

Abstract
Ulipristal acetate (UPA), a progesterone receptor (PR) modulator, is used as an emergency contraceptive in women. Here, using a mouse model, we investigated the mechanism of action of UPA as an ovulation blocker. In mice, ovulation is induced ~12 hours following the treatment with exogenous gonadotropins, including human chorionic gonadotropin (hCG), which mimics the action of luteinizing hormone (LH). When administered within 6 hours of hCG treatment, UPA is a potent blocker of ovulation. However, UPA’s effectiveness declined significantly when it was given at 8 hours post hCG. Our study revealed that, when administered within 6 hours of hCG, UPA blocks ovulation by inhibiting PR-dependent pathways intrinsic to the ovary. At 8 hours post hCG, when the PR signaling has already occurred, UPA is unable to block ovulation efficiently. Collectively, these results indicated that UPA, when administered within a critical time window following the LH surge, blocks PR-dependent pathways in the ovary to function as an effective antiovulatory contraceptive.
Keywords: ovulation, progesterone receptor, contraception, ulipristal acetate
Introduction
Despite the availability of a wide variety of contraceptive methods, many women experience unintended pregnancy as a result of either a lack of preparedness or contraceptive failure. Emergency contraception (EC) is an effective option after unprotected intercourse to prevent an unwanted conception. It is only during a limited period, from 5 days prior to and 1 day after ovulation, that unprotected intercourse may result in a pregnancy.1–4 Blockade of ovulation during this crucial 6-day period form the basis of an effective EC treatment. Studies over the past 2 decades have led to the development of EC measures that can be used after a single unprotected act of sexual intercourse. Currently, the progestin levonorgestrel (LNG) is the most widely used oral drug for EC.5–8 If administered at least 2 days prior to the luteinizing hormone (LH) surge, LNG is able to cause either a delay or an inhibition of the LH surge, thereby preventing ovulation in women.9–11 However, LNG is unable to prevent ovulation if administered when the LH level has already started to rise.12
Ulipristal acetate (UPA), also referred to as VA/CDB-2914, is a new and promising emergency contraceptive.13–16 Ulipristal acetate is a progesterone receptor (PR) modulator that has been reported to be efficacious for EC, if it is used within 120 hours after an unprotected intercourse.17,18 The drug has been approved by Food and Drug Administration for clinical use and is available as a 30-mg tablet. Compared to mifepristone, a prototype of PR antagonist, UPA displays improved specificity and efficacy as a PR antagonist due to its lower antiglucocorticoid activity and better binding affinity to PR.19 When used for EC, UPA has been shown to be associated with a lower pregnancy rate compared to LNG.14 Administration of UPA prior to the LH rise completely blocked follicular rupture as in the case of LNG. However, unlike LNG, UPA also has the capacity to prevent ovulation when administered immediately after the onset of LH signaling in women.20,21 A recent study indicated that UPA is effective as an emergency contraceptive within a limited window relative to the time of LH surge. The mechanisms that determine this critical window of effectiveness of UPA remain unclear.
Our previous study, using a mouse model, reported the potential of UPA as a blocker of ovulation.22 In mice, ovulation can be induced in a time-dependent manner upon treatment with exogenous gonadotropins.23 In this paradigm, mice are initially treated with pregnant mare serum gonadotropin (PMSG), which mimics follicle-stimulating hormone, to promote follicular development; subsequent administration of human chorionic gonadotropin (hCG), which mimics the LH, induces ovulation. Typically, ovulation occurs 11 to 12 hours following hCG treatment.23,24 This experimentally induced ovulation offers a unique opportunity to investigate the efficacy of a potential antiovulatory drug in a preclinical animal model. Since previous studies have shown that UPA blocks LH release,20 the ovulation protocol that depends on exogenous gonadotropins is particularly useful in addressing the direct effect of this PR antagonist on the ovary. We showed that when mice are treated with UPA prior to the onset of hCG/LH signaling, this drug functions an effective blocker of ovulation.22
In the present study, we investigated the potential of UPA in blocking ovulation after the onset of hCG/LH signaling. Our studies revealed that UPA completely blocked ovulation when administered 2 hours after hCG. When it was given 4 or 6 hours after hCG, it was more than 90% effective in blocking ovulation. However, the efficacy of this drug as an ovulation blocker declined sharply when it was given 8 hours following the initiation of hCG signaling. We, therefore, identified a critical time window following the LH/hCG surge during which UPA is effective in blocking ovulation. Further mechanistic studies revealed that UPA blocked ovulation by inhibiting PR-dependent pathways that are intrinsic to the ovary.
Materials and Methods
Reagents
Ulipristal acetate was a generous gift from the Population Council (New York). Both PMSG and hCG were purchased from Sigma Chemical Co (St Louis, Missouri).
Animals
All experiments involving animals were conducted in accordance with the National Institutes of Health (NIH) standards for the use and care of animals and were approved by the Institutional Animal Care and Use Committee at the University of Illinois at Urbana–Champaign.
Ovulation Induction and Tissue Collection
To experimentally induce ovulation, the immature CD-1 mice (24-28 days old) were injected intraperitoneally (ip) with 5 IU of PMSG and 48 hours later by 5 IU of hCG. To assess the effect of UPA on ovulation in mice (n = 20), the compound was dissolved in sesame oil and administered ip at 40 mg/kg body weight (BW) at various time points after hCG injection. The dose of 40 mg/kg BW of UPA was chosen because in a previous study we showed that administration of 40 mg/kg BW dose of UPA to mice was more effective than 20 mg/kg BW in preventing ovulation.22 For histological analysis, ovaries were collected from mice (n = 6) and fixed in neutral buffered formalin. After paraffin embedding, ovarian sections were subjected to hematoxylin and eosin staining (Sigma Chemical Co). Oocytes were counted at 18 hours after administration of hCG as described previously.23 Briefly, oviducts were removed and placed in the Hank Balanced Salt Solution containing 0.1% hyaluronidase. The ampulla was opened with a fine forceps to release the cumulus–oocyte complexes and the oocytes were counted under stereo zoom microscope. The ovaries were also collected for histological analysis.
Real-Time PCR Analysis
Total RNA was isolated from ovarian tissue by using TRIZOL reagent according to manufacturer instructions (Invitrogen, Carlsbad, California) and converted to complementary DNA (cDNA) by using high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, California). The cDNA was amplified by real-time polymerase chain reaction (PCR) to quantify gene expression using gene-specific primers and SYBR using ABI prism 7500 sequence detector system (Applied Biosystems). The expression level of the gene for Rplp0, a gene encoding a ribosomal protein, was used as a loading control. For a given treatment, the mean Ct was calculated from individual Ct values obtained from 3 replicates of a sample. The normalized mean Ct was computed as ▵Ct by subtracting the mean Ct of Rplp0 from the Ct of a target gene for each treatment. ▵▵Ct was then calculated as the difference between the ▵Ct values of a control and each treatment group. The n-fold change in gene expression relative to a control was computed as 2–▵▵Ct. The PCR primers used were as follows: PR, CTAAATGAGCAGAGGATGAAGGAG (forward) and TGGGCAACTGGGCAGCAATAAC (reverse) peroxisome proliferator–activated receptorγ (PPARγ), CGTGAAGCCCATCGAGGACATC (forward) and TGGAGCACCTTGGCGAACAG (reverse); a disintegrin and metalloprotease with thrombospondin motif 1 (ADAMTS-1), TGCTCCAAGACATGCGGCTCAG (forward) and TGGTACTGGCTGGCTTCACTTCC (reverse); endothelin 2 (ET-2), CCTGTGCTACCTTCTGCCATC (forward) and CCCTCAGCAGTCCACATCTTG (reverse); ankyrin repeat domain–containing protein 1 (Ankrd1), CTGGGGAGTTCAGAAATGGA (forward) and AGGTTCTGGCTCCTTCACAA (reverse); ERBB receptor feedback inhibitor 1 (Errfi1), TGCGGTGTAACCATAGACCA(forward) and ATGTTACAGGCCTGGTGGAG (reverse); suppressor of cytokine signaling 3 (Socs3), CCTTTGACAAGCGGACTCTC (forward) and GCCAGCATAAAAACCCTTCA (reverse); calponin 3 (Cnn3), CTAGCAGGTCTGGCGAAAAC (forward) and AATCGTGGTCTGGTCAAAGG; brain-derived neurotrophic factor (Bdnf), GCGGCAGATAAAAAGACTGC (forward) and CTTATGAATCGCCAGCCAAT (reverse); runt-related transcription factor 1 (Runx1), TACCTGGGATCCATCACCTCG (forward) and GACGGCAGAGTAGGGAACTG (reverse); hypoxia-inducible factor 1α (Hif1α), TCTCCAAGCCCTCCAAGTATG (forward) and CATCAGTGGTGGCAGTTGTG (reverse); cysteine rich protein 61 (Cyr61), CAAGAAATGCAGCAAGACCA (forward) and TTCTGGTCTGCAGAGGTGTG (reverse); and Rplp0, CGACATCACAGAGCAGGC (forward) and CACCGAGGCAACAGTTGG (reverse).
DNA Microarray Analysis
Microarray analyses were performed using total RNA samples prepared from vehicle or UPA-treated mice (n = 8). Mice were treated with PMSG for 48 hours followed by hCG. Two hours after hCG treatment, mice were administered with UPA or vehicle. Ovaries were collected at 11 hours and total RNA was analyzed using Affymetrix mouse arrays (GeneChip Mouse Genome 430 2.0 Array; Affymetrix, Inc, Santa Clara, California) following the Affymetrix protocol as described previously.
Statistical Analysis
Statistical analysis was performed by Student t test or analysis of variance. Values of P < .05 were considered significant.
Results
To examine whether UPA is able to block ovulation after the initiation of hCG/LH signaling, we used an experimentally induced ovulation protocol (Figure 1A). Mice were treated with PMSG and 48 hours later with hCG. At 2, 6, and 8 hours after hCG injection, vehicle or UPA was administered ip and the number of released oocytes were counted 18 hours post hCG to evaluate the ovulatory response. As shown in Figure 1B, administration of UPA 2 hours after hCG treatment led to a complete blockade of ovulation. Upon treatment with UPA 6 hours after hCG, we observed more than 90% decrease in the number of released oocytes when compared to the vehicle-treated mice. The efficacy of UPA as an antiovulatory agent declined markedly (less than 40%) when the drug was administered 8 hours after hCG injection. These results indicated that the maximum antiovulatory effect of UPA is exerted within a critical time window of 0 to 6 hours following the hCG/LH surge. Furthermore, the antiovulatory effect of UPA in the presence of exogenously added hCG indicated that this activity is independent of hCG/LH surge and is intrinsic to the ovary.
Figure 1.

Ulipristal acetate (UPA) inhibits ovulation after the initiation of luteinizing hormone (LH) signaling. A, Schematic diagram depicts the superovulatory protocol used in our experiments. In this paradigm, mice are treated with pregnant mare serum gonadotropin (PMSG) for 48 hours, mimicking follicle-stimulating hormone (FSH), to foster follicular development; subsequent administration of human chorionic gonadotropin (hCG), mimicking the effects of the LH, induces ovulation. Typically, ovulation occurs 11 to 12 hours following hCG treatment. The total number of oocytes is counted 18 hours later to evaluate the ovulatory response. B, Vehicle or UPA was administered intraperitoneally at 2, 6, or 8 hours after hCG injection, and the number of oocytes was counted 18 hours later to evaluate the ovulatory response. The data are represented as mean ± standard error of the mean (SEM).
We next analyzed the ovarian phenotype of UPA-treated mice to gain insights into the mechanism of UPA action as an antiovulatory agent. In this experiment, mice were treated with or without UPA 2 hours post hCG and the ovaries were collected at 11 hours post hCG. As expected, hemotoxylin and eosin staining of ovarian sections from control vehicle-treated mice at 5 hours after hCG injection showed that cumulus expansion has occurred (Figure 2A, panels a and b). At 11 hours, the ovarian sections exhibited a few corpora lutea and many expanded unruptured follicles that are destined to ovulate (Figure 2A, panel c). Further analysis of ovarian sections at 18 hours showed many corpora lutea, indicating rupture of expanded follicles and successful ovulation (Figure 2A, panel d). Histological analysis of ovarian sections collected from UPA-treated mice revealed that cumulus expansion is unaffected (Figure 2B, panels a and b). At 11 hours, prior to impending ovulation, the ovarian sections showed many preovulatory follicles (Figure 2B, panel c). However, these follicles failed to rupture even at 18 hours after hCG injection, indicating impaired ovulatory response in UPA-treated mice (Figure 2B, panel d). Collectively, these results established that UPA does not affect follicular development or maturation but precisely targets certain steps in the ovulatory pathway to control follicular rupture and release of oocytes.
Figure 2.
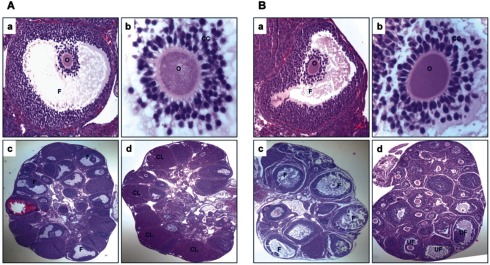
Ulipristal acetate (UPA) does not affect cumulus expansion and follicular development but inhibits follicular rupture. A, Hematoxylin and eosin (H&E) staining of ovarian sections from vehicle-treated mice at 5 hours after hCG injection shows cumulus expansion (panels a and b), at 11 hours the ovarian sections show expanded follicles (panel c), and at 18 hours the ovarian sections show many corpora lutea (panel d). B, H&E staining of ovarian sections from UPA treated mice at 5 hours after hCG injection shows cumulus expansion (panels a and b), at 11 hours the ovarian sections show expanded follicles (panel c), and at 18 hours the ovarian sections show unruptured follicles (panel d). The UPA was administered 2 hours after hCG. CC indicates cumulus cells; CL, corpus luteum; F, follicle; O, oocyte; UF, unruptured follicles.
Previous studies have shown that PR is a critical regulator of ovulation.22,25,26 The development of PR-null mice by Lydon et al revealed that mice lacking PR failed to ovulate due to a failure in the rupture of the preovulatory follicles. We hypothesized that perhaps UPA, a known ligand of PR, blocks ovulation by inhibiting PR-dependent pathways that are critical for follicular rupture. To test this hypothesis, we undertook the identification of the ovarian genes whose expression is altered in the ovary in response to UPA treatment.
In this experiment, mice were subjected to experimentally induced ovulation and 2 hours after hCG injection, vehicle or UPA was administered. Ovaries were collected at 11 hours; messenger RNAs (mRNAs) were isolated from vehicle or UPA-treated mice and subjected to microarray analysis. We applied a threshold of a 2-fold change in expression level between UPA-treated samples and untreated controls. Applying this cutoff, we identified a total of ~7000 mRNAs corresponding to known genes whose expression altered significantly in the ovary at the time of ovulation in response to UPA (GEO accession #GSE37383). In a previous study, we identified the genes that are expressed downstream of PR during ovulation by comparing the mRNA expression profiles of the ovaries of wild-type (WT) and PR-null mice at 11 hours post hCG.27 Interestingly, we noted that the vast majority of the genes that are expressed downstream of PR in the ovary during ovulation are inhibited in response to UPA treatment (Table 1).
Table 1.
Genes Downregulated in the PR-Null Ovaries and in Response to UPA Treatment at 11 Hours Post-hCG/LH.
| Biological Process | PRKOa | UPAb | Genebank# |
|---|---|---|---|
| Signal transduction | |||
| Cell communication | |||
| ECM protein-mediated signaling | |||
| ADAMTS-1 (a disintegrin and metalloprotease with thrombospondin motif 1) | −4.6 | −6.9 | D67076 |
| Tnfaip6 (tumor necrosis factor alpha induced protein 6) | −2.3 | −4.2 | NM_009398 |
| Ligand-mediated signaling | |||
| ET-2 (endothelin 2: Edn2) | −52.0 | −25.2 | NM_007902 |
| Areg (amphiregulin) | −4.3 | −22.7 | NM_009704 |
| Cell adhesion–mediated signaling | |||
| ADAMTS-4 (a disintegrin and metalloprotease with thrombospondin motif 4) | −3.7 | −3.2 | BB443585 |
| Ptpre (protein tyrosine phosphatase, receptor type, E) | −1.6 | −1.8 | U35368 |
| Cell surface receptor–mediated signaling | |||
| Ereg (epiregulin) | −2.8 | −7.3 | NM_007950 |
| Efnb2 (ephrin B2) | −2.6 | −3.2 | BB453355 |
| EGFR (epidermal growth factor receptor) | −2.6 | −2.5 | U03425 |
| Osmr (oncostatin M receptor) | −3.7 | −2.3 | AB015978 |
| Lepr (leptin receptor) | −1.3 | −2.3 | U42467 |
| Intracellular signaling cascade | |||
| Socs3 (suppressor of cytokine signaling 3) | −5.3 | −4.5 | NM_007707 |
| cGKII (cGMP-dependent protein kinase II; Prkg2) | −5.3 | −5.7 | BB823350 |
| Rras2 (related RAS viral [r-ras] oncogene homolog 2) | −2.5 | −2.8 | BE200500 |
| Sh2b2 (SH2B adaptor protein 2) | −2.6 | −7.5 | NM_018825 |
| Srgap3 (SLIT-ROBO Rho GTPase-activating protein 3) | −1.7 | −1.9 | AF481964 |
| Dusp2 (dual specificity phosphatase 2) | −29.9 | −5.2 | L11330 |
| Developmental process | |||
| Runx1 (Runt-related transcription factor 1) | −4.0 | −1.9 | BI696226 |
| Gas7 (growth arrest specific 7) | −36.8 | −1.6 | NM_008088 |
| Dystrophin | −1.4 | −2.3 | BB085328 |
| Hif1α (hypoxia inducible factor 1α) | −7.5 | −2.1 | BB269715 |
| Hif2α (EPAS1 [endothelial PAS domain protein 1]) | −3.2 | −2.2 | U81983 |
| Hhip (hedgehog-interacting protein) | −1.6 | −7.7 | BB773386 |
| Ifrd1 (interferon-related developmental regulator 1) | −1.3 | −2.0 | NM_013562 |
| Immunity and defense | |||
| Ptgs2 (prostaglandin-endoperoxide synthase 2) | −29.7 | −11.1 | M94967 |
| CXCR4 (C-X-C motif receptor 4) | −4.6 | −1.5 | D87747 |
| IL-6 (interleukin 6) | −13.9 | −21.2 | NM_031168 |
| Clcf1 (cardiotrophin-like cytokine factor 1) | −6.5 | −3.0 | NM_019952 |
| LIF (leukemia inhibitory factor) | −3.3 | −3.0 | BB235045 |
| IL-1r1 (interleukin 1 receptor, type I) | −1.4 | −2.1 | NM_008362 |
| PPARγ (peroxisome proliferator–activated receptor-γ) | −1.3 | −1.5 | NM_011146 |
| Plaur (plasminogen activator, urokinase receptor) | −2.6 | −1.8 | X62701 |
| Prdx2 (peroxiredoxin 2) | −2.3 | −2.0 | AK011963 |
| F3 (coagulation factor III) | −3.5 | −9.5 | BC024886 |
| Ier3 (immediate early response 3) | −2.8 | −3.1 | NM_133662 |
| Xbp1 (X-box binding protein 1) | −2.1 | −1.6 | NM_013842 |
| Klf6 (Kruppel-like factor 6) | −2.1 | −1.8 | BG800611 |
| Nfil3 (nuclear factor, interleukin 3, regulated) | −1.9 | −2.9 | AY061760 |
| Protein modification and metabolism | |||
| Map3k14 (mitogen-activated protein kinase | −2.5 | −1.8 | BG072756 |
| Eif4g1 (eukaryotic translation initiation factor 4, gamma 1) | −4.9 | −1.9 | BB531220 |
| Proteolysis | |||
| MMP19 (matrix metalloproteinase 19) | −7.0 | −2.1 | AV375008 |
| Ctsl (cathepsin L) | −55.7 | −1.5 | AV023994 |
| Usp43 (Ubiquitin specific peptidase 43) | −1.7 | −7.1 | BB831130 |
| Cell structure and motility | |||
| Vinculin | −11.3 | −1.6 | NM_009502 |
| Cldn1 (claudin 1) | −2.5 | −4.0 | AV227581 |
| Versican (Cspg2: chondroitin sulphate proteoglycan 2) | −2.5 | −4.3 | NM_019389 |
| Actn3 (actinin alpha 3) | −4.0 | −2.1 | NM_013456 |
| Pdlim1 (PDZ and LIM domain 1 (elfin)) | −1.5 | −2.1 | NM_016861 |
| Cgn (cingulin) | −2.3 | −1.8 | BI455486 |
| Cell proliferation and differentation | |||
| Sphk1b (sphingosine kinase) | −4.9 | −2.9 | AF068749 |
| Epgn (epithelial mitogen) | −13.0 | −7.0 | NM_053087 |
| C/EBP delta (CCAAT enhancer binding protein, delta) | −3.5 | −3.1 | BB831146 |
| Intracellular protein traffic | |||
| SNAP25 (snaptosomal-associated protein, 25 kD) | −21.1 | −58.6 | BC018249 |
| Vps26a (vacuolar protein sorting 26 homolog A [yeast]) | −1.6 | −1.7 | NM_133672 |
| Arl4a (ADP-ribosylation factor-like 4A) | −1.6 | −1.5 | AK006286 |
| Transport | |||
| Slc2a1 (solute carrier family 2 [facilitated glucose transporter], member 1; GLUT1) | −1.5 | −2.0 | M23384 |
| Slc7a8 (solute carrier family 7 [cationic amino acid transporter, y+ system], member 8) | −2.0 | −1.7 | NM_016972 |
| Slc7a11 (solute carrier family 7 [cationic amino acid transporter, y+ system], member 11) | −3.7 | −3.9 | NM_011990 |
| Apoa1 (apolipoprotein A-I) | −4.3 | −3.4 | NM_009692 |
| Clcn6 (chloride channel 6) | −6.1 | −2.1 | NM_011929 |
| Cacna2d1 (calcium channel, voltage-dependent, alpha2/delta subunit 1) | −3.2 | −2.7 | BB629558 |
| Apoptosis | |||
| Sphk1 (sphingosine kinase 1) | −4.9 | −2.9 | AF068749 |
| Axud1 (AXIN1 upregulated 1) | −2.8 | −4.3 | BG070296 |
| Cell adhesion | |||
| Loxl2 (lysyl oxidase-like protein 2) | −2.3 | −2.1 | AF117951 |
| Jam3 (junction adhesion molecule 3) | −1.7 | −2.7 | AK013156 |
| Pkp2 (plakophilin 2) | −1.5 | −2.0 | AK005020 |
| Porcn (porcupine homolog [Drosophila]) | −4.0 | −2.3 | AB036749 |
| Cell cycle | |||
| Cyr61 (cysteine rich protein 61) | −4.3 | −4.5 | BM202770 |
| Hira (histone cell cycle regulation defective homolog A) | −2.5 | −1.6 | NM_010435 |
| Junb (Jun-B oncogene) | −17.2 | −1.9 | NM_008416 |
| Dusp4 (Dual specificity phosphatase 4) | −1.6 | −3.2 | AK012530 |
| Top1 (topoisomerase [DNA] I) | −1.4 | −1.6 | BB127876 |
| Hmga1 (high mobility group AT-hook 1) | −3.0 | −2.3 | NM_016660 |
| Fos (FBJ osteosarcoma oncogene) | −2.6 | −3.1 | AV026617 |
| Unclassified | |||
| IL-11 (interleukin 11) | −8.6 | −7.5 | NM_008350 |
| Mt2 (metallothionein 2) | −5.3 | −3.5 | AA796766 |
| Scara5 (scavenger receptor class A, member 5 [putative]) | −3.5 | −2.4 | BC016096 |
| Tnfrsf12a (tumor necrosis factor receptor superfamily, member 12a) | −3.5 | −2.1 | NM_013749 |
a Wild-type and PR-null mice (PRKO) were superovulated and ovaries were collected at 11 hours post-hCG. Ovarian RNAs were analyzed by microarrays (GeneChip Mouse Genome 430 2.0 array, Affymetrix, Santa Clara, California).27 The GEO accession number for the microarray data is GSE37383. The values are individual fold changes with “–” sign, representing a decrease in gene expression in PR-null ovaries compared to that of wild type. The genes were classified according to known biological functions (Panther: http://www.pantherdb.org/).
b Mice were superovulated as described in Materials and Methods section. They were treated with vehicle or ulipristal acetate (UPA) at 2 hours post-hCG and ovaries were collected at 11 hours post-hCG. Ovarian RNAs were analyzed by microarrays (GeneChip). The GEO accession number for the microarray data is GSE37383. The values are individual fold changes with “–” sign representing a decrease in gene expression in response to UPA treatment compared to that of vehicle. The genes were classified according to known biological functions (Panther: http://www.pantherdb.org/).
To further confirm the results of our microarray analysis, we carried out real-time PCR analysis, using RNA isolated from the ovaries of vehicle and UPA-treated mice that were subjected to experimentally induced ovulation. As shown in Figure 3A, the expression of gene-encoding factors that are well known to operate downstream of PR, namely ADAMTS-1,28 ET-2,22 PPARγ,23 and Hif1α27 was significantly downregulated in the ovary in response to UPA administration. Additionally, the expression of several genes, which are downregulated in PR-null ovaries compared to WT ovaries, was also repressed upon UPA treatment. These included Ankrd1, Bdnf, Cnn3, Cyr61, Errfi1, Runx1, and Socs3 (Figure 3B). These results strongly supported our hypothesis that UPA exerts its antiovulatory effects by inhibiting PR-dependent pathways.
Figure 3.

Ulipristal acetate (UPA) inhibits the expression of progesterone receptor (PR)-regulated genes during ovulation. Mice (n = 12) were subjected to superovulation as described in Materials and Methods section. Vehicle or UPA was administered intraperitoneally 2 hours after human chorionic gonadotropin (hCG) injection. Ovaries were collected at 11 hours after hCG injection and the total RNA was isolated. A, Real-time polymerase chain reaction (PCR) was performed to analyze the expression of PR-regulated genes, ET-2, ADAMTS-1, PPARγ, and Hif1α, in response to vehicle and UPA treatments. The level of Rplp0 was used as an internal control to normalize the gene expression. The values are presented as the mean fold induction ± standard error of the mean (SEM), *P < .01, **P < .001, and ***P < .0001. B, Real-time PCR was performed to analyze the expression of Ankrd1, Bdnf, Cnn3, Cyr61, Errfi1, Runx1, and Socs3, in response to vehicle and UPA treatments. The level of Rplp0 was used as an internal control to normalize gene expression. The values are presented as the mean fold induction ± standard error of the mean (SEM), *P < .01, **P < .001, and ***P < .0001.
Our studies showed that UPA was partially effective in blocking ovulation when it was administered at 8 hours following hCG treatment. It is conceivable that by this time the PR signaling is well underway and the majority of the downstream effects of PR are already expressed in the ovary, thereby reducing the impact of the inhibitory effects of UPA. To address this possibility, we monitored the temporal expression of mRNAs corresponding to PR target genes in ovaries of mice at 0, 4, 8, and 11 hours post hCG. As shown in Figure 4A, low levels of mRNAs encoding PR, Bdnf, Socs3, and Cyr61 were detectable in the ovaries prior to hCG administration. The ovarian expression of PR mRNA was markedly elevated at 4 hours post hCG. The expression of PR mRNA declined at 8 hours and remained low at 11 hours. A similar pattern of expression was observed for Bdnf, Socs3, and Cyr61 mRNAs at 4, 8, and 11 hours post hCG (Figure 4A). Previous studies indicated that PR-dependent expression of PPARγ also peaks at 4 to 5 hours following hCG administration.23
Figure 4.

Expression profile of progesterone receptor (PR) and its target genes before and after human chorionic gonadotropin/luteinizing hormone (hCG/LH) treatment. A, Mice (n = 20) were subjected to superovulation as described in Materials and Methods sections. Ovaries were collected at 0 hours (without hCG), 4, 8, and 11 hours after hCG injection and the total RNA was isolated. Real-time PCR was performed to analyze the expression of PR, Bdnf, Socs3, and Cyr61. The level of Rplp0 was used as internal control to normalize the gene expression. The messenger RNA (mRNA) levels are expressed as mean fold induction ± standard error of the mean (SEM). B, Mice (n = 21) were subjected to superovulation as described in Materials and Methods section. Vehicle or ulipristal acetate (UPA) was administered intraperitoneally at 8 hours after hCG injection. Ovaries were collected at 11 hours after hCG injection and the total RNA was isolated. Real-time PCR was performed to analyze the expression of PR-regulated genes Bdnf, Cyr61, Errfi1, Runx1, Socs3, and PPARγ in response to vehicle and UPA treatments. The level of Rplp0 was used as an internal control to normalize the gene expression.
We reasoned that administration of UPA at 8 hours will not be able to block the induction of the majority of the PR-regulated genes, thereby achieving only partial inhibition of ovulation. To test this possibility, we treated mice with or without UPA at 8 hours post hCG and analyzed the expression of Bdnf, Socs3, Cyr61, Runx1, Errfi1, and PPARγ mRNAs. Our results indicated that UPA failed to inhibit the expression of Bdnf, Runx1, and PPARγ mRNAs, while exerting only partial inhibitory effects on Socs3, Cyr61, and Errfi1 mRNAs (Figure 4B). Taken together, these results indicated that for UPA to exert its maximal inhibitory effect on PR-dependent pathways that control ovulation, it needs to be administered within a critical time period, that is, 0 to 6 hours following the LH/hCG surge. Additionally, these studies led to the identification of certain ovarian factors as potential targets of UPA during ovulation. However, the specific roles of these factors during ovulation need further investigation.
Discussion
Since the first estrogen-progestin EC29 described more than 3 decades ago, only a handful of hormonal emergency contraceptives have been developed. Currently, the most widely used EC regimen is the progestin LNG, which is effective only when administered within 72 hours after unprotected intercourse. The LNG-EC regimen prevents pregnancy by regulating the feedback mechanism of hypothalamo–pituitary–gonadal axis, thereby preventing or delaying the LH surge.10,11 Thus, the ability of LNG to interfere with the ovulatory process is limited to its administration during a time interval preceding the onset of the LH surge. Once the ovulatory process is triggered upon the LH surge, LNG is unable to prevent ovulation.12 In the present study, using mice, we make the important observation that, unlike LNG, UPA is able to block ovulation even after the LH surge if administered within a critical time period following the surge.
Recent clinical studies indicated that UPA prevents conception in women when administered up to 5 days following unprotected intercourse and this time window of protection is significantly longer than that seen with LNG.14 It was observed that a single dose (30 mg) of UPA administered immediately before ovulation delays or inhibits ovulation in comparison to placebo-treated cycles.20 When administered before the onset of the LH surge, UPA, like LNG, delayed the LH peak and ovulation in all cycles. However, when administered after the LH level begins to rise but before it reaches the peak, LNG is ineffective as an ovulation blocker. However, UPA is an effective inhibitor of ovulation within this time period. In humans, the time interval from the rise of LH to its peak level is 30 to 36 hours. It appears that UPA needs to be administered during this time window in order to be maximally effective as an ovulation blocker. Once the LH reaches its peak, the UPA’s effect in blocking ovulation declines sharply.20 The ability of UPA to interfere with follicular rupture, therefore, appears to depend on the time when the drug is administered in relation to LH levels. Therefore, it is important to understand the cellular mechanisms that this drug controls, which in turn determines its window of effectiveness.
The LH-mediated signaling cascade that leads to the rupture of ovarian follicle is tightly controlled by PR-dependent pathways. In rodents, LH rapidly and selectively induces PR expression in mural granulosa cells of the preovulatory follicles within 4 hours.28 The development of PR-null mice firmly established the essential role of PR in ovulation.26 In these mutant mice, the follicles mature to the preovulatory stage in response to exogenous gonadotropins, but, in the absence of PR signaling, they fail to rupture and release the oocyte.26 Cumulus expansion, which is critical for oocyte maturation, is not affected in the follicles of PR-null ovaries. Our study showed that the ovaries of UPA-treated mice mimic the anovulatory phenotype of PR-null mice. While the cumulus expansion was unaltered upon UPA treatment, this drug suppressed follicular rupture, presumably by repressing the production of PR-dependent factors critical for ovulation.22 These results are consistent with the previous reports, which suggested direct ovarian antiovulatory effects of other PR antagonists.30–34
The direct inhibition of ovarian function by UPA was further confirmed when we analyzed its effects on the gene networks underlying PR function during ovulation. Our studies revealed a remarkable similarity in the ovulatory pathways that are downregulated in PR-null ovaries and in WT ovaries in response to UPA. Treatment with UPA drastically reduced the expression of several genes, such as ET-2,22 ADAMTS-1,28 PPARγ,23 and Hif1α,27 which have previously been described as targets of regulation by PR in the ovary and critical for ovulation. In addition, we noted that the expression of several other genes, such as Ankrd1, Bdnf, Cnn3, Cyr61, Errfi1, Runx1, and Socs3, is blocked upon UPA treatment. As PR expression rises following the LH surge, the expression of these downstream genes is induced in the ovary prior to ovulation. When administered early in the ovulatory period, between 0 and 6 hours following hCG/LH, UPA is able to act as a PR antagonist to effectively block the expression of these genes, thereby preventing ovulation. We believe that by 8 hours following hCG/LH, the majority of these PR-regulated genes are already expressed and have promoted synthesis of secondary targets to advance the ovulatory pathway. Administration of UPA at this time fails to achieve its inhibitory effects on PR function and prevents follicular rupture. Hence, timing of UPA administration after the onset of LH signaling is critical for effective contraceptive capacity of this compound. While it is clear that the UPA exerts its antiovulatory effect by acting directly on the ovary, we also need to consider the possibility that UPA has the potential to act at other sites including the endometrium.
In humans, PR is expressed in the granulosa cells of the dominant follicle downstream of LH signaling.35–37 Following ovulation, PR expression is maintained in the corpus luteum during the extended luteal phase in the human.35–37 A recent study examined the expression and localization of PR isoforms in human granulosa cells during the periovulatory period.38 It revealed that the expression of the A isoform of PR is induced in granulosa cells following the LH surge, presumably via cyclic adenosine monophosphate–dependent signaling triggered by the binding of LH to its receptor. Interestingly, gene knockout studies in mice revealed that PR-A is the critical PR isoform during ovulation.39 The study further indicated that maximal expression of PR mRNA in primary human granulosa cell cultures is reached within 6 hours of hCG treatment.38 These findings revealed a remarkable similarity in the PR expression pattern downstream of LH/hCG in rodent and human granulosa cells. These results are also consistent with our hypothesis that UPA suppresses ovulation in the human by blocking the induction of PR-dependent pathways downstream of LH action in the ovary as it does in the mouse. We propose that this direct inhibitory effect of UPA in the ovary allows it to function as an ovulation blocker in humans during the time interval when the LH level has begun to rise, but the essential PR signaling is yet to occur.20
In summary, this study, using mouse as a preclinical model, establishes that UPA is able to block ovulation when administered after the onset of LH signaling. This effect results from its inhibition of PR-dependent pathways that are intrinsic to the ovary and is critical for the identification of potential targets, which are mediating the ovulatory process during follicular rupture. These results have obvious clinical relevance for EC, since it is likely that UPA inhibits follicular PR-dependent pathways downstream of LH to prevent ovulation in the human. Most importantly, this study provides insights into a plausible mechanism that allows UPA to function as an antiovulatory agent when administered in the advanced follicular phase, a time when other EC agents, such as LNG, is no longer effective in inhibiting ovulation.
Acknowledgments
The PRKO mice were kindly provided by F. J. DeMayo of Baylor College of Medicine, Houston, Texas.
Footnotes
Authors' Note: The authors Shanmugasundaram Nallasamy and Jaeyeon Kim contributed equally to this work.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: a grant from the NICHD/NIH U54 HD 29990. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR16515-01 from the National Center for Research Resources, National Institutes of Health (I.C.B.).
References
- 1.Danielsson KG, Meng CX. Emergency contraception: potential role of ulipristal acetate. Int J Womens Health. 2010;2:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trussell J, RodrIguez G, Ellertson C. New estimates of the effectiveness of the Yuzpe regimen of emergency contraception. Contraception. 1998;57(6):363–369. [DOI] [PubMed] [Google Scholar]
- 3.Wilcox AJ, Day Baird D, Dunson DB, McConnaughey DR, Kesner JS, Weinberg CR. On the frequency of intercourse around ovulation: evidence for biological influences. Hum Reprod. 2004;19(7):1539–1543. [DOI] [PubMed] [Google Scholar]
- 4.Wilcox AJ, Weinberg CR, Baird DD. Timing of sexual intercourse in relation to ovulation — effects on the probability of conception, survival of the pregnancy, and sex of the baby. N Engl J Med. 1995;333(23):1517–1521. [DOI] [PubMed] [Google Scholar]
- 5.Guillebaud J. Time for emergency contraception with levonorgestrel alone. Lancet. 1998;352(9126):416–417. [DOI] [PubMed] [Google Scholar]
- 6.Ho PC, Kwan MSW. A prospective randomized comparison of levonorgestrel with the Yuzpe regimen in post-coital contraception. Hum Reprod. 1993;8(3):389–392. [DOI] [PubMed] [Google Scholar]
- 7.Marions L, Hultenby K, Lindell I, Sun X, Ståbi B, Danielsson KG. Emergency contraception with mifepristone and levonorgestrel: mechanism of action. Obstet Gynecol. 2002;100(1):65–71. [DOI] [PubMed] [Google Scholar]
- 8.Piaggio G, von Hertzen H, Grimes DA, Look PFAV. Timing of emergency contraception with levonorgestrel or the Yuzpe regimen. Lancet. 1999;353(9154):721–721. [DOI] [PubMed] [Google Scholar]
- 9.Durand M, del Carmen Cravioto M, Raymond EG, et al. On the mechanisms of action of short-term levonorgestrel administration in emergency contraception. Contraception. 2001;64(4):227–234. [DOI] [PubMed] [Google Scholar]
- 10.Hapangama D, Glasier AF, Baird DT. The effects of peri-ovulatory administration of levonorgestrel on the menstrual cycle. Contraception. 2001;63(3):123–129. [DOI] [PubMed] [Google Scholar]
- 11.Marions L, Cekan SZ, Bygdeman M, Gemzell-Danielsson K. Effect of emergency contraception with levonorgestrel or mifepristone on ovarian function. Contraception. 2004;69(5):373–377. [DOI] [PubMed] [Google Scholar]
- 12.Croxatto HB, Brache V, Pavez M, et al. Pituitary-ovarian function following the standard levonorgestrel emergency contraceptive dose or a single 0.75-mg dose given on the days preceding ovulation. Contraception. 2004;70(6):442–450. [DOI] [PubMed] [Google Scholar]
- 13.Gainer EE, Ulmann A. Pharmacologic properties of CDB(VA)-2914. Steroids. 2003;68(10-13):1005–1011. [DOI] [PubMed] [Google Scholar]
- 14.Glasier AF, Cameron ST, Fine PM, et al. Ulipristal acetate versus levonorgestrel for emergency contraception: a randomised non-inferiority trial and meta-analysis. Lancet. 2010;375(9714):555–562. [DOI] [PubMed] [Google Scholar]
- 15.Nichols MI. Ulisprisal acetate: a novel molecule and 5-day emergency contraceptive. Obstet Gynecol. 2010;116(6):1252–1253. [DOI] [PubMed] [Google Scholar]
- 16.Blithe DL, Nieman LK, Blye RP, Stratton P, Passaro M. Development of the selective progesterone receptor modulator CDB-2914 for clinical indications. Steroids. 2003;68(10-13):1013–1017. [DOI] [PubMed] [Google Scholar]
- 17.Jadav SP, Parmar DM. Ulipristal acetate, a progesterone receptor modulator for emergency contraception. J Pharmacol Pharmacother. 2012;3(2):109–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson AR, Maltz FN. Ulipristal acetate: review of the efficacy and safety of a newly approved agent for emergency contraception. Clin Ther. 2012;34(1):24–36. [DOI] [PubMed] [Google Scholar]
- 19.Hild SA, Reel JR, Hoffman LH, Blye RP. CDB-2914: anti-progestational/anti-glucocorticoid profile and post-coital anti-fertility activity in rats and rabbits. Hum Reprod. 2000;15(4):822–829. [DOI] [PubMed] [Google Scholar]
- 20.Brache V, Cochon L, Jesam C, et al. Immediate pre-ovulatory administration of 30 mg ulipristal acetate significantly delays follicular rupture. Hum Reprod. 2010;25(9):2256–2263. [DOI] [PubMed] [Google Scholar]
- 21.Stratton P, Hartog B, Hajizadeh N, et al. A single mid-follicular dose of CDB-2914, a new antiprogestin, inhibits folliculogenesis and endometrial differentiation in normally cycling women. Hum Reprod. 2000;15(5):1092–1099. [DOI] [PubMed] [Google Scholar]
- 22.Palanisamy GS, Cheon YP, Kim J, et al. A novel pathway involving progesterone receptor, endothelin-2, and endothelin receptor B controls ovulation in mice. Mol Endocrinol. 2006;20(11):2784–2795. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Sato M, Li Q, et al. Peroxisome proliferator-activated receptor γ is a target of progesterone regulation in the preovulatory follicles and controls ovulation in mice. Mol Cell Biol. 2008;28(5):1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Bagchi IC, Bagchi MK. Control of ovulation in mice by progesterone receptor-regulated gene networks. Mol Hum Reprod. 2009;15(12):821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loutradis D, Bletsa R, Aravantinos L, Kallianidis K, Michalas S, Psychoyos A. Preovulatory effects of the progesterone antagonist mifepristone (RU486) in mice. Hum Reprod. 1991;6(9):1238–1240. [DOI] [PubMed] [Google Scholar]
- 26.Lydon JP, DeMayo FJ, Funk CR, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Bagchi IC, Bagchi MK. Signaling by hypoxia-inducible factors is critical for ovulation in mice. Endocrinology. 2009;150(7):3392–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robker RL, Russell DL, Espey LL, Lydon JP, O'Malley BW, Richards JS. Progesterone-regulated genes in the ovulation process: ADAMTS-1 and cathepsin L proteases. Proc Natl Acad Sci U S A. 2000;97(9):4689–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuzpe AA, Thurlow HJ, Ramzy I, Leyshon JI. Post coital contraception—a pilot study. J Reprod Med. 1974;13(2):53–58. [PubMed] [Google Scholar]
- 30.Loutradis D, Bletsa R, Aravantinos L, Kallianidis K, Michalas S, Psychoyos A. Preovulatory effects of the progesterone antagonist mifepristone (RU486) in mice. Hum Reprod. 1991;6(9):1238–1240. [DOI] [PubMed] [Google Scholar]
- 31.Brännström M. Inhibitory effect of mifepristone (RU 486) on ovulation in the isolated perfused rat ovary. Contraception. 1993;48(4):393–402. [DOI] [PubMed] [Google Scholar]
- 32.Hamoda H, Ashok PW, Stalder G, Flett GMM, Kennedy E, Templeton A. A randomized trial of mifepristone (10 mg) and levonorgestrel for emergency contraception. Obstet Gynecol. 2004;104(6):1307–1313. [DOI] [PubMed] [Google Scholar]
- 33.Croxatto HB, Salvatierra AM, Fuentealba B, Zurth C, Beier S. Effect of the antiprogestin onapristone on follicular growth in women. Hum Reprod. 1994;9(8):1442–1447. [DOI] [PubMed] [Google Scholar]
- 34.Donath J, Michna H, Nishino Y. The antiovulatory effect of the antiprogestin onapristone could be related to down-regulation of intraovarian progesterone (receptors). J Steroid Biochem Mol Biol. 1997;62(1):107–118. [DOI] [PubMed] [Google Scholar]
- 35.Iwai T, Nanbu Y, Iwai M, Taii S, Fujii S, Mori T. Immunohistochemical localization of oestrogen receptors and progesterone receptors in the human ovary throughout the menstrual cycle. Virchows Arch A Pathol Anat Histopathol. 1990;417(5):369–375. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T, Sasano H, Kimura N, et al. Immunohistochemical distribution of progesterone, androgen and oestrogen receptors in the human ovary during the menstrual cycle: relationship to expression of steroidogenic enzymes. Hum Reprod. 1994;9(9):1589–1595. [DOI] [PubMed] [Google Scholar]
- 37.Revelli A, Paahioni D, Cassoni P, Bussolati G, Massobrio M. In situ hybridization study of messenger RNA for estrogen receptor and immunohistochemical detection of estrogen and progesterone receptors in the human ovary. Gynecol Endocrinol. 1996;10(3):177–186. [DOI] [PubMed] [Google Scholar]
- 38.García V, Kohen P, Maldonado C, et al. Transient expression of progesterone receptor and cathepsin-l in human granulosa cells during the periovulatory period. Fertil Steril. 2012;97(3):707–713. [DOI] [PubMed] [Google Scholar]
- 39.Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B-isoform. Science. 2000;289(5485):1751–1754. [DOI] [PubMed] [Google Scholar]