

Abstract
Exogenous brain-derived neurotrophic factor (BDNF) can regulate behavioral sensitization and conditioned place preference (CPP) when animals are exposed to repeated cocaine administration. However, it is unclear whether BDNF signaling through the TrkB receptor can mediate these behavioral responses when animals are given a single cocaine exposure. Because TrkB knockout mice die as neonates, we engineered a transgenic mouse that expressed a dominant negative form of TrkB (dnTrkB) in a conditional and reversible manner. We assessed also activation of endogenous TrkB by quantifying levels of phosphorylated TrkB (p-TrkB) in the nucleus accumbens (NAc). We found that a single exposure to cocaine was sufficient to increase p-TrkB within the NAc 9–12 h after administration. Expression of the dnTrkB transgene not only prevented the acute cocaine-induced increase in p-TrkB, but it also prevented behavioral sensitization and CPP following a single cocaine injection. These findings demonstrate that TrkB activation is required both for behavioral sensitization and CPP to a single cocaine exposure. The fact that enhanced TrkB activation is induced within 9 h of a single injection of cocaine suggests that inhibition of TrkB signaling commencing hours after cocaine exposure may prevent at least the initial antecedents to the sensitizing and reinforcing effects of this psychostimulant.
Keywords: Transgenic mice, TrkB receptor, Brain-derived neurotrophic factor, Cocaine, Behavioral sensitization, Conditioned place preference
1. Introduction
The actions of brain-derived neurotrophic factor (BDNF) are exerted through the tropomyosin-related tyrosine kinase B (TrkB) and p75NTR receptors (Kalb, 2005; Reichardt, 2006). Most BDNF-mediated events related to neural plasticity are conveyed through the TrkB receptor, while activation of p75 is concerned primarily with cell survival or apoptosis. Activation of the TrkB receptor has been implicated in various neuropsychiatric conditions that include epileptogenesis (He et al., 2004), depression (Berton et al., 2006; Nibuya et al., 1995; Siuciak et al., 1997), and drug abuse (Butovsky et al., 2005; Lu et al., 2004; McGough et al., 2004; Graham et al., 2007, 2009).
Over the years, considerable evidence has accrued showing that cocaine can influence the levels of BDNF (Pierce and Bari, 2001; Russo et al., 2009). For instance, acute or repeated administration of cocaine increases the concentrations of BDNF protein in rat neostriatum and/or nucleus accumbens (NAc) (Berglind et al., 2007; Filip et al., 2006; Zhang et al., 2002). The elevation in the NAc appears to be region-specific since levels of BDNF are increased in the shell but not in the core (Graham et al., 2007). By comparison, levels of TrkB receptor protein are enhanced in both areas (Graham et al., 2009). Withdrawal from cocaine further potentiates the concentrations of BDNF in the NAc (Grimm et al., 2003; Pu et al., 2006). Additionally, withdrawal facilitates long-term potentiation in the ventral tegmental area and this effect can be blocked with a TrkB receptor scavenger or tyrosine kinase inhibitor (Pu et al., 2006). Infusion of BDNF into rat NAc is sufficient to augment behavioral sensitization and self-administration of cocaine (Horger et al., 1999). This enhancement in self-administration persists for at least one month after the BDNF infusions have terminated (Grimm et al., 2003; Horger et al., 1999). Similarly, overexpression of BDNF or TrkB by lentiviral infection of the rat NAc potentiates cocaine-induced behavioral sensitization and conditioned place preference (CPP), and the infections prolong CPP extinction and increase CPP reinstatement (Bahi et al., 2008). By comparison, reductions of BDNF by genetic manipulation or by siRNA procedures serve to reduce cocaine-responsiveness in the open field and CPP (Graham et al., 2009; Hall et al., 2003; Horger et al., 1999). Importantly, infusion of BDNF antiserum into the NAc attenuates cocaine self-administration and decreases relapse (Graham et al., 2007). From these reports, we have hypothesized that a single injection of cocaine will enhance TrkB activation within the NAc and that this response is required for the appearance of both behavioral sensitization and CPP. To test these hypotheses, we have engineered a transgenic mouse that expresses a dominant negative form of TrkB within subpopulations of forebrain CNS neurons in a temporally controlled manner. We have utilized a behavioral sensitization paradigm that involves a single exposure to cocaine (see Grignaschi et al., 2004; Kalivas and Alesdatter, 1993; Vanderschuren et al., 1999) followed by a challenge injection 28 days later because this procedure permits an investigation of the initial molecular and cellular mechanisms that underlie early stages of addictive behaviors that are not confounded by the additional changes that may occur with repeated cocaine exposure.
2. Methods
2.1. Generation of transgenic mice
Transgenic mice were generated that express a dominant negative mutation of the TrkB receptor (accession number X17647; National Center for Biotechnology, Bethesda, Maryland). Briefly, the 5′ portion of TrkB (bases 20–1408) was cloned by PCR from a mouse brain cDNA library (Clontech, Mountain View, CA) using a forward primer: 5′ATGATATCCGTTAGAGCCCAGTCGCTGCTTCAGC-3′; and reverse primer: 5′-GTGATCTGAGGTTGGAGACTCGAG-3′. To render TrkB kinase-inactive, GeneEditor kit (Promega, Madison, WI) was used to modify base pairs 2222–2224 from AAG (Lys) to CTA (Leu) using the primer 5′-CTGGTGGCTGTGGCTACGCTGAAGGAC-3′; thereby, creating plasmid pSP73-dnTrkB (dominant-negative TrkB). This construct was excised and cloned into pMM400 (a kind gift of Dr. Mark Mayford, Scripps Research Institute, San Diego, CA), placing dnTrkB under control of the tet-operon (tetO) promoter (Mayford et al., 1996). DNA sequencing confirmed the sequences were correct. The plasmid was digested, the insert containing the tet-O promoter and dnTrkB construct was excised, purified, and microinjected into B6SJLF1/J single-cell embryos (Duke University Transgenic Facility, Durham, NC). Mice expressing the tetracycline transactivator (tTA) gene under control of the CAMKIIα promoter and maintained on a C57BL/6 background were provided by Dr. Mark Mayford (Mayford et al., 1996). These mice were crossed to founder dnTrkB mice to generate double-transgenic (dtg) animals. Mice used in this study were age-matched 3–5 month old males with littermate controls where minimal numbers of mice were used to reach statistical significance. Standard laboratory chow and water were provided ad libitum. Animals were housed in a humidity- and temperature-controlled room under a 14:10 h light/dark cycle (lights on 0700 h). To regulate transgenic expression in vivo, 100 ng/ml doxycycline hyclate (dox; Sigma, St. Louis, MO) was provided in the drinking water at least 14 days prior to study. Experiments were conducted in accordance with an approved protocol from the Duke University Institutional Animal Care and Use Committee. All investigations were performed to minimize any potential pain or discomfort to the animals and to utilize alternatives to in vivo experiments.
2.2. Generation of dnTrkB-specific antibody
Rabbits were immunized with a keyhole limpet hemocyanin-linked dnTrkB-FLAG antigen (i.e., PVYLDILG-DYKDDDDK) in complete Freund’s adjuvant and boosted with antigen in Freund’s incomplete adjuvant 2, 6, and 8 wks later. Serum was collected 10 wks after the initial injection and total IgG was concentrated using the ImmunoPure IgG Purification Kit (Pierce, Rockford, IL).
2.3. DnTrkB immunohistochemistry
Brain sections were prepared as described (Danzer et al., 2002). To reduce cross-reactivity, the dnTrkB primary antibody (1:1,000 dilution) was pre-absorbed with 1 mg wild type (WT) brain homogenate per ml antibody, slides were washed, incubated with horseradish peroxidase (HRP) conjugated goat anti-rabbit antibody (1:1,000 dilution; Jackson Immunoresearch, West Grove, PA), and rinsed. Color was developed using the SigmaFast diaminobenzadine reaction (Sigma).
2.4. Western blot
Mice were euthanized and heads immediately immersed in liquid nitrogen to cool but not freeze the brain. The brain was removed and cortex, hippocampus, NAc, dorsal striatum, brainstem, and/or cerebellum were dissected. Tissues were homogenized in ice-cold lysis buffer [20 mM Tris (pH 7.4), 137 mM NaCl, 1% Igepal, 10% glycerol, 1 mM PMSF, and 1.5 mM sodium orthovanadate with Complete Protease Inhibitor (Roche, Indianapolis, IN)], mixed 30 min at 4°C, and cleared by centrifugation at 16,000 × g at 4°C for 10 min. Homogenates were diluted in SDS sample buffer and boiled. Homogenates (15 μg) were resolved by SDS-PAGE and electrophoretically transferred to Immobilon-P membranes (Millipore, Billerica, MA). Blots were blocked with 3% non-fat dry milk and 0.05% Tween-20 in TBS (25mM Tris base, 136 mM NaCl, 2.7 mM KCl, pH = 7.4). The top half of each blot was incubated with anti-dnTrkB pre-absorbed antibody (1:2,500 dilution). To control for loading and transfer, the bottom blot was incubated with anti-β-actin antibody (1:10,000 dilution; Sigma). Blots were washed, incubated with secondary antibody [1:2,000 dilution HRP-conjugated goat anti-rabbit IgG (Jackson Immunoresearch) for anti-dnTrkB and 1:5,000 dilution HRP-conjugated donkey anti-mouse IgG (Chemicon, Temecula, CA) for β-actin antisera]. Blots were washed, incubated with SuperSignal West Pico Chemiluminescent Substrate (Pierce), and exposed to Biomax MR film (Kodak, Rochester, NY). Band intensity was quantified using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
2.5. Immunoprecipitation
Naïve mice were treated with saline or 20 mg/kg cocaine (i.p.). At various times after injection, mice were anesthetized with pentobarbital and decapitated. Heads were placed into liquid nitrogen to cool, but not freeze, the brain. A coronal section (approximately 0.95–1.95 mm bregma) was taken. The NAc was dissected from this section, homogenized in ice-cold lysis buffer, and homogenates were pooled from 2 mice in each group. The sample was diluted to 1 mg/ml in lysis buffer. For immunoprecipitation, 3 μg anti-phosphotyrosine antibody (p-Tyr clone 4G10; Upstate, Waltham, MA) and 60 μl protein G agarose (Roche) were added, and mixed for 1 h at 4°C. Tubes were centrifuged at 16,000 × g for 2 min at 4°C, and the pellets were washed 3X in lysis buffer, re-suspended in sample buffer [63 mM Tris-HCl (pH 6.8), 75 mM dithiothreitol, 2% SDS, 7.5% glycerol, 0.005% bromophenol blue] and boiled. Western blot was performed as described above using the anti-TrkB antibody (1:1,000 dilution; Upstate) and HRP-labeled goat anti-mouse IgG1 secondary antibody (1:1,000 dilution; Jackson Immunoresearch).
2.5. PC12 cell assay
PC12 cells stably expressing TrkB (a kind gift of Dr. Moses Chao, New York University Medical Center, New York, NY) were transfected with gold particles coated with one of the following plasmid combinations: 0.25 μg GFP (pEGFP-IRESneo; a generous gift of Dr. Nancy Ip, Hong Kong University of Science and Technology, Hong Kong, China) + 1.75 μg pcDNA3/μg gold or 0.25 μg GFP + 1.75 μg dnTrkB/μg gold (see Danzer et al., 2002). Immediately after transfection, vehicle, 50 ng/mL BDNF (Sigma), or 50 ng/mL neurotrophic growth factor (NGF; Sigma) was added to medium + serum. Three days later, cells were fixed in 4% paraformaldehyde in PBS, washed with methanol, and rinsed with PBS. Cells were blocked with PBS + 3% BSA, 10% normal goat serum, and 0.5% Triton-X100. Slides were incubated with mouse anti-FLAG primary antibody (1:1,000 dilution; Sigma), followed by secondary antibody (1:1,000 dilution, Alexa Fluor 594 goat anti-mouse IgG; Invitrogen). Cells were visualized under a fluorescent microscope. A GFP-fluorescent cell was defined as “differentiated” if it extended two or more neurites, each longer than twice the width of the cell body. To ensure detection of dnTrkB expression in transfected cells, only those cells containing both GFP fluorescence and FLAG immunoreactivities were counted. For cells transfected with GFP + pcDNA3, no cells exhibited FLAG immunoreactivity and all fluorescent cells were counted.
2.7. Neurophysiological status
General neurophysiological status was assessed as described (Rodriguiz et al., 2008) by an observer blinded to genotype.
2.8. Morris water maze
This test was conducted as described (Rodriguiz and Wetsel, 2006; Rodriguiz et al., 2008). Total swim distance, latency to locate the platform, and swim velocity were calculated from Ethovision (Noldus Information Technology, Leesburg, VA, USA).
2.9. Locomotor activity
Mice were evaluated in the open field (see Pogorelov et al., 2005). Locomotor (distance traveled) and stereotypical activities (repeated beam-breaks <1 s) were monitored (AccuScan Instruments, Columbus, OH). Naïve mice were habituated to the open field for 60 min, injected (i.p.) with saline or cocaine (Sigma), and immediately returned to the open field for 120 min. Afterwards, mice were returned to their home cages where they remained for 28 days. At the end of this time, all mice were challenged with a single injection of 20 mg/kg cocaine (i.p.) as described above.
2.10. CPP
Training and testing were conducted in a mouse CPP apparatus consisting of black and white chambers (16.8 × 12.7 × 12.7 cm), separated by manually-operated doors connected to a gray compartment (7.2 × 12.7 × 12.7 cm) (Med Associates, St. Albans, VT). Testing took place over 8 days. Mice were allowed to explore the apparatus for 30 min over 4 consecutive days. On day 5, mice were randomly given saline or cocaine and confined to either the black or white chamber for 30 min. On day 6, mice that were given cocaine on day 5 received saline and vice versa; each mouse was confined to the chamber opposite that on day 5. On day 8 mice were permitted free access to the entire apparatus for 30 min. The data are presented as the time spent in the saline- and cocaine-paired chambers; the percent increase in time spent in the cocaine-paired chamber from pre-training to testing, and as preference scores represented as the time spent in the saline chamber subtracted from the time spent in the cocaine chamber divided by the total time spent in both chambers.
2.11. Statistical analyses
The data are expressed as means ± SEM. All analyses were conducted with the Statistical Package for the Social Sciences, Version 11.0 (Chicago, IL). PC12 cell differentiation was evaluated by chi-square analysis. Results from all biochemical experiments with mice represent data from individual animals from experiments. Western blot band intensities were evaluated by Student’s t-test or ANOVA. For the cocaine dose-response study in the open field, behavioral sensitization, and CPP experiments, the data were analyzed by repeated measures ANOVA (RMANOVA). These data were also analyzed by ANOVA where locomotor activity in first 20 min following injection was summed for each genotype and dose and for the CPP results expressed as the percent change in time spent in the cocaine-paired chamber from pre-training to test and as preference scores. Post-hoc analyses were by Bonferroni corrected pair-wise comparisons. Significance was set at p<0.05.
3. Results
3.1. Cocaine exposure increases p-TrkB contents in the NAc
Previous reports implicating a role for BDNF/TrkB signaling in animal models of drug abuse (Bahi et al., 2008; Graham et al., 2007,2009; Grimm et al., 2003; Hall et al., 2003; Horger et al., 1999) raise the possibility that drug exposure may increase TrkB activation within the NAc. To test this idea, we examined the phosphorylation status of TrkB, a surrogate measure of TrkB activation, in WT mice euthanized at various times after a single injection of saline or 20 mg/kg cocaine (i.p.). Levels of p-TrkB in the NAc were analyzed by immunoprecipitation with a p-Tyr antibody and Western blot for TrkB. Compared to saline, cocaine treatment induced a transient increase in p-TrkB that was maximal 9–12 h after injection, and this effect was largely reversed within 24 h (Fig. 1A). Quantitative analysis of p-TrkB immunoreactivity 9 h after injection demonstrated that cocaine significantly increased p-TrkB levels compared to saline (Fig. 1B). By contrast, Western blot for total TrkB revealed immunoreactivities for the saline and cocaine-treated groups to be similar at each time-point (Fig. 1C). Together, these results demonstrate that a single exposure to cocaine induces a selective but transient increase in p-TrkB within the NAc.
Fig. 1.
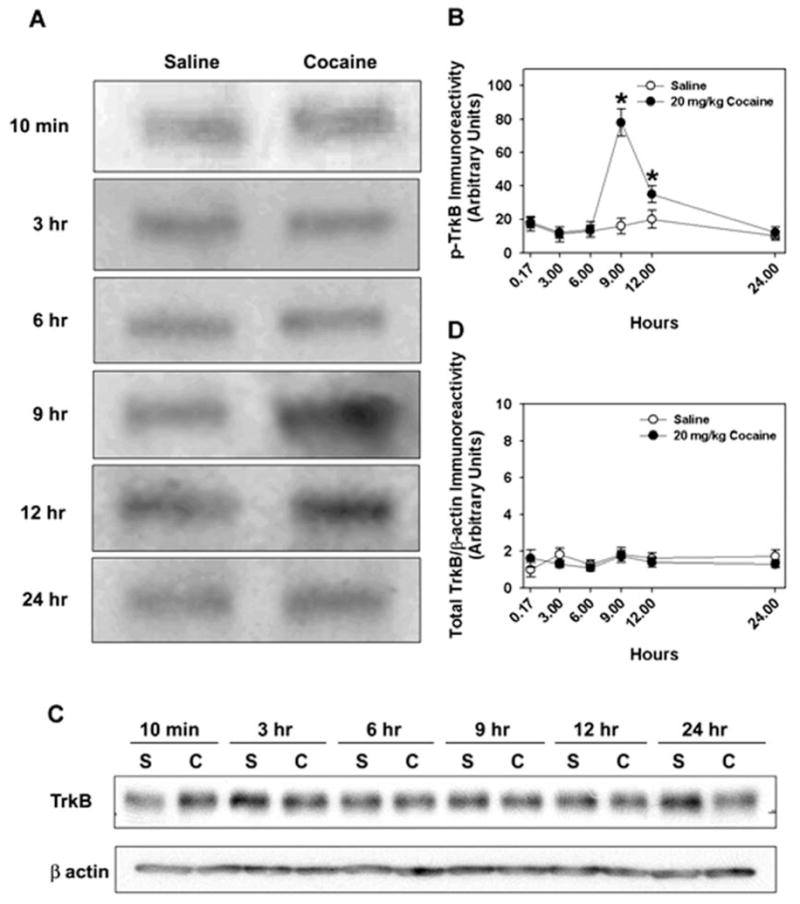
Western blot analysis of total TrkB and p-Trk-B. (A) Western blot of immunoprecipitable p-Trk-B from NAc homogenates from WT mice given saline or cocaine and euthanized 10 min to 24 h later. Cocaine exposure induces an increase in p-TrkB immunoreactivity in the NAc that is maximal 9 h after injection. (B) Cocaine significantly increased p-TrkB immunoreactivity 9 and 12 h after injection [F1,35 = 5.46, p < 0.024]. N=3 experiments. (C) Western blot analysis of total TrkB expression in NAc homogenates from mice euthanized 10 min to 24 h following saline (S) or cocaine (C) administration; β-actin was used as a loading control. (D) Densitometric analysis of total TrkB/β-actin immunoreactivity over time. N=3 experiments.
3.2. Mutant TrkB selectively inhibits TrkB signaling in PC12 cells
Our initial finding demonstrated that a single exposure to a moderate dose of cocaine is sufficient to increase p-TrkB levels in the NAc. To examine the functional significance of this event, we engineered a mutant mouse in which TrkB activation could be selectively inhibited in adulthood. To this purpose, we introduced a single aminoacid substitution (K571L) into the TrkB receptor cDNA, a mutation that renders the protein kinase inactive. To determine whether this construct (dnTrkB) would exert dominant inhibitory action on endogenous TrkB signaling, we examined its effects on TrkB-mediated responses in a pheochromocytoma (PC12) cell line. These cells stably express endogenous TrkB in addition to endogenous TrkA receptors and elaborate neurites in response to BDNF or NGF treatments (Fig. 2A). Proliferating PC12 cells were transfected with plasmids containing either GFP + pcDNA containing the empty vector or GFP + pcDNA containing the dnTrkB. Cells were treated with vehicle, NGF, or BDNF for three days. At the end of this time, expression of dnTrkB was detected by immunocytoreactivity of its FLAG epitope tag, and neurite outgrowth was examined by GFP immunocytochemistry. Vehicle alone induced no differentiation (Fig. 2A, Left; Fig. 2B, Left). As expected, both NGF and BDNF induced striking differentiation to equivalent extents in cells transfected with the empty vector (Fig. 2A, Middle and Right; Fig. 2B, Middle and Right, pcDNA3-mediated responses to NGF and BDNF). Expression of dnTrkB selectively eliminated BDNF-induced differentiation (Fig. 2B, Right, dnTrkB-mediated response), but had no effect on NGF-induced differentiation (Fig. 2B, Middle, dnTrkB-mediated response). These results demonstrate the efficacy and selectivity of dnTrkB inhibition of wild type TrkB.
Fig. 2.

DnTrkB selectively abolishes response to BDNF in PC12 cells. (A) PC12 cells stably expressing TrkB lack neurites in the absence of neurotrophin treatment (far left panel), but differentiate in response to NGF (middle panel) or BDNF (right panel). (B) PC12 cells endogenously expressing the TrkB receptor were transfected with GFP + pcDNA3 (empty vector; open bars) or GFP + dnTrkB (gray bars) plasmids and were treated with vehicle, 50 ng/mL NGF, or 50 ng/mL BDNF for 3 days. Expression of dnTrkB exerted no effect on the response to vehicle or NGF (empty vector, 30.0% differentiation; dnTrkB, 35.7% differentiation; χ2 = 0.188, n.s.), but it eliminated the BDNF response (empty vector, 32% differentiation; dnTrkB, 0% differentiation; χ2 = 5.803, p<0.05). N=2 experiments.
3.3. Transgenic mice express dnTrkB in a region-specific and doxycycline-regulated manner
To selectively inhibit TrkB activation in forebrain, we engineered mice in which expression of dnTrkB was controlled by the tet-off system (Gossen et al., 1992). In this system, expression of the dnTrkB transgene was placed under the control of the tetO promoter, which contained a minimal cytomeglovirus promoter that alone was insufficient to cause transcription. Transcription was activated when the product of the second transgene, the tetracycline transactivator (tTA), bound to the tetO promoter and was inhibited when a tetracycline was administered to the animal.
We first developed 10 different lines of dnTrkB transgenic mice. Animals from each of these lines were crossed with mice that express tTA under the control of the CAMKIIα promoter (Mayford line B mice; Mayford et al., 1996) to generate bitransgenic (double-transgenic or “dtg”) mice that express dnTrkB in a tetracycline-regulated manner. The CAMKIIα promoter restricts expression of tTA and, therefore, of dnTrkB to postmitotic neurons that express CAMKIIα in the forebrain (Burgin et al., 1990; Hanson and Schulman, 1992; Miller and Kennedy, 1986). Immunohistochemical analyses using an antibody specific to the dnTrkB protein revealed that dnTrkB expression was restricted to the forebrain in dtg mice with no detectable dnTrkB immunoreactivity in WT animals (Fig. 3A–D). Within dtg forebrain, the highest expression of dnTrkB was found throughout the striatum with lower levels in cortex and hippocampus (Fig. 3B–D). The dnTrkB immunoreactivity was absent in dtg midbrain, including the ventral tegmental area (data not shown). These results were confirmed by Western blot analyses of the dissected brain regions (Fig. 3E). The line of mice (line 7) with the highest level of expression was used in this study.
Fig. 3.

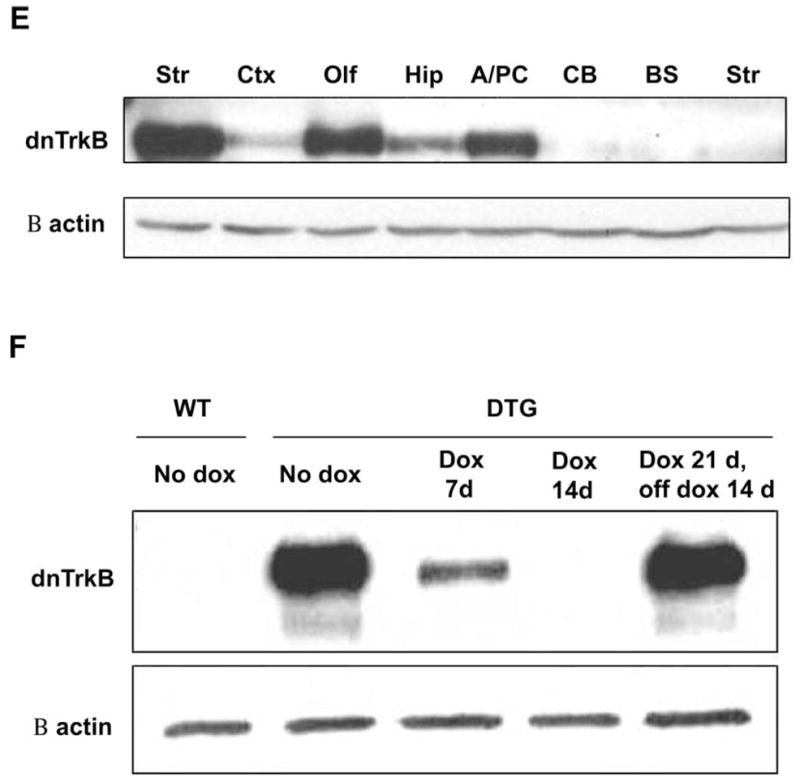
DnTrkB is expressed throughout the forebrain of dtg mice and is regulated by doxycycline (dox). (A–D) Horizontal sections of dtg (left panels) and WT (right panels) brain that were processed for anti-dnTrkB immunoreactivity demonstrate expression of dnTrkB throughout the forebrain only in dtg mice. Expression is highest in striatum (Str) and nucleus accumbens (NAc) (B), with lower levels in cortex (Ctx) (C) and hippocampus (Hip) (D). N=2 experiments. (E) Western blot of homogenates from various brain regions of dnTrkB mice shows relative levels of dnTrkB expression in Str (lane 1), Ctx (lane 2) olfactory bulb (Olf; lane 3), Hip (lane 4), and amygdala/piriform cortex (A/PC; lane 5). As expected, expression is absent in cerebellum (CB; lane 6) and brainstem (BS; lane 7) of dtg mice and is absent from Str of WT mice (lane 8). N=3 experiments. (F) Dox administration for 7 days exerts no effect on dnTrkB expression in Str from WT mice (lane 1) because the transgene is not expressed in these animals. However, in dnTrkB mice dox treatment for 7 days reduces dnTrkB expression (lane 3) and after 14 days of treatment, dnTrkB is no longer detectable (lane 4). Expression of the dnTrkB transgene is restored following removal of dox from the drinking water for 14 days (lane 5). N=3 experiments.
In the tet-off system, expression of the transgene is prevented by dox, a tetracycline derivative which inhibits the tTA-tetO interaction. To test dox-responsiveness in our animals, adult WT and dtg mice raised in the absence of dox were provided 100 ng/ml dox in their drinking water and dnTrkB expression in striatum was examined by Western blot. Expression of dnTrkB was reduced after 7 days of treatment and was completely eliminated at 14 days (Fig. 3F). When dox was withdrawn subsequently, dnTrkB expression was restored to basal levels within 14 days. Taken together, these findings show that the dnTrkB transgene is expressed selectively in the forebrain and its expression can be regulated by dox treatment.
3.4. Transgenic mice appear behaviorally normal
All mice described in this experiment were raised in the absence of dox, which permits expression of dnTrkB in the forebrain beginning between E15 and P0 as determined by expression in this line of CaMKIIα-tTA mice (Krestel et al., 2001; Yamamoto et al., 2000). In previous studies, reduced expression of the TrkB receptor beginning in embryonic development resulted in behavioral disorders and physical impairments including runting, obesity, motor neuron deficiencies, and, in some instances, increased mortality (He et al., 2004; Klein et al., 1993; Luikart et al., 2005; Xu et al., 2003). We were concerned that inhibition of TrkB signaling mediated by dnTrkB expression could produce a similar phenotype. For this purpose, neurophysiological assessments were conducted in adult WT and dtg mice raised in the absence of dox. No overt differences between WT and dtg animals were detected in general appearance, body weight, postural and righting reflexes, balance, coordination, strength, or in the elevated zero maze or forced swim tests (Table S1). Likewise, no differences between WT and mutant mice were found in the Morris water maze (Supplementary Fig S1). Together, these data demonstrate that expression of dnTrkB in a subset of principal neurons in forebrain beginning in late embryonic life and persisting into adulthood results in no detectable general behavioral impairments.
3.5. The dnTrkB transgenic mice respond normally to cocaine in the open field
Previous studies have indicated that TrkB signaling may play a role in locomotor activity after acute cocaine administration (Horger et al., 1999). As an initial screen for potential alterations in response to acute cocaine exposure, locomotor activity was examined in WT and dtg mice that had not been treated with dox. In this experiment, mice were habituated to the open field for 1 h. Separate naïve groups of animals were given saline or 10, 15, or 20 mg/kg cocaine (i.p.) and immediately returned to the open field for an additional 2 h. Importantly, pre-injection locomotor activity was not different between the genotypes, and WT and dtg mice showed increased activity in a dose-dependent manner to acute cocaine injection (Fig. 4A–D). Administration of 15 or 20 mg/kg, but not 0 or 10 mg/kg, resulted in an immediate increase in locomotion in both genotypes and this activity returned to baseline within 2 h. To more readily compare locomotor activities across genotypes and cocaine doses, activity was collapsed over the first 20 min following injection (Fig. 4B,D). Overall, increasing the acute cocaine dose resulted in similar increases in locomotor activity in both WT and dtg mice.
Fig. 4.

Locomotor activity after acute cocaine treatment. Mice were habituated to the open field for 1 h, given saline or cocaine (i.p.), and immediately returned to the open field for 2 h. Effects of saline, 10, 15, or 20 mg/kg cocaine on open field activity in WT (A,B) or dtg (C,D) mice. Both WT (A) and dtg (C) mice display dose-dependent increases in locomotor activity following administration of cocaine. RMANOVA for distance traveled for the first h before drug injection was not significant. RMANOVA over the 2 h post-injection period revealed significant main effects of time [F23,851 = 47.528, p<0.001] and a significant time by treatment interaction [F69,851 = 10.612, p<0.001]. No significant time by genotype or time by genotype by treatment interactions were discerned. Bonferroni corrected pair-wise comparisons for the time by treatment effects revealed that WT and dtg mice had enhanced locomotor responses to 10, 15, and 20 mg/kg cocaine at 5, 10, 15, and 20 min post-injection (ps<0.04) relative to the vehicle controls. Cumulative total distance traveled during 20 min before and following drug injection is presented for WT (B) and dtg (D) mice. RMANOVA for distance traveled 20 min pre- and post-injection revealed significant effects of time [F1,37 = 91.633, p<0.001] and a significant time by dose interaction [F3,37 = 35.421, p<0.001]. No effects of genotype were observed. Bonferroni corrected pair-wise comparisons confirmed that WT and dtg animals did not differ in locomotor activities prior to or following cocaine administration. Behavioral responses to both 15 and 20 mg/kg cocaine were augmented relative to animals given vehicle (ps<0.02) or 10 mg/kg cocaine (ps<0.04). At 0 mg/kg cocaine: N=12 WT and 4 dtg mice, 10 mg/kg cocaine: N=4 WT and 4 dtg mice, 15 mg/kg cocaine: N=4 WT and 5 dtg mice, 20 mg/kg cocaine: N=5 WT and 7 dtg mice were tested; +p<0.05, within genotype from vehicle.
3.6. Expression of dnTrkB prevents acute cocaine sensitization
Repeated administration of cocaine induces behavioral sensitization and this response is thought to be due to alterations in neuronal plasticity that occur over the course of multiple exposures to the psychostimulant (Robinson and Berridge, 2001). However, sensitization can also occur following a single injection of cocaine (Grignaschi et al., 2004; Kalivas and Alesdatter, 1993; Vanderschuren et al., 1999). Although TrkB activation has been implicated in diverse neuronal plasticities (Black and Lessmann, 1999; Bramham and Messaoudi, 2005; Lessmann, 1998), it is unclear whether TrkB signaling is required for behavioral sensitization to a single injection of cocaine. To test this possibility and because p-TrkB immunoreactivity was evident 9–12 h after cocaine administration, the single injection procedure for behavioral sensitization was adopted (Grignaschi et al., 2004). On the first day of the experiment, WT and dtg mice (raised in the absence of dox) were placed into the open field for 1 h to habituate them to the novel environment. Animals were given vehicle or 20 mg/kg cocaine (i.p.) and immediately returned to the open field for 2 h. When cumulative locomotor activity was examined during the 20 min period before drug administration and compared to the 20 min immediately after injection, cocaine was found to produce a robust increase in locomotion for both WT and dtg mice (Fig. 5A,B). Importantly, the increase in locomotion after cocaine administration was similar for WT and dtg mice. Four weeks later, all animals were challenged with a single injection of 20 mg/kg cocaine (Fig. 5A,B). WT mice showed marked sensitization to the locomotor-stimulating effects of the single previous administration of cocaine, whereas dtg mice gave no evidence of sensitization as the magnitude of locomotion to cocaine stimulation on day 28 was less than that at the time of the first cocaine exposure. These findings demonstrate that expression of dnTrkB prevents behavioral sensitization to a single injection of cocaine.
Fig. 5.
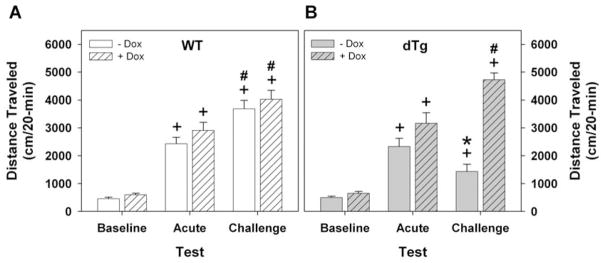
Expression of the dnTrkB transgene prevents sensitization to a single injection of cocaine. Mice were tested as described in Figure 4. The data are displayed as activity 20 min just prior to and immediately after drug injection. (A) WT mice treated without (open bars) or with dox (hatched bars) have similar low levels of locomotion at baseline. Activity is increased upon first exposure to 20 mg/kg cocaine (acute) and is further augmented when challenged with the same dose 28 days later (challenge). (B) Relative to baseline, dtg mice treated without dox (filled bars) display similar increases in locomotor activity in response to the first injection of cocaine but activity is not further enhanced at challenge 28 days later. Dox treatment of dtg mice (filled-hatched bars) rescues the behavioral response at 28 days. RMANOVA revealed significant main effects of condition (baseline, acute, challenge) [F2,84 = 121.877, p<0.001] and significant interactions between condition and genotype [F2,84 = 3.166, p<0.05], condition and treatment (i.e., with or without dox) [F2,84 = 9.717, p<0.001], and among condition, treatment, and genotype [F2,84 = 8.542, p<0.001]. Bonferroni corrected pairwise comparisons confirmed that acute cocaine exposure augmented locomotion (ps<0.001) over that of baseline activity for WT and dtg mice, regardless of dox status. WT mice with or without dox treatment demonstrated further increases in locomotion at challenge relative to activity levels at baseline (ps<0.001) as well as upon first exposure to cocaine (ps<0.005). By comparison, dtg mice without dox treatment failed to show increased locomotion at cocaine challenge relative to activity levels at first exposure to cocaine; mutants given dox had marked increases in locomotion relative to levels at baseline (p<0.001) and upon acute cocaine exposure (p<0.03), demonstrating that dox rescued behavioral sensitization in the dtg animals. Notably, WT and dtg mice maintained on dox only differed in their behavioral responses to cocaine at the time of challenge at 28 days (p<0.001). For the non-dox groups N=17 WT and 8 dtg mice, for dox-treated groups N=8 WT and 6 dtg mice; *p<0.05, WT versus dtg mice; +p<0.05, within genotype comparison of acute or challenge versus baseline activity; #p<0.05, within genotype comparison of acute exposure versus challenge.
As dnTrkB is expressed beginning in late embryonic development and persists throughout adulthood, the absence of behavioral sensitization to acute cocaine administration led us to question whether the failure of dtg mice to show sensitization was a result of inhibition of TrkB activation by the dnTrkB transgene at the time of the experiment or was a result of developmental impairment from early and lifelong dnTrkB expression. To address this question, we selectively inhibited dnTrkB expression in adulthood by providing dox in the drinking water. Accordingly, WT and dtg mice were raised in the absence of dox and were first placed on dox as adults beginning 14 days prior to the first cocaine exposure. Dox exerted no effect on WT responses to cocaine either during the acute or challenge phases of the experiment because these animals exhibited robust cocaine sensitization (Fig. 5A, Supplementary Fig. S2). Interestingly, suppression of expression of the dnTrkB transgene in adult dtg mice alleviated the blockade of behavioral sensitization in these mutants because sensitization to a single cocaine injection was readily apparent in the dox-treated dtg mice (Fig. 5B, Supplementary Fig. S2). Together with the findings described above, this experiment demonstrates that expression of the dnTrkB transgene in adulthood is required to prevent sensitization to an acute exposure to cocaine. Importantly, expression of the transgene beginning in late embryonic development and persisting through postnatal development alone is not sufficient to inhibit one-day cocaine sensitization in adulthood.
3.7. Expression of dnTrkB prevents CPP
Most drugs of abuse have some reward potential (Johanson, 1978). To determine whether dnTrkB expression interfered with the rewarding properties of cocaine in our single injection protocol, WT and dtg mice were tested in CPP. This experiment was conducted over 8 days and comprised 3 phases: pre-training (4 days) where mice were acclimated to the conditioning/test apparatus, training (2 days) where animals received single exposures to vehicle and cocaine, a 1 day vehicle/cocaine-free hiatus, and a 1 day test where CPP was assessed. The WT and dtg untreated mice (i.e., no Dox condition) spent similar amounts of time in each of the two conditioning chambers at pre-training (Fig. 6A–B, Left). An examination of the results at the time of testing revealed that the WT mice developed CPP to cocaine (Fig. 6C, Left), whereas dtg animals failed to display this behavior (Fig. 6D, Left).
Fig. 6.

Expression of the dnTrkB transgene prevents CPP to a single exposure to cocaine. (A,B) The data are presented as the time spent in the cocaine- and saline-paired chambers. During pre-training, WT (A) and dtg (B) mice without or with dox spent similar amounts of time in the chambers to be paired subsequently with cocaine or saline. An omnibus RMANOVA found no significant within subjects effects of chamber pairings, genotype, or dox treatment, or interactions between/among these effects at pre-training. (C) At testing, WT mice without (Left) or with dox (Right) demonstrate CPP by spending more time in the cocaine- compared to saline-paired chamber. (D) By comparison, dtg mice without dox (Left) spent similar amounts to time in both chambers; dox-treated dtg mice (Right) spent more time in the cocaine-paired chamber. An omnibus RMANOVA found within subjects effects of test chamber (saline or cocaine) [F1,37 = 25.335, p<0.001] and the test chamber by genotype [F1,37 = 5.996, p<0.019] and the test chamber by genotype by treatment interactions [F1,37 = 14.537, p<0.001] to be significant. Bonferroni corrected pairwise comparisons revealed that WT mice spent more time in the cocaine-compared to the saline-paired chamber, regardless of dox treatment status (ps<0.040). WT mice without or with dox were not different from each other. By contrast, untreated dtg mice spent similar amounts of time in both chambers; however, they spent more time in the cocaine-paired chamber when given dox (p<0.001). Notably, the amount of time the dox-treated dtg animals spent in this chamber was similar to that of WT controls. (E) The data are presented as the percent change in time spent in the cocaine-paired chamber from pre-training to test. Although the percent time spent in the cocaine-paired chamber was increased and was not different between WT mice without or with dox, only dtg mice given dox showed this same change. ANOVA noted main effects of treatment [F1,37 = 7.882, p<0.001] and the treatment by genotype interaction [F1,37 = 10,825 p<0.002] to be significant. Bonferroni analyses showed that regardless of dox status, WT mice had a greater than a 35% increase in time spent in the cocaine-paired chamber at test compared to that at pre-training. By contrast, dtg animals without dox at test failed to show any increase in the percent time spent in the cocaine-paired chamber compared to pre-training and this was significantly reduced below that of the WT controls (p<0.001). Dox treatment in dtg animals markedly increased this percent time relative to the non-treated mutants (p<0.001) and this was not different from the WT mice without or with dox. (E) Preference scores represented as the time spent in the saline chamber subtracted from the time spent in the cocaine chamber divided by the total time spent in both chambers. Only dtg mice without dox displayed no preference for the cocaine-paired chamber at testing. ANOVA observed significant main effects of genotype [F1,37 = 12.586, p<0.001] and treatment [F1,37 = 3.746, p<0.047], and a significant genotype by treatment interaction [F1,37 = 17.442, p<0.001]. Bonferroni comparisons showed that preference for the cocaine-paired chamber among WT mice did not differ relative to dox status. By contrast, dtg mice without dox showed reduced preference for the cocaine-paired chamber relative to WT controls (p<0.001) and the mutants given dox (p<0.001). The preference in these latter animals did not differ from those of WT controls without or with dox. For the non-dox groups N=13 WT and 12 dtg mice, for dox-treated groups N=8 WT and 8 dtg mice; *p<0.05, WT versus dtg mice within dox treatment condition; +p<0.05, no dox versus dox within genotype; ^p<0.05, compared to the cocaine treated chamber within genotype.
To determine whether expression of the dnTrkB transgene was responsible for eliminating CPP in dtg mice, additional groups of naïve WT and dtg animals were raised in the absence of dox then given dox in adulthood 14 days prior to CPP testing. Dox administration to WT and dtg mice exerted no effects on the times spent in the to be conditioned saline- and cocaine-paired chambers at pre-training (Fig. A–B, Right). At the time of testing, dox did not influence performance of WT animals, as they still demonstrated CPP to cocaine (Fig. 6C, Right). In the absence of dnTrkB expression, dtg mice now displayed CPP to cocaine (Fig. 6D, Right) to a similar extent as WT controls (Fig. 6C, Right). Since at pre-test both WT and dtg mice spent some time in the chamber that would be paired subsequently with cocaine, the proclivity to remain in this chamber at testing following the single pairing with cocaine was examined in terms of percent change in time spent in this location. Regardless of dox status, WT mice showed similar increases in the percent time spent in this chamber from pre-training to testing following the single cocaine exposure (Fig. 6E). By comparison, the dtg mice without dox spent similar percent times spent in the cocaine-paired chamber at pre-training and at testing; however, mutants given dox showed increased percent time in the cocaine-paired chamber at testing and this was comparable to that of the WT controls. These findings were further confirmed when the data were examined in terms of preference scores (Fig. 6F). Together, these results demonstrate that TrkB activation in adulthood is required for the development of CPP to a single exposure to cocaine.
3.8. The dnTrkB transgene prevents cocaine-induced TrkB activation
The absence of behavioral sensitization and CPP to acute cocaine exposure in dtg mice suggested that expression of the dnTrkB transgene prevented the cocaine-induced enhancement of TrkB activation. To address this question, WT and dtg mice were euthanized 9 h after exposure to saline or 20 mg/kg cocaine (i.p.) and levels of p-TrkB in the NAc were analyzed by immunoprecipitation and Western blot. Administration of cocaine to WT mice resulted in an approximate 3-fold increase in p-TrkB in the NAc (Fig. 7, top). By contrast, no increase in p-TrkB immunoreactivity was detected following cocaine administration to the dtg mice (Fig. 7, bottom).
Fig. 7.
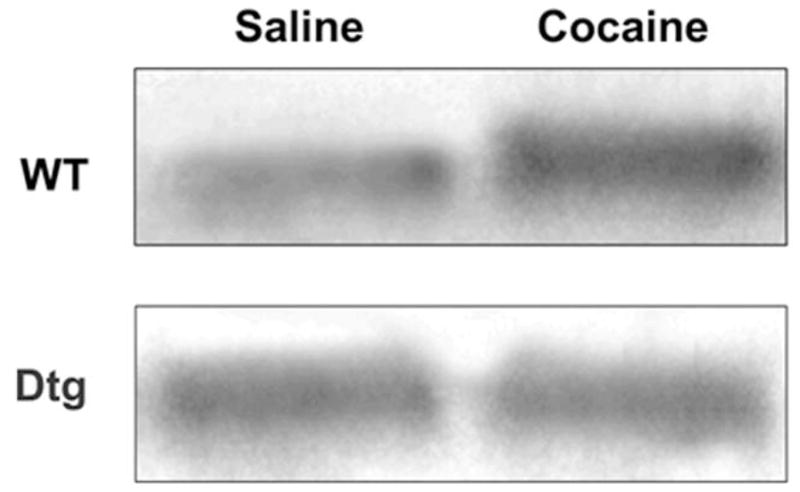
Expression of the dnTrkB transgene prevents cocaine-induced TrkB activation in the NAc. Cocaine increases p-TrkB in the NAc in WT mice (top), but not in dtg mice (bottom). N=3 experiments.
4. Discussion
Since local injection of BDNF into the NAc promotes behavioral sensitization to cocaine (Horger at al., 1999) and because BDNF infusions into this brain area enhance self-administration of cocaine for at least for one month after BDNF administration has terminated (Grimm et al., 2003; Horger et al., 1999), we have hypothesized that a single cocaine injection may be sufficient to activate endogenous TrkB within the NAc and lead to CPP and behavioral sensitization. To test this idea we have used a transgenic mouse expressing a conditional dominant negative form of TrkB (dnTrkB) and have assessed cocaine-induced biochemical and behavioral responses in this mutant. Four principal findings have emerged. First, a single exposure to cocaine is sufficient to increase TrkB activation within the NAc as evidenced by enhanced levels of p-TrkB within the NAc 9–12 h after cocaine administration. Second, selective expression of dnTrkB in adulthood prevents the locomotor sensitizing effects of acute cocaine exposure. Third, expression of the dnTrkB transgene blocks CPP to a single cocaine exposure. Fourth, expression of the dnTrkB abrogates the cocaine-induced increase in p-TrkB within the NAc. These findings demonstrate that TrkB activation is required both for behavioral sensitization and for enhanced CPP to a single exposure to cocaine.
Previous studies have demonstrated that perturbations in BDNF/TrkB signaling can alter behavioral responses to cocaine (Bahi et al., 2008; Graham et al., 2007, 2009; Grimm et al., 2003; Hall et al., 2003; Horger et al., 1999; Pu et al., 2006). These findings raise the possibility that cocaine exposure may lead to enhanced TrkB signaling which, in turn, may mediate some of the molecular and cellular events that underlie the development of addictive-like behaviors. In support of this idea, we find that a single exposure to cocaine is sufficient to enhance levels of p-TrkB -- a surrogate measure of TrkB activation -- within the NAc. This increase is first detected at 9 h and is still evident at 12 h, but it returns to baseline within 24 h after cocaine administration. Although we observed no changes in the levels of total TrkB protein in murine NAc over the 24 h period, levels of total TrkB or the downstream are reported to be increased in rat NAc within 4 h after the end of a session of cocaine self-administration (Graham et al., 2007, 2009). While the discrepancy between the experiments may be due to the species of rodent tested and/or the route of administration, both findings indicate that the BDNF-TrkB system in the NAc is highly responsive to cocaine.
The fact that a single injection of cocaine is sufficient to increase endogenous levels of p-TrkB within the NAc strengthens the possibility that TrkB activation may promote changes in neural plasticity associated with this drug. Evaluating this idea in adult mice with preexisting genetic modifications is potentially confounded by the effects of TrkB signaling during neuronal development. To address this point, we developed a dnTrkB mouse where the transgene is expressed in restricted regions of the forebrain in an inducible and repressible manner. Expression of dnTrkB continuously from the late embryonic period throughout life abolished the development of both behavioral sensitization and CPP to a single exposure to cocaine. Nevertheless, expression of dnTrkB during development alone was not sufficient to mediate these effects, because eliminating dnTrkB expression just prior to and during the period of cocaine exposure in adulthood restored the ability of the dtg mice to develop behavioral sensitization and CPP. Thus, expression of the dnTrkB transgene at the time of cocaine exposure eliminates the development of sensitization and CPP to a single injection of cocaine. In our study we wanted to evaluate the initial biochemical changes to a single administration of cocaine rather than the cumulative effects of multiple cocaine exposures on various signaling pathways (c.f., Hope et al., 1994). Nonetheless, it may be anticipated that dtg mice may eventually develop behavioral sensitization with repeated cocaine dosing since this response is delayed in BDNF heterozygous mice (Horger at al., 1999).
The question arises as to the anatomical location of cocaine-induced TrkB activation required for the development of behavioral sensitization and CPP. We found a detectable increase in p-TrkB in the NAc beginning 9 h after cocaine administration. This cocaine-induced increase in TrkB activation, together with restriction of dnTrkB expression to the forebrain and the relatively low expression of dnTrkB outside the striatum in our transgenic mice, supports the idea that TrkB activation within the NAc is critical for behavioral sensitization and CPP to a single cocaine injection. Indeed, this same brain area has also been reported to be important for supporting these same behaviors as well as self-administration when animals are given repeated cocaine injections (Bahi et al., 2008; Graham et al., 2009). Nevertheless, because in our own experiments the transgene is expressed in forebrain areas (even at relatively low levels) besides the NAc, a possibility exists that TrkB inactivation in these additional areas may also play some role in the behavioral responses of the dtg mice to acute cocaine administration.
The mechanism by which the dnTrkB transgene prevents sensitization and CPP to a single cocaine exposure may involve abrogation of TrkB activation, a presumption strengthened by direct biochemical studies in the dnTrkB mice. Under normal circumstances, activation of TrkB occurs through dimerization and subsequent phosphorylation of each monomer by its dimer partner. The phosphorylated state of both members of the homodimer is thought to be required for the conformational change leading to the binding and subsequent phosphorylation of interacting proteins including shc, phosphoinositol 3-kinase, and phospholipase C-γ(see Reichardt, 2006). The dnTrkB transgene inhibits activation of TrkB by forming inactive heterodimers (WT-TrkB:dnTrkB) or homodimers (dnTrkB:dnTrkB). Although the dnTrkB monomer can presumably serve as a substrate for its WT-TrkB partner, the absence of the ATP binding site in the dnTrkB protein eliminates its kinase function and precludes phosphorylation of its WT-TrkB partner -- the net effect being a functionally inactive heterodimer. Elimination of BDNF-induced neurite outgrowth of dnTrkB-transfected PC12 cells supports this idea. Likewise eliminating the cocaine-induced increase of p-TrkB in the NAc is consistent with this notion. The slight increase in p-TrkB levels in saline-treated dnTrkB animals compared to saline-treated WT-TrkB mice presumably reflects phosphorylation of dnTrkB members of inactive heterodimers by the WT-TrkB partners.
The cellular and molecular consequences by which enhanced TrkB activation may contribute to these cocaine-mediated events are uncertain. One possibility involves increased expression of the dopamine D3 receptor. A selective D3 receptor antagonist blocks the development and expression of cocaine CPP (Vorel et al., 2002). Interestingly, D3 and TrkB receptor mRNAs are largely co-localized in neurons within the NAc and BDNF promotes increased expression of D3 receptor mRNA within this brain area as evident by studies of BDNF heterozygous mice and by infusions of BDNF directly into the NAc (Guillin et al., 2001). A single exposure to cocaine is sufficient to induce increased D3 receptor mRNA and binding in the NAc and increased BDNF mRNA contents in frontal and medial prefrontal cortex within 2–3 h (Le Foll et al., 2005). Together these findings raise the possibility that acute cocaine administration induces increased expression of BDNF mRNA in these neocortical areas where it is translated and subsequently transported to the NAc (Altar et al., 1997) to be released to activate TrkB. Alternatively, the recent discovery of BDNF mRNA intrinsic to the NAc (Filip et al., 2006; Liu et al., 2006) raises the possibility that increased transcription and translation of BDNF locally within the NAc might underlie the cocaine-induced enhanced activation of TrkB. In this regard, phospholipase C-γ, a downstream target of TrkB activation, has been reported to be phosphorylated in response to cocaine self-administation (Graham et al., 2007). Additionally, enhanced activation of TrkB on dopamine axon terminals within the NAc may promote increased release of this monoamine (see Seroogy et al., 1994); thereby contributing to the cocaine-induced behavioral changes (Grimm et al., 2003; Guillin et al., 2001; Horger et al., 1999). Apart from these potential molecular consequences, increased TrkB signaling has been linked to dendritic changes in a variety of preparations (Danzer et al., 2002; Horch and Katz, 2002; Yacoubian and Lo, 2000) that are similar to those described on medium spiny neurons within the NAc in association with changes induced by sensitization to psychostimulants (Robinson and Kolb, 1997, 1999).
In summary, our data demonstrate that although expression of the dnTrkB transgene at the time of acute cocaine exposure exerts no effects on the initial cocaine-induced response, it abolishes the acute cocaine-induced activation of TrkB in the NAc and prevents the development of behavioral sensitization and CPP in dtg mice. Together our findings demonstrate that TrkB activation plays an essential, not merely regulatory, role in these single-injection animal models of addiction, and they focus the search for mechanisms on the structural and functional consequences of TrkB activation. Our rsults suggest that TrkB and its downstream signaling pathways may be attractive molecular targets for therapies aimed at preventing addiction.
Supplementary Material
Acquisition training in the Morris water maze for WT and dtg mice. These animals do not differ in their escape latencies across training days in the hidden platform version of the maze. Both WT (open circles) and dtg (filled circles) mice exhibit a time-dependent decrease in the latency to escape the maze. RMANOVA revealed a significant main effect of time [F7,105 = 18.780, p<0.001]. No significant effects of genotype or time by genotype interaction were discerned. N=9 WT and 8 dtg mice.
Cocaine-induced stereotypical activities WT and dtg mice in the open field. The data are displayed as stereotypical activity (repeated beam-breaks <1 sec apart) measured 20 min immediately after drug injection. (A) WT mice treated without (open bars) or with dox (hatched bars) display moderate stereotypy in response to the acute cocaine injection and the challenge 28 days later. (B) Stereotypical activity of dtg mice following cocaine administration is similar to that of WT controls. Although a RMANOVA revealed significant main effect of time (acute, challenge) [F1,45 = 21.827, p<0.001], no significant time by genotype [F1,45 = 1.288, p=n.s.], time by treatment (±dox) [F1,45 = 3.122, p=n.s.], or time by genotype by treatment interactions [F1,45 = 1.948, p=n.s.] emerged for the within subjects test. The between-subjects test for the RMANOVA identified a main effect of treatment [F1,45 = 12.021, p<0.001]. However, no genotype or treatment by genotype interactive effect was evident. Bonferroni corrected pair-wise comparisons demonstrated that dox increased stereotypical activity during the challenge test for dtg mice (p<0.02), with a marginal effect on WT mice (p<0.053). For the non-dox groups N=17 WT and 8 dtg mice, for dox-treated groups N=8 WT and 6 dtg mice; #p<0.05, non-dox versus dox treatment on the challenge test.
Results of neurophysiological screen for WT and dtg mice. WT and dtg mice do not differ with respect to appearance, weight, or performance on tests of postural and righting reflexes, balance, coordination, strength, and anxiety- and depressive-like behaviors. N=9 WT and 8 dtg mice.
Acknowledgments
This work was supported by NIH grants NS17771 and NS056217 (J.O.M.) and Unrestricted Funds (W.C.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Bahi A, Boyer F, Chandrasekar V, Dreyer JL. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology (Berl) 2008;199:169–182. doi: 10.1007/s00213-008-1164-1. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Black IB, Lessmann V. Trophic regulation of synaptic plasticity: Neurotrophin-dependent modulation of glutamatergic synaptic transmission in the mammalian CNS. J Neurobiol. 1999;41:108–118. [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Progr Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Burgin KE, Waxham MN, Rickling S, Westgate SA, Mobley WC, Kelly PT. In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci. 1990;10:1788–1798. doi: 10.1523/JNEUROSCI.10-06-01788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky E, Juknat A, Goncharov I, Elbaz J, Eilam R, Zangen A, Vogel Z. In vivo up-regulation of brain-derived neurotrophic factor in specific brain areas by chronic exposure to delta-tetrahydrocannabinol. J Neurochem. 2005;93:802–811. doi: 10.1111/j.1471-4159.2005.03074.x. [DOI] [PubMed] [Google Scholar]
- Danzer SC, Crooks KR, Lo DC, McNamara JO. Increased expression of brain-derived neurotrophic factor induces formation of basal dendrites and axonal branching in dentate granule cells in hippocampal explant cultures. J Neurosci. 2002;22:9754–9763. doi: 10.1523/JNEUROSCI.22-22-09754.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filip M, Faron-Gorecka A, Kusmider M, Golda A, Frankowska M, Dziedzicka-Wasylewska M. Alterations in BDNF and trkB mRNAs following acute or sensitizing cocaine treatments and withdrawal. Brain Res. 2006;1071:218–225. doi: 10.1016/j.brainres.2005.11.099. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Graham DL, Krishnan V, Larson EB, Graham A, Edwards S, Bachtell RK, Simmons D, Gent LM, Berton O, Bolanos CA, DiLeone RJ, Parada LF, Nestler EJ, Self DW. Tropomycin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biol Psychiatry. 2009;65:696–701. doi: 10.1016/j.biopsych.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignaschi G, Burbassi S, Zennaro E, Bendotti C, Cervo L. A single high dose of cocaine induces behavioural sensitization and modifies mRNA encoding GluR1 and GAP-43 in rats. Eur J Neurosci. 2004;20:2833–2837. doi: 10.1111/j.1460-9568.2004.03712.x. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for Incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. 2001;411:86–89. doi: 10.1038/35075076. [DOI] [PubMed] [Google Scholar]
- Hall FS, Drgonova J, Goeb M, Uhl GR. Reduced behavioral effects of cocaine in heterozygous brain-derived neurotrophic factor (BDNF) knockout mice. Neuropsychopharmacology. 2003;28:1485–1490. doi: 10.1038/sj.npp.1300192. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5:1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE. Drugs as reinforcers. In: Blackman DE, Sanger DJ, editors. Contemporary Research in Behavioral Pharmacology. Plenum Press; New York: 1978. pp. 325–390. [Google Scholar]
- Kalb R. The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends Neurosci. 2005;28:5–11. doi: 10.1016/j.tins.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Alesdatter JE. Involvement of N-methyl-D-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;267:486–495. [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel R. A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res. 2001;29:E39. doi: 10.1093/nar/29.7.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll BCA, Diaz J, Sokoloff P. A single cocaine exposure increases BDNF and D3 receptor expression: implications for drug-conditioning. Neuroreport. 2005;16:175–178. doi: 10.1097/00001756-200502080-00022. [DOI] [PubMed] [Google Scholar]
- Lessmann V. Neurotrophin-dependent modulation of glutamatergic synaptic transmission in the mammalian CNS. Gen Pharmacol. 1998;31:667–674. doi: 10.1016/s0306-3623(98)00190-6. [DOI] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Bari AA. The role of neurotrophic factors in psychostimulant-induced behavioral and neuronal plasticity. Rev Neurosci. 2001;12:95–110. doi: 10.1515/revneuro.2001.12.2.95. [DOI] [PubMed] [Google Scholar]
- Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, Wetsel WC. Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology. 2005;30:1818–1831. doi: 10.1038/sj.npp.1300724. [DOI] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo M-m. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotropin-regulated signaling pathways. Phil Trans R Soc B. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction. 2001;96:103–114. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 1997;17:8491–8497. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Rodriguiz RM, Wetsel WC. Assessments of cognitive deficits in mutant mice. In: Levin EB, Buccafusco JJ, editors. Animal Models of Cognitive Impairment. CRC Press; Boca Raton, FL: 2006. pp. 223–284. [PubMed] [Google Scholar]
- Rodriguiz RM, Gadnidze K, Ragnauth A, Dorr N, Yanagisawa M, Wetsel WC, Devi LA. Animals lacking endothelin converting enzyme-2 are deficient in learning and memory. Genes Brain Behav. 2008;7:418–426. doi: 10.1111/j.1601-183X.2007.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56:73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol. 1994;342:321–334. doi: 10.1002/cne.903420302. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF) Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Schmidt ED, De Vries TJ, Van Moorsel CA, Tilders FJ, Schoffelmeer AN. A single exposure to amphetamine is sufficient to induce long-term behavioral, neuroendocrine, and neurochemical sensitization in rats. J Neurosci. 1999;19:9579–9586. doi: 10.1523/JNEUROSCI.19-21-09579.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorel SR, Ashby CR, Jr, Paul M, Liu X, Hayes R, Hagan JJ, Middlemiss DN, Stemp G, Gardner EL. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J Neurosci. 2002;22:9595–9603. doi: 10.1523/JNEUROSCI.22-21-09595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian TA, Lo DC. Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci. 2000;3:342–349. doi: 10.1038/73911. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang L, Lou DW, Nakabeppu Y, Zhang J, Xu M. The dpamine D1 receptor is a critical mediator for cocaine-induced gene expression. J Neurochem. 2002;82:1453–1464. doi: 10.1046/j.1471-4159.2002.01089.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Acquisition training in the Morris water maze for WT and dtg mice. These animals do not differ in their escape latencies across training days in the hidden platform version of the maze. Both WT (open circles) and dtg (filled circles) mice exhibit a time-dependent decrease in the latency to escape the maze. RMANOVA revealed a significant main effect of time [F7,105 = 18.780, p<0.001]. No significant effects of genotype or time by genotype interaction were discerned. N=9 WT and 8 dtg mice.
Cocaine-induced stereotypical activities WT and dtg mice in the open field. The data are displayed as stereotypical activity (repeated beam-breaks <1 sec apart) measured 20 min immediately after drug injection. (A) WT mice treated without (open bars) or with dox (hatched bars) display moderate stereotypy in response to the acute cocaine injection and the challenge 28 days later. (B) Stereotypical activity of dtg mice following cocaine administration is similar to that of WT controls. Although a RMANOVA revealed significant main effect of time (acute, challenge) [F1,45 = 21.827, p<0.001], no significant time by genotype [F1,45 = 1.288, p=n.s.], time by treatment (±dox) [F1,45 = 3.122, p=n.s.], or time by genotype by treatment interactions [F1,45 = 1.948, p=n.s.] emerged for the within subjects test. The between-subjects test for the RMANOVA identified a main effect of treatment [F1,45 = 12.021, p<0.001]. However, no genotype or treatment by genotype interactive effect was evident. Bonferroni corrected pair-wise comparisons demonstrated that dox increased stereotypical activity during the challenge test for dtg mice (p<0.02), with a marginal effect on WT mice (p<0.053). For the non-dox groups N=17 WT and 8 dtg mice, for dox-treated groups N=8 WT and 6 dtg mice; #p<0.05, non-dox versus dox treatment on the challenge test.
Results of neurophysiological screen for WT and dtg mice. WT and dtg mice do not differ with respect to appearance, weight, or performance on tests of postural and righting reflexes, balance, coordination, strength, and anxiety- and depressive-like behaviors. N=9 WT and 8 dtg mice.