activated complement, and inflammatory cell infiltration. The ideal model, which so far has not been developed despite considerable effort, should manifest an AQP4-IgG autoimmune response with spontaneous development of NMO pathology in spinal cord and optic nerve. Short of this goal, our lab has developed ex vivo and in vivo mouse models of NMO that have been useful in studying NMO pathogenesis and testing new therapies.
4.1. Spinal cord and optic nerve culture models
As an ex vivo model of NMO, 300 μm-thick vibratome-cut transverse slices of mouse spinal cord were cultured on transwell porous supports (Fig. 3A) (Zhang et al., 2011). Spinal cord cellular structure, including astrocytes, microglia, neurons and myelin, were preserved in culture. After 7 days in culture, spinal cord slices were exposed to NMO inducers, such as AQP4-IgG and complement, for 2–3 days and analyzed by immunofluorescence. Slices exposed to AQP4-IgG and complement showed marked loss of GFAP, AQP4 and myelin (Fig. 3B), as well as deposition of activated complement and microglial cell activation. Lesions were not seen with AQP4-IgG or complement alone, or in spinal cord slices from AQP4 null mice exposed to AQP4-IgG and complement together. The slice culture model has been useful in examining the roles of specific cell types and soluble factors in NMO pathogenesis. For example, in slice cultures treated with submaximal AQP4-IgG and complement, lesion severity was increased with inclusion of neutrophils, eosinophils or macrophages, or the soluble factors TNFα, IL-6, IL-1β or interferon-γ (Zhang et al., 2011, in press). Interestingly, lesions without myelin loss were produced by exposure of spinal cord slides to AQP4-IgG and NK-cells in the absence of complement. Further studies using an in vivo mouse model described below implicated the involvement of neutrophils in early NMO lesions and provided evidence for the potential utility of the neutrophil elastase inhibitor Sivelestat for NMO therapy (Saadoun et al., 2012). The spinal cord slice model was also used to demonstrate efficacy of monoclonal antibody (Tradtrantip et al., 2012b) and small molecule (Tradtrantip et al., 2012a) blockers, as discussed further below. A similar ex vivo model of NMO optic neuritis was accomplished by optic nerve culture for 1 day (Fig. 3C), in which NMO lesions were produced by incubation of optic nerve cultures with AQP4-IgG and complement (Zhang et al., 2011).
Fig. 3. Ex vivo.

organ culture models of NMO. (A) Schematic showing spinal cord slices cultured on a semi-porous membrane at an air-medium interface. After 7 days in culture, spinal cord slices were incubated with human complement (HC) and/or AQP4-IgG for 2–3 days. (B) Immunofluorescence for GFAP (green), AQP4 (red) and myelin basic protein (MBP) (red) in wildtype (AQP4+/+)and AQP4 knockout (AQP4−/−) mice.‘Control’ indicates no added AQP4-IgG or HC. (C) Schematic of optic nerve culture model showing 24 h incubation of freshly isolated optic nerves.
Adapted from Zhang et al., (2011).
4.2. Rodent models
Rodent models have provided important data supporting the conclusion that AQP4-IgG is pathogenic in NMO. Initial studies showed that peripheral administration of AQP4-IgG exacerbates CNS lesions in rats with pre-existing experimental autoimmune encephalomyelitis (Bennett et al., 2009; Bradl et al., 2009; Kinoshita et al., 2009) and in rats pre-treated with complete Freund’s adjuvant (Kinoshita et al., 2010). In these models, AQP4-IgG caused perivascular astrocyte destruction with loss of AQP4 and GFAP, as well as perivascular deposition of IgG and activated complement. Pathology was not seen in rats administered IgG from non-NMO patients. However the background hyper-inflammatory environment and presence of myelin protein-reactive T lymphocytes in these models preclude unambiguous interpretation of data in terms of a pathogenic role of AQP4-IgG in NMO.
Direct evidence of a pathogenic role of AQP4-IgG came from intracerebral injection of AQP4-IgG and human complement in mice, which produced the major features of human NMO lesions, including loss of AQP4 and GFAP (Fig. 4), inflammatory cell infiltration, loss of myelin, and perivascular deposition of activated complement (Saadoun et al., 2010). Loss of myelin was seen as early as 12 h after administration of AQP4-IgG and human complement. In these experiments human complement was used because of the relatively inefficient mouse complement system. The inflammatory cell infiltrate in this model includes primarily neutrophils at early time and macrophages at later times. Interestingly, NMO pathology, including demyelination, was reduced in neutropenic mice and increased in neutrophilic mice, providing evidence for involvement of neutrophils in NMO pathology, particularly early in lesion generation (Saadoun et al., 2012). NMO pathology in this mouse model was not seen in key controls, including injection of complement with IgG from non-NMO humans or with AQP4-IgG depleted NMO serum, injection of AQP4-IgG in the absence of complement or with complement together with complement inhibitor, or injection of AQP4-IgG and complement in AQP4-knockout mice (Fig. 4).
Fig. 4.

Intracerebral injection of AQP4-IgG and human complement produces NMO lesions in mice. Wildtype (AQP4+/+) and AQP4 knockout (AQP4−/−) mice were killed 4 days after injection of AQP4-IgG and human complement (HC). Brain sections were immunostained for AQP4, GFAP and myelin basic protein (MBP). Yellow lines represent needle tracts. White dashed lines demarcate lesions.
To date, there is no good animal model of NMO based on peripheral administration of AQP4-IgG. We found that intravenously administered AQP4-IgG accesses and binds rapidly to AQP4 in peripheral organs (kidney, stomach, trachea and skeletal muscle), but does not measurably penetrate the CNS except in circumventricular organs, such as the area postrema, which have a leaky blood-brain barrier (Ratelade et al., 2011b). Therefore reduced accessibility of AQP4-IgG to AQP4 cannot explain why AQP4-expressing peripheral organs are spared in NMO. Similarly, passive transfer of AQP4-IgG to naïve rats did not induce NMO pathology (Bradl et al., 2009), even in circumventricular organs or when AQP4-IgG was administered to young rats with a leaky blood-brain barrier. Perhaps inefficient activation of rodent complement in these models is responsible for the absence of pathology, or perhaps structural or functional differences between rodent and human astrocytes. Human astrocytes have a substantially greater volume than their rodent counterparts and a higher ratio of glia to neurons (Oberheim et al., 2006). The greater complexity of the astrocyte network in humans may translate to increased susceptibility to NMO pathology. Development of improved animal models of NMO is a high priority.
5. Evaluation of NMO pathogenesis mechanisms
As diagrammed in Fig. 5, it is thought that NMO pathogenesis involves binding of AQP4-IgG to AQP4 on astrocyte end-feet, which activates complement, leading to formation of membrane attack complex (MAC) and primary astrocyte injury. This initial event is followed by recruitment of inflammatory cells, first granulocytes (neutrophils and eosinophils) and later macrophages, which further disrupt the blood-brain barrier. Primary astrocyte injury and initiation of an inflammatory reaction (such as neutrophil degranulation) are thought to damage oligodendrocytes and neurons secondarily. Evidence for this chronology comes from observations in human lesions in which AQP4 and GFAP can be diminished without myelin loss (Misu et al., 2007), and from animal models showing myelin loss after initial astrocyte injury (Saadoun et al., 2010). Though CDC is probably the major initiating mechanism in NMO, other pathogenic mechanisms triggered by AQP4-IgG have been proposed as discussed in this section.
Fig. 5.

NMO pathogenesis mechanism. In the normal CNS, AQP4 is expressed at astrocyte end-feet facing the blood-brain barrier formed by endothelial cells connected by tight junctions (labeled ‘1’). In NMO, by an unknown mechanism, circulating AQP4-IgG crosses the blood-brain barrier and binds AQP4 on astrocytes (2). This leads to recruitment and activation of complement and deposition of the membrane attack complex (MAC), producing astrocyte damage (3). Complement activation and cytokine secretion by astrocytes recruit inflammatory cells (eosinophils, neutrophils and macrophages), which further disrupt the blood-brain barrier, allowing more entry of AQP4-IgG (4). Degranulating inflammatory cells (5) and astrocyte damage secondarily cause oligodendrocyte injury, myelin loss and axon damage (6).
5.1. Role of ADCC
As mentioned above, AQP4-IgG together with NK-cells can cause death of AQP4-transfected cells and astrocyte cultures. Binding of NK-cells to the Fc region of AQP4-IgG leads to their degranulation and release of perforin and granzymes that cause astrocyte injury (Fig. 6A). We found that intracerebral injection of AQP4-IgG and NK-cells produces NMO-like lesions in mice with astrocyte injury, but without myelin loss (Ratelade et al., 2012). Minimal lesions were observed when AQP4-IgG was injected with NK-cells and an excess of a mutant AQP4-IgG lacking ADCC effector function, suggesting that ADCC is the pathogenic mechanism in this model. However, the relevance of ADCC to NMO pathogenesis is not clear. Though NK-cells are seen infrequently in human NMO lesions, their short life following activation precludes any conclusion about their involvement in NMO pathogenesis.
Fig. 6.

Alternative proposed NMO pathogenesis mechanisms based on AQP4-IgG binding to AQP4. (A) Antibody-dependent cell-mediated cytotoxicity. (B) Glutamate excitotoxicity. (C) Preferential M1-AQP4 internalization. (D) AQP4 water transport inhibition. See text for explanations.
Several leukocyte types besides NK-cells express Fc receptors and can mediate ADCC, including macrophages, neutrophils and eosinophils, which are abundantly found in NMO lesions. We found in spinal cord slice cultures that macrophages and neutrophils exacerbate NMO lesions caused by submaximal AQP4-IgG and complement, which probably involves an ADCC mechanism (Zhang et al., 2011). Recent work supports an important pathogenic role of eosinophils in NMO, through complement-dependent and independent cell-mediated cytotoxicity mechanisms (Zhang et al., in press). Definitive quantification of involvement of ADCC in human NMO, which will be quite challenging, is of clinical importance in determining the need for NMO therapies targeting ADCC.
5.2. Role of glutamate excitotoxicity
Hinson et al. (2008) reported decreased glutamate uptake in astrocytes exposed to human NMO serum and internalization of the glutamate transporter EAAT2 together with AQP4 in transfected HEK293 cells following exposure to NMO serum. They postulated a direct interaction of EAAT2 with AQP4. In addition, NMO lesions in human spinal cord show loss of AQP4 and EAAT2 immunostaining. The authors proposed that NMO pathogenesis involves glutamate excitotoxicity by a mechanism involving AQP4-IgG-induced internalization of glutamate transporter EAAT2 on astrocytes and consequent impairment in glutamate uptake from the extracellular space following neuroexcitation, leading to oligodendrocyte damage and myelin loss (Fig. 6B). Supporting this hypothesis, Marignier et al. (2010) reported reduced oligodendrocyte loss in mixed primary glial cultures exposed to AQP4-IgG and a competitive NMDA receptor antagonist. Based on this mechanism, ceftriaxone, which upregulates EAAT2 (Rothstein et al., 2005), was proposed as a therapy for NMO.
In re-evaluation of the glutamate excitotoxicity mechanism, we found no significant internalization of glutamate transporter EAAT2 in astrocytes after exposure to high concentrations of AQP4-IgG, nor was glutamate uptake reduced (Ratelade et al., 2011a). The loss of EAAT2 immunoreactivity in NMO lesions reported by Hinson et al. (2008) is probably related to astrocyte injury and loss, as AQP4 and GFAP are lost as well. We conclude that glutamate excitotoxicity is not involved in NMO pathogenesis and thus EAAT2 upregulation by ceftriaxone would not have therapeutic utility in NMO. Mice deficient in EAAT2 show lethal spontaneous seizures and increased susceptibility to acute cortical injury due to elevated glutamate in brain (Tanaka et al., 1997). EEAT2 internalization, if it occurred, would be predicted to produce a seizure phenotype, which is not seen clinically in NMO.
5.3. Role of differential M1-AQP4 vs. M23-AQP4 internalization
Hinson et al. (2012) recently proposed a new mechanism of AQP4-IgG pathogenesis in which M1-AQP4 is rapidly internalized by astrocytes upon AQP4-IgG exposure, whereas M23-AQP4 resists internalization. They concluded that NMO pathogenesis involves AQP4-IgG-induced internalization of M1-AQP4, resulting in increased OAP size and enhanced CDC (Fig. 6C). If correct, their mechanism has important implications regarding the initiating events in NMO pathogenesis and new treatment strategies. However, differential internalization of M1-AQP4 and M23-AQP4 following AQP4-IgG exposure is quite unlikely a priori, as M1-AQP4 and M23-AQP4 form tight heterotetramers in cell plasma membranes (Crane et al., 2009; Neely et al., 1999). Differential internalization of M1-AQP4 and M23-AQP4 conflicts with an earlier report by Hinson et al. (2008) showing full internalization of AQP4 in astrocytes, and data from our laboratory showing rapid internalization of M1-AQP4 and M23-AQP4 in transfected cells (Ratelade et al., 2011a). Additional studies of internalization mechanisms in transfected cells show that M1-AQP4 is internalized slower than M23-AQP4 when expressed separately, but equally when coexpressed (Phuan et al., 2012; Rossi et al., 2012). These findings, though interesting, are probably not relevant to NMO pathogenesis because, as mentioned above, our prior study indicated minimal internalization of AQP4-IgG in vivo (Ratelade et al., 2011a). Hinson et al. (2012) also proposed that rapid internalization of AQP4-IgG bound to M1-AQP4 protected M1-AQP4 transfected cells from CDC. However, as discussed above, we found that the reduced CDC for M1-AQP4 is not due to preferential internalization of M1-AQP4 vs. M23-AQP4, but to assembly of M23-AQP4 in OAPs and efficient, multivalent binding of C1q to clustered AQP4-IgG (Phuan et al., 2012). Also, Hinson et al. (2012) reported that a consequence of preferential M1-AQP4 internalization was increased OAP size. By super-resolution imaging, we found that AQP4-IgG did not affect OAP size in astrocyte cultures (Rossi et al., in press), though clustering could be induced by non-selective toxic insults that may account for the results in Hinson et al. (2012). We conclude that preferential internalization of M1-AQP4 does not occur and is thus not involved in NMO pathogenesis.
5.4. Role of AQP4 water transport inhibition
Hinson et al. (2012) noted that intramyelinic edema can be seen near some brain lesions in NMO. They interpreted the edema in terms of reduced AQP4 water permeability by AQP4-IgG binding and/or AQP4 internalization, disrupting water homeostasis and leading to local edema (Fig. 6D). As discussed above, however, our lab and others do not find altered AQP4 water permeability or internalization in response to AQP4-IgG binding. Further, AQP4 knockout mice do not manifest baseline abnormalities in brain water content (Manley et al., 2000; Papadopoulos et al., 2004). The intramyelinic edema observed in some NMO lesions is likely a non-specific finding that is also seen in other CNS diseases, such as Alexander Disease, where AQP4-IgG is absent (Balbi et al., 2010).
6. NMO therapy based on blocking AQP4-IgG binding to AQP4
Fig. 7A summarizes the central features of NMO pathogenesis, showing AQP4-IgG generation and binding to AQP4 on astrocytes, which initiates CDC and ADCC, inflammation and demyelination. NMO therapies, as shown, can target various steps in the scheme. Currently used therapies include general immunosuppression (e.g. steroids, azathioprine), immunomodulation (e.g. B-cell depletion by rituximab) and plasmapheresis. An ongoing open-label clinical trial is in progress of a monoclonal antibody complement inhibitor (eculizumab). Theoretically, therapies directed at reducing cell surface AQP4 expression or OAP assembly may also be beneficial.
Fig. 7.

NMO therapy based on blocking of AQP4-IgG binding to AQP4 on astrocytes. (A) Overview of NMO pathogenesis showing AQP4-IgG production by lymphocytes, binding to AQP4 on astrocytes, CDC and (potentially) ADCC, and initiation of a cascade of inflammatory events. Therapeutic approaches shown in black boxes. (B) Schematic of AQP4-IgG antibody showing heavy (VH) and light (VL) chain variable regions, light chain constant region (CL), and heavy chain constant regions (CH1-CH3). Locations of amino acid mutations introduced in the CH2 domain of AQP4-IgG to reduce CDC (K322A), ADCC (K326W/E333S) or both (L234A/L235A). (C) Aquaporumab (AQmab) prevents CDC following exposure to AQP4-IgG and complement as shown by live/dead cell assay.
Adapted with permission from Tradtrantip et al. (2012b).
Because AQP4-IgG binding to AQP4 is likely the initiating event in NMO, we introduced a strategy to block, using monoclonal antibodies or small molecules, AQP4-IgG binding to AQP4. In the monoclonal antibody strategy, a non-pathogenic, high-affinity monoclonal antibody (‘aquaporumab’) was generated from a recombinant monoclonal NMO antibody that binds tightly to AQP4 by mutation of its Fc region to eliminate CDC and ADCC effector functions (Tradtrantip et al., 2012b) (Fig. 7B). Aquaporumab prevented binding of AQP4-IgG in NMO patient sera, as well as cytotoxicity in AQP4-expressing cell cultures (Fig. 7C), spinal cord slice cultures, and mice receiving intraparenchymal AQP4-IgG and complement in vivo. Because aquaporumab targeting of AQP4 is highly selective, minimal toxicity is anticipated. Aquaporumab therapy may be useful during acute disease exacerbation to stop disease progression when the blood-brain barrier at the lesion site is disrupted. In this context aquaporumab could prevent astrocyte killing by AQP4-IgG and complement, and allow migration of reactive astrocytes into the already formed lesion, with consequent glial scarring and prevention of further inflammation. Aquaporumab might also be used as a maintenance therapy to reduce the frequency and severity of exacerbations. In an alternative approach, high-throughput screening identified several small-molecule drugs and natural products, including the antiviral agent arbidol, that bind to AQP4 and sterically prevent AQP4-IgG binding (Tradtrantip et al., 2012a). Further pre-clinical evaluation and optimization of monoclonal antibody and small-molecule blockers is in progress.
7. Remaining questions and directions
Though substantial progress has been made recently in understanding the involvement of AQP4-IgG in NMO pathogenesis and the central role of CDC, major questions remain. It is not known why NMO lesions localize mainly in spinal cord and optic nerve rather than in brain, and why peripheral, AQP4-expressing organs are unaffected. It is not known how AQP4-IgG, which has been assumed to be produced peripherally, initially enters the CNS. The identification of AQP4-IgG-producing plasma cells in the cerebrospinal fluid raises the possibility of AQP4-IgG production in the CNS, perhaps as the initiating event in NMO lesion formation. The roles of specific inflammatory cell types and cytokines in NMO pathogenesis are largely unknown and have potentially important therapeutic implications. As the involvement of neutrophils in NMO has led to consideration of a neutrophil elastase inhibitor for NMO therapy, definition of the roles of eosinophils and macrophages may lead as well to new therapeutic strategies. New therapies are needed for NMO, as disease morbidity remains high with current therapies. Blocker therapy has considerable potential as it selectively targets the disease-initiating event without causing generalized immunosuppression. Perhaps therapies could be developed in which a patient’s own pathogenic AQP4-IgG could be converted into therapeutic blocking antibodies, or in which tolerization to AQP4 prevents AQP4-IgG generation.
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- AQP
aquaporin
- AQP4
aquaporin-4
- AQP4-IgG
anti-AQP4 immunoglobulin NMO autoantibody
- BN-PAGE
blue-native polyacrylamide gel electrophoresis
- CDC
complement-dependent cytotoxicity
- CNS
central nervous system
- CSF
cerebrospinal fluid
- EAAT2
excitatory amino acid transporter 2
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescent protein
- LDH
lactate dehydrogenase
- MAC
membrane attack complex
- MBP
myelin basic protein
- NK-cell
natural-killer cell
- NMDA
N-methyl-D-aspartate
- NMO
neuromyelitis optica
- OAP
orthogonal arrays of particles
- TIRFM
total internal reflection fluorescence microscopy
References
- Antoine JC, Camdessanche JP, Absi L, Lassabliere F, Feasson L. Devic disease and thymoma with anti-central nervous system and antithymus antibodies. Neurology. 2004;62:978–80. doi: 10.1212/01.wnl.0000115168.73299.88. [DOI] [PubMed] [Google Scholar]
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, et al. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB Journal. 2007;21:108–16. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Balbi P, Salvini S, Fundaro C, Frazzitta G, Maestri R, Mosah D, et al. The clinical spectrum of late-onset Alexander disease: a systematic literature review. Journal of Neurology. 2010;257:1955–62. doi: 10.1007/s00415-010-5706-1. [DOI] [PubMed] [Google Scholar]
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Annals of Neurology. 2009;66:617–29. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–6. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Bloch O, Auguste KI, Manley GT, Verkman AS. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. Journal of Cerebral Blood Flow and Metabolism. 2006;26:1527–37. doi: 10.1038/sj.jcbfm.9600306. [DOI] [PubMed] [Google Scholar]
- Bloch O, Papadopoulos MC, Manley GT, Verkman AS. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. Journal of Neurochemistry. 2005;95:254–62. doi: 10.1111/j.1471-4159.2005.03362.x. [DOI] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Annals of Neurology. 2009;66:630–43. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Capel PJ, van de Winkel JG, van den Herik-Oudijk IE, Verbeek JS. Heterogeneity of human IgG Fc receptors. Immunomethods. 1994;4:25–34. doi: 10.1006/immu.1994.1004. [DOI] [PubMed] [Google Scholar]
- Crane JM, Bennett JL, Verkman AS. Live cell analysis of aquaporin-4 M1/M23 interactions and regulated orthogonal array assembly in glial cells. Journal of Biological Chemistry. 2009;284:35850–60. doi: 10.1074/jbc.M109.071670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. Journal of Biological Chemistry. 2011a;286:16516–24. doi: 10.1074/jbc.M111.227298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Rossi A, Gupta T, Bennett JL, Verkman AS. Orthogonal array formation by human aquaporin-4: Examination of neuromyelitis optica-associated aquaporin-4 polymorphisms. Journal of Neuroimmunology. 2011b;236:93–8. doi: 10.1016/j.jneuroim.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Van Hoek AN, Skach WR, Verkman AS. Aquaporin-4 dynamics in orthogonal arrays in live cells visualized by quantum dot single particle tracking. Molecular Biology of the Cell. 2008;19:3369–78. doi: 10.1091/mbc.E08-03-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Verkman AS. Determinants of aquaporin-4 assembly in orthogonal arrays revealed by live-cell single-molecule fluorescence imaging. Journal of Cell Science. 2009a;122:813–21. doi: 10.1242/jcs.042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Verkman AS. Reversible, temperature-dependent supramolecular assembly of aquaporin-4 orthogonal arrays in live cell membranes. Biophysical Journal. 2009b;97:3010–8. doi: 10.1016/j.bpj.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducray F, Roos-Weil R, Garcia PY, Slesari J, Heinzlef O, Chatelain D, et al. Devic’s syndrome-like phenotype associated with thymoma and anti-CV2/CRMP5 antibodies. Journal of Neurology, Neurosurgery and Psychiatry. 2007;78:325–7. doi: 10.1136/jnnp.2006.097972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio R, Radaelli M, Furlan R. Neuromyelitis optica: concepts in evolution. Journal of Neuroimmunology. 2011;231:100–4. doi: 10.1016/j.jneuroim.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Frigeri A, Gropper MA, Turck CW, Verkman AS. Immunolocalization of the mercurial-insensitive water channel and glycerol intrinsic protein in epithelial cell plasma membranes. Proceedings of the National Academy of Sciences of the United States of America. 1995a;92:4328–31. doi: 10.1073/pnas.92.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeri A, Gropper MA, Umenishi F, Kawashima M, Brown D, Verkman AS. Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. Journal of Cell Science. 1995b;108:2993–3002. doi: 10.1242/jcs.108.9.2993. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi Y, Mitsuoka K, de Groot BL, Philippsen A, Grubmuller H, Agre P, et al. Structure and function of water channels. Current Opinion in Structural Biology. 2002;12:509–15. doi: 10.1016/s0959-440x(02)00355-x. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M, Verkman AS. Roles of aquaporin-3 in the epidermis. Journal of Investigative Dermatology. 2008;128:2145–51. doi: 10.1038/jid.2008.70. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Ma T, Skach W, Matthay MA, Verkman AS. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. Journal of Biological Chemistry. 1994;269:5497–500. [PubMed] [Google Scholar]
- Hinson SR, McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Pittock SJ. Prediction of neuromyelitis optica attack severity by quantitation of complement-mediated injury to aquaporin-4-expressing cells. Archives of Neurology. 2009;66:1164–7. doi: 10.1001/archneurol.2009.188. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–31. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, et al. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. Journal of Experimental Medicine. 2008;205:2473–81. doi: 10.1084/jem.20081241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1245–50. doi: 10.1073/pnas.1109980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA, et al. Crystal structure of human aquaporin 4 at 1.8A and its mechanism of conductance. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7437–42. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe N, Yonekawa T, Matsushita T, Kawano Y, Masaki K, Yoshimura S, et al. Quantitative assays for anti-aquaporin-4 antibody with subclass analysis in neuromyelitis optica. Multiple Sclerosis. doi: 10.1177/1352458512443917. in press. [DOI] [PubMed] [Google Scholar]
- Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nature Reviews Neuroscience. 2010;6:383–92. doi: 10.1038/nrneurol.2010.72. [DOI] [PubMed] [Google Scholar]
- Jin BJ, Rossi A, Verkman AS. Model of aquaporin-4 supramolecular assembly in orthogonal arrays based on heterotetrameric association of M1-M23 isoforms. Biophysical Journal. 2011;100:2936–45. doi: 10.1016/j.bpj.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri SR, Illes Z, Srivastava R, Cree B, Menge T, Bennett JL, et al. Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Archives of Neurology. 2010;67:1201–8. doi: 10.1001/archneurol.2010.269. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, et al. Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochemical and Biophysical Research Communications. 2009;386:623–7. doi: 10.1016/j.bbrc.2009.06.085. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, et al. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochemical and Biophysical Research Communications. 2010;394:205–10. doi: 10.1016/j.bbrc.2010.02.157. [DOI] [PubMed] [Google Scholar]
- Landis DM, Reese TS. Arrays of particles in freeze-fractured astrocytic membranes. Journal of Cell Biology. 1974;60:316–20. doi: 10.1083/jcb.60.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. Journal of Experimental Medicine. 2005;202:473–7. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Patil RV, Verkman AS. Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin-4 water channels. Investigative Ophthalmology and Visual Science. 2002;43:573–9. [PubMed] [Google Scholar]
- Li J, Verkman AS. Impaired hearing in mice lacking aquaporin-4 water channels. Journal of Biological Chemistry. 2001;276:31233–7. doi: 10.1074/jbc.M104368200. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang H, Varrin-Doyer M, Zamvil SS, Verkman AS. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB Journal. 2011;25:1556–66. doi: 10.1096/fj.10-177279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Zhang H, Zador Z, Verkman AS. Impaired olfaction in mice lacking aquaporin-4 water channels. FASEB Journal. 2008;22:3216–23. doi: 10.1096/fj.07-104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Lee MD, Smith BL, Jung JS, Agre P, Verdijk MA, et al. The human AQP4 gene: definition of the locus encoding two water channel polypeptides in brain. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10908–12. doi: 10.1073/pnas.93.20.10908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nature Medicine. 2000;6:159–63. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin-Doyer M, et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain. 2010;133:2578–91. doi: 10.1093/brain/awq177. [DOI] [PubMed] [Google Scholar]
- Matiello M, Kim HJ, Kim W, Brum DG, Barreira AA, Kingsbury DJ, et al. Familial neuromyelitis optica. Neurology. 2010;75:310–5. doi: 10.1212/WNL.0b013e3181ea9f15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matiello M, Schaefer-Klein JL, Hebrink DD, Kingsbury DJ, Atkinson EJ, Weinshenker BG. Genetic analysis of aquaporin-4 in neuromyelitis optica. Neurology. 2011;77:1149–55. doi: 10.1212/WNL.0b013e31822f045b. [DOI] [PubMed] [Google Scholar]
- Melamud L, Fernandez JM, Rivarola V, DiGiusto G, Ford P, Villa A, et al. Neuromyelitis optica immunoglobulin G present in sera from neuromyelitis optica patients affects aquaporin-4 expression and water permeability of the astrocyte plasma membrane. Journal of Neuroscience Research. 2012;90:1240–8. doi: 10.1002/jnr.22822. [DOI] [PubMed] [Google Scholar]
- Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130:1224–34. doi: 10.1093/brain/awm047. [DOI] [PubMed] [Google Scholar]
- Neely JD, Christensen BM, Nielsen S, Agre P. Heterotetrameric composition of aquaporin-4 water channels. Biochemistry. 1999;38:11156–63. doi: 10.1021/bi990941s. [DOI] [PubMed] [Google Scholar]
- Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, et al. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. 2009;57:1363–73. doi: 10.1002/glia.20855. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. Journal of Neuroscience. 1997;17:171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends in Neuroscience. 2006;29:547–53. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB Journal. 2004;18:1291–3. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 assembly in orthogonal arrays. Journal of Biological Chemistry. 2012;287:13829–39. doi: 10.1074/jbc.M112.344325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani F, Mastrototaro M, Rossi A, Nicchia GP, Tortorella C, Ruggieri M, et al. Identification of two major conformational aquaporin-4 epitopes for neuromyelitis optica autoantibody binding. Journal of Biological Chemistry. 2011;286:9216–24. doi: 10.1074/jbc.M110.123000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittock SJ, Lennon VA. Aquaporin-4 autoantibodies in a paraneoplastic context. Archives of Neurology. 2008;65:629–32. doi: 10.1001/archneur.65.5.629. [DOI] [PubMed] [Google Scholar]
- Rash JE, Staehelin LA, Ellisman MH. Rectangular arrays of particles on freeze-cleaved plasma membranes are not gap junctions. Experimental Cell Research. 1974;86:187–90. doi: 10.1016/0014-4827(74)90670-3. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11981–6. doi: 10.1073/pnas.95.20.11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Evidence against cellular internalization in vivo of NMO-IgG, aquaporin-4, and excitatory amino acid transporter 2 in neuromyelitis optica. Journal of Biological Chemistry. 2011a;286:45156–64. doi: 10.1074/jbc.M111.297275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One. 2011b;6:e27412. doi: 10.1371/journal.pone.0027412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathologica. 2012;123:861–72. doi: 10.1007/s00401-012-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek A, Praetorius J, Frokiaer J, Nielsen S, Fenton RA. A current view of the mammalian aquaglyceroporins. Annual Review of Physiology. 2008;70:301–27. doi: 10.1146/annurev.physiol.70.113006.100452. [DOI] [PubMed] [Google Scholar]
- Rossi A, Moritz TJ, Ratelade J, Verkman AS. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell membranes. Journal of Cell Science. doi: 10.1242/jcs.109603. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS. Consequences of NMO-IgG binding to aquaporin-4 in neuromyelitis optica. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1511. doi: 10.1073/pnas.1203463109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. Journal of Cell Science. 2005;118:5691–8. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intracerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–61. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Macdonald C, Bell BA, Vincent A, Verkman AS, et al. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Annals of Neurology. 2012;71:323–33. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC, et al. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB Journal. 2012a;26:2197–208. doi: 10.1096/fj.11-201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, et al. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Annals of Neurology. 2012b;71:314–22. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbavatz JM, Ma T, Gobin R, Verkman AS. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. Journal of Cell Science. 1997;110:2855–60. doi: 10.1242/jcs.110.22.2855. [DOI] [PubMed] [Google Scholar]
- Verkman AS. Aquaporins at a glance. Journal of Cell Science. 2011;124:2107–12. doi: 10.1242/jcs.079467. [DOI] [PubMed] [Google Scholar]
- Verkman AS. Aquaporins in clinical medicine. Annual Review of Medicine. 2012;63:303–16. doi: 10.1146/annurev-med-043010-193843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T, Saikali P, Cayrol R, Roth AD, Bar-Or A, Prat A, et al. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. Journal of Immunology. 2008;181:5730–7. doi: 10.4049/jimmunol.181.8.5730. [DOI] [PubMed] [Google Scholar]
- Walz T, Fujiyoshi Y, Engel A. The AQP structure and functional implications. Handbook of Experimental Pharmacology. 2009:31–56. doi: 10.1007/978-3-540-79885-9_2. [DOI] [PubMed] [Google Scholar]
- Wang H, Dai Y, Qiu W, Zhong X, Wu A, Wang Y, et al. HLA-DPB1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern Han Chinese. Journal of Neuroimmunology. 2011;233:181–4. doi: 10.1016/j.jneuroim.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. The Lancet Neurology. 2007;6:805–15. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Wolburg-Buchholz K, Fallier-Becker P, Noell S, Mack AF. Structure and functions of aquaporin-4-based orthogonal arrays of particles. International Review of Cell and Molecular Biology. 2011;287:1–41. doi: 10.1016/B978-0-12-386043-9.00001-3. [DOI] [PubMed] [Google Scholar]
- Yang B, Brown D, Verkman AS. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. Journal of Biological Chemistry. 1996;271:4577–80. [PubMed] [Google Scholar]
- Yang B, Ma T, Verkman AS. cDNA cloning, gene organization, and chromosomal localization of a human mercurial insensitive water channel. Evidence for distinct transcriptional units. Journal of Biological Chemistry. 1995;270:22907–13. doi: 10.1074/jbc.270.39.22907. [DOI] [PubMed] [Google Scholar]
- Yu X, Green M, Gilden D, Lam C, Bautista K, Bennett JL. Identification of peptide targets in neuromyelitis optica. Journal of Neuroimmunology. 2011;236:65–71. doi: 10.1016/j.jneuroim.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zephir H, Fajardy I, Outteryck O, Blanc F, Roger N, Fleury M, et al. Is neuromyelitis optica associated with human leukocyte antigen. Multiple Sclerosis. 2009;15:571–9. doi: 10.1177/1352458508102085. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Annals of Neurology. 2011;70:943–54. doi: 10.1002/ana.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ratelade J, Verkman AS. Eosinophil pathogenicity in neuromyelitis optica by ADCC and CDCC mechanisms: potential therapeutic efficacy of over-the counter eosinophil stabilizing drugs. ECTRIMS. in press. [Google Scholar]