

Abstract
We studied obesity-related differences in the relation of maternal levels of leptin to levels of soluble fms-like tyrosine kinase 1 (sFlt1), an antiangiogenic protein that influences placentation and risk of adverse pregnancy outcomes. In a prospective cohort of 286 gravidas, we measured maternal serum levels of sFlt1 and leptin at 5 time points across pregnancy. Analyses stratified on prepregnancy body mass index (<25 vs ≥25) were done using mixed linear models. The mean leptin concentrations were significantly higher in overweight/obese compared to normal-weight women, while mean sFlt1 levels in second and third trimester were significantly higher in normal weight compared to overweight/obese women. The relationship between sFlt1 and leptin differed between the 2 strata. After controlling for maternal weight, a 1 ng/mL increase in leptin was associated with an 19.4 pg/mL increase in sFlt1 (P = .01) in normal-weight women, while leptin was not associated with sFlt1 (β = 1.1, P = .75) in overweight/obese women. Such differences suggest that metabolic differences in overweight/obese women compared to their normal weight peers may differentially impact the physiologic changes during pregnancy.
Keywords: angiogenic factors, obesity, Flt1 protein, longitudinal analysis, leptin, pregnancy
Introduction
Maternal obesity is associated with the dysregulation of metabolic and physiologic factors that may affect the course of pregnancy and lead to adverse outcomes. However, the underlying pathways mediating the effects of obesity on pregnancy outcomes are not well characterized. Normal pregnancy outcomes depend on the establishment of the maternal–fetal vascular interface early in pregnancy and ongoing placental development over the duration of gestation.1 Accordingly, disruption of placental production of angiogenic factors contributes to the pathophysiology of many adverse perinatal outcomes.2 For example, soluble fms-like tyrosine kinase 1 (sFlt1), an important antiangiogenic protein, has been strongly associated with disorders of placentation, including preeclampsia and intrauterine growth restriction.2,3 We have recently shown that maternal serum sFlt1 concentrations are associated with an increased risk of preterm delivery independent of preeclampsia.4 Maternal obesity may affect these factors via a number of adipokine-mediated pathways.5 However, the relationship between maternal obesity, adipokines, and angiogenic factors is poorly characterized in human pregnancies.
Beyond its well-known role in the regulation of energy metabolism, the adipokine leptin has well-documented effects on many other physiological processes, including angiogenesis.6–8 For example, several studies suggest that leptin may act directly on the vascular endothelium to stimulate angiogenesis.8–10 Thus, endothelial cells have been shown to express functional leptin receptors and leptin has been demonstrated to stimulate endothelial cell proliferation and to induce the formation of capillary-like tubes in vitro.10 Leptin has also been shown to stimulate new blood vessel formation in the chick chorioallantoic membrane.10 In fact, the angiogenic effect of leptin is comparable to that of vascular endothelial growth factor.10
The regulation of maternal leptin during pregnancy is complex. As in nonpregnant adults, serum leptin levels are related to adipose tissue mass.11 Moreover, in both overweight/obese and normal-weight women, serum leptin concentrations significantly increase during the course of pregnancy.11–13 Such changes are thought to be involved in optimizing the availability of substrates necessary for fetal growth, particularly by mobilizing maternal fat stores.14 However, factors other than fat mass alone contribute to this increase. In humans, placental production of leptin contributes significantly to maternal serum levels.14–16
Our prior work demonstrates that variation in maternal serum leptin levels across pregnancy differs in overweight and obese women when compared to their normal-weight counterparts.17 After accounting for maternal weight gain, overweight/obese gravidas do not show the progressive increases in leptin production per unit of body mass that are seen in normal-weight women. Such differences suggest altered regulation of leptin production in obese/overweight women, perhaps due to metabolic disturbances arising from adipocyte and adipose tissue dysfunction. The metabolic factors that result in such differences in leptin regulation may also result in different effects of leptin on angiogenesis, beyond that expected from just a difference in concentration. Such metabolic differences may ultimately be involved in differences in pregnancy outcomes between the 2 groups.
The goal of this study was to estimate the influence of variation in maternal serum leptin levels during pregnancy on variation in maternal sFlt1 levels in women with overweight/obese prepregnancy body mass index (BMI ≥25.0) and normal prepregnancy BMI (<25.0). We hypothesized that leptin would influence sFlt1 levels differently in overweight/obese compared to normal-weight women, potentially disrupting the balance of angiogenic and antiangiogenic factors in these 2 groups.7 Such differences may contribute to the risk of adverse pregnancy outcomes in obese women.
Methods
Study Sample
Data were collected as part of the Gestational Regulators of Weight study, a prospective cohort study of pregnant women who presented for early prenatal care at the University of Michigan Health System. The study was approved by the institutional review board at the University of Michigan Health System. Women were enrolled between 6 and 10 weeks’ gestation. Women were eligible for inclusion if they were between 18 and 45 years of age, had a singleton pregnancy, and intended to deliver at the study hospital. Informed consent was obtained at the initial study visit (6-10 weeks’ gestation). Participants were seen for 4 additional study visits at 10 to 14, 16 to 20, 22 to 26, and 32 to 36 weeks’ gestation.
Data Collection and Study Variables
At each of the 5 study visits, questionnaire data, anthropometric measurements, and biological samples were collected. Baseline maternal characteristics were obtained by questionnaire and medical record review. Data elements collected included maternal age, self-reported race and ethnicity, education, marital status, pregnancy history, pregnancy complications, and tobacco smoke exposure. Standing height was measured at baseline using a wall-mounted stadiometer. Weight was measured in street clothes, without shoes, on a calibrated electronic scale (Scale-Tronix, White Plains, New York). Maternal prepregnancy weight was collected at the initial visit by maternal self-report. Prepregnancy BMI was calculated using height and prepregnancy weight (BMI = weight/height2) and classified as normal (BMI <25 kg/m2) or overweight/obese (BMI ≥25 kg/m2).18
Maternal blood samples were collected in serum separator tubes and were allowed to clot for 30 minutes prior to centrifugation (15 minutes at 1000g). The serum samples were aliquoted and stored at −80°C until analysis. Concentration of maternal serum sFlt1 was determined in duplicate using a commercially available solid-phase enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, Minnesota). Serum leptin concentration was also measured in duplicate using a standard commercial radioimmunoassay kit (Linco Research, St Charles, Missouri). Laboratory assays were performed in the chemistry laboratory of the Michigan Diabetes Research and Training Center. Laboratory personnel were blind to all patient identifiers as well as pregnancy characteristics and outcomes.
Statistical Analysis
All analyses were performed using SAS version 9.1 (SAS Institute, Cary, North Carolina). Demographic and health-related characteristics of the study population were compared using t test, chi-square test, and Fisher exact test as appropriate. Leptin and sFlt1 were treated as continuous variables in all analyses.
Our analysis included repeated measures of sFlt1 and leptin over the course of pregnancy. Therefore, mixed linear models were used to assess the impact of leptin on sFlt1 while considering the longitudinal structure of the data. Unlike conventional linear regression models, mixed linear regression models allow the data to exhibit correlation and nonconstant variability by including both fixed effect and covariance parameters. The covariance structure of the repeated measurements can be modeled to increase efficiency so that the estimates and standard errors can be efficiently generated. In addition, the mixed linear modeling procedure used here implements a likelihood-based estimation method so that all available data are used in the analysis without excluding participants with data missing at one or more time points. For these analyses, repeated measures of leptin across pregnancy were analyzed using a model in which prepregnancy BMI stratum (normal vs overweight/obese) was a fixed factor that varied between participants. Gestational age at each leptin measurement was a repeated factor that varied within participants. All tests of significance in main effects models were 2-tailed with a type 1 error rate fixed at 5%.
In pregnant women, leptin is strongly correlated with maternal fat mass and placental size. During pregnancy, maternal weight includes contributions from maternal tissue and the products of conception, including the fetus, placenta, and amniotic fluid. Therefore, in order to model the effects of leptin independent of maternal and placental tissue mass, we included the adjusted maternal weight (maternal weight at each visit minus estimated fetal weight) at each visit19 as a covariate. Estimated fetal weight was determined by ultrasound biometry using the method of Hadlock.20
Results
Sociodemographic and health characteristics of the 286 women included in the study are presented in Table 1. Half of the study participants were overweight or obese (N = 143). Normal-weight and overweight/obese women were similar with respect to age, marital status, parity, and prenatal smoking. A greater proportion of normal-weight women were white, whereas a greater proportion of overweight/obese women were black (P = .04). When compared to normal-weight women, a greater proportion of overweight/obese women had a college education or less (P = .002).
Table 1.
Study Sample Characteristics by BMI Group.a
| Normal-Weight Women, BMI <25 kg/m2 | Overweight/Obese Women, BMI ≥25 kg/m2 | |||
|---|---|---|---|---|
| N = 143 | N = 143 | |||
| N | % | N | % | |
| Raceb | ||||
| White | 120 | 83.9 | 113 | 79.0 |
| Black | 4 | 2.8 | 14 | 9.8 |
| Asian | 12 | 8.4 | 6 | 4.2 |
| Other | 7 | 4.9 | 10 | 7.0 |
| Maternal age | ||||
| ≤30 | 53 | 37.1 | 58 | 40.6 |
| >30 | 90 | 62.9 | 85 | 59.4 |
| Prenatal smokingc | ||||
| Yes | 13 | 9.4 | 11 | 7.8 |
| No | 126 | 90.7 | 130 | 92.2 |
| Education leveld | ||||
| ≤College | 67 | 46.9 | 94 | 65.7 |
| >College | 76 | 53.2 | 49 | 34.3 |
| Incomeb | ||||
| ≤$ 80 000 | 57 | 39.9 | 79 | 55.2 |
| >$ 80 000 | 81 | 56.6 | 59 | 41.3 |
| Unknown or chose not to report | 5 | 3.5 | 5 | 3.5 |
| Marital status | ||||
| Married | 127 | 88.8 | 118 | 82.5 |
| Unmarried | 16 | 11.2 | 25 | 17.5 |
| Parity | ||||
| Primipara | 59 | 41.3 | 42 | 29.4 |
| Multipara | 84 | 58.7 | 101 | 70.6 |
a Percentages have been rounded and may not total 100.
b P < .05.
c Smoking status was unknown for 6 women.
d P < .01.
There were very few missing data points for our analyses. There were only 42 missing measurements of maternal sFlt1 (2.9% of the measures) and 54 missing measurements of maternal leptin (3.8% of the measures). Only 2 women were missing more than 1 measure of sFlt1 (missing 2 each); while 7 women were missing more than 1 measure of leptin (range 1-3). Covariates were reliably measured. Six women were missing smoking status. There were no other missing covariates. The missing data appeared to be random. However, the number of missing observations was too small to model the distribution of missingness for evidence of bias. The mixed linear regression models described below are generally robust to missing at random data.
The mean maternal serum leptin and sFlt1 concentration at each study visit for each BMI stratum are presented in Table 2. The mean leptin concentration differed by BMI group at each visit (P < .0001 at each visit). As previously reported, leptin was higher in overweight/obese women over the entire course of gestation.17 In contrast, mean sFlt1 levels were higher for normal weight as opposed to overweight/obese women at each time point, although not all differences were statistically significant or substantial (Table 2). Differences in sFlt concentration were statistically significant at visit 2 between 10 to 14 weeks’ gestation (P = .003), visit 3 between 16 to 20 weeks’ gestation (P < .0001), and visit 4 between 22 to 26 weeks’ gestation (P = .002). The relationship between sFlt1 and gestational age for each BMI group is presented in Figure 1. The trajectory of sFlt1 increased significantly more quickly for normal-weight women compared to their overweight/obese peers (β = −5.68; 95% confidence interval [CI] −8.82 to −2.54).
Table 2.
Maternal Serum sFlt1 Concentrations at Each Visit by Obesity Status.
| Visit | sFlt1 Concentrations (pg/mL) | ||
|---|---|---|---|
| Normal-Weight Women (mean ± SD) | Overweight/Obese Women (mean ± SD) | P a | |
| Baseline | 2327.3 ± 1305.8 | 2089.8 ± 1187.9 | .11 |
| Visit 1 | 2580.4 ± 1366.1 | 2291.9 ± 1237.2 | .07 |
| Visit 2 | 2602.1 ± 1894.4 | 2037.1 ± 1193.9 | <.01 |
| Visit 3 | 2741.4 ± 1754.0 | 1959.2 ± 1290.8 | <.0001 |
| Visit 4 | 5470.1 ± 4268.5 | 4107.3 ± 2765.8 | <.01 |
Abbreviations: SD, standard deviation; sFlt1, soluble fms-like tyrosine kinase 1.
a P value for differences in mean sFlt1 by obesity status.
Figure 1.
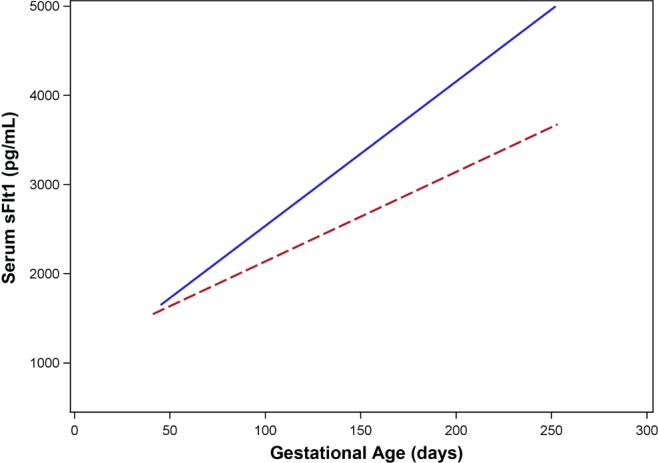
The relationship between soluble fms-like tyrosine kinase 1 (sFlt)1 and gestational age for normal-weight and overweight/obese women. Data for normal-weight women are represented by the solid line, whereas the dashed line represents overweight/obese women.
We used mixed linear models to examine the longitudinal relation between maternal serum leptin and sFlt1 concentrations across pregnancy. After adjusting for maternal weight, these models suggest that the relationship between leptin and sFlt1 differs by BMI strata (Table 3). A 1 ng/mL increase in leptin was associated with an 19.4 pg/mL increase in sFlt1 (95% CI 4.7 to 34.0) in normal-weight women, whereas leptin did not impact sFlt1 levels (β = 1.1; 95% CI −5.9 to 8.1) in overweight/obese women. The observed interaction between BMI and leptin was statistically significant (P = .01). We also include results after adjusting for several important maternal covariates (maternal race, education, age, and prenatal smoking). None of the covariates examined (maternal race, education, age, and prenatal smoking) appreciably changed our results or conclusions. However, as reported in Table 3, the β estimates for leptin changed slightly more than 5% when race was added to the model. Unfortunately, our sample contains too few non-white participants to appropriately test the effects of race on these relations. To evaluate the possible influence of other placental disorders on our results, we reanalyzed the data excluding women with preeclampsia or suspected preeclampsia (N = 5). Excluding these cases did not impact our results (data not shown).
Table 3.
The Relationship of Maternal Serum sFlt1 With Leptin Stratified by BMI Group.
| Normal-Weight Women | Overweight/Obese Women | P b | |||
|---|---|---|---|---|---|
| βleptin a | 95% CI | βleptin a | 95% CI | ||
| Model 1c | 19.4 | 4.7-34.0 | 1.1 | −5.9-8.1 | .01 |
| Model 2d | 20.6 | 5.8-35.4 | 0.6 | −6.3-7.5 | .01 |
Abbreviations: BMI, body mass index; CI, confidence interval; sFlt1, soluble fms-like tyrosine kinase 1.
a β estimates for linear mixed models regressing leptin on sFlt1 for each BMI group.
b P value for interaction between BMI group and leptin on sFlt1 by obesity status.
c Model 1: Controls for adjusted maternal weight (maternal weight at each visit-estimated fetal weight).
d Model 2: Model 1 plus race.
Discussion
Although obese women may have a higher risk of placental vascular dysfunction, few studies have investigated the pathophysiological factors that may be related to this predisposition. There is growing evidence that the regulation and effects of metabolic systems in overweight and obese individuals is substantially different from their normal-weight counterparts.21–24 This work is among the first to demonstrate that obesity-related factors may influence factors related to angiogenesis differently in overweight/obese compared to normal-weight women during pregnancy. Specifically, we found that while maternal sFlt1 increased significantly across pregnancy for both normal-weight and overweight/obese women, its trajectory was significantly different in the 2 groups. Moreover, our analyses demonstrate that the relationship between maternal sFlt1 and leptin concentrations across pregnancy differs between overweight/obese and normal-weight women. Leptin was associated with substantially higher sFlt1 levels in normal-weight women, whereas in overweight/obese women, leptin did not impact sFlt1 levels.
The possible relation of maternal leptin to placental vascular dysfunction has been recognized for some time. A number of prior studies suggest that increased maternal plasma leptin levels may be associated with preeclampsia,25–28 although there have been conflicting reports.29,30 Similarly, leptin levels in the placenta were higher for women with severe preeclampsia compared to normotensive pregnancies associated with intrauterine growth restriction as well as controls.31 Moreover, the placental leptin level was positively associated with the umbilical artery resistance index, suggesting that placental insufficiency may be associated with an increase in placental leptin.31 Interestingly, in at least 2 prior studies that stratified on BMI, second trimester serum leptin levels were significantly higher in preeclamptic compared to normotensive women only in the normal weight (BMI <25 kg/m2) stratum.27,32 There was no difference in overweight (BMI ≥25 kg/m2) women. The difference in the relation we observed between leptin and sFlt1 in these 2 strata suggests a physiologic correlation underlying this prior observation.
The only prior human study that reported on the relationship between maternal leptin and sFlt1 levels was a small case–control study (N = 68) of preeclampsia in Japanese women in which a single cross-sectional maternal blood level was obtained during the second or third trimester. Among the preeclamptic cases, leptin was negatively associated with sFlt-1, while there was no correlation among the controls. Furthermore, BMI was significantly higher for those preeclamptic women with leptin/sFlt1 ratios above the median compared to those below the median; no difference in BMI was seen in comparison with controls.33 In contrast to this prior study, our study had a more robust study design that documented maternal leptin and sFlt1 levels at multiple time points starting in early pregnancy and more appropriately modeled their relationship using longitudinal methods.
Our analyses demonstrate that levels of maternal serum sFlt1 differ between overweight/obese and normal-weight women across gestation, even in the absence of preeclampsia. Moreover, its relationship with maternal serum leptin is altered in overweight/obese women compared to their normal-weight counterparts. The mechanisms through which maternal obesity and maternal leptin may influence sFlt1 are poorly understood. However, differences between the 2 strata may be related to the dysregulation of the normal metabolic systems found in obese/overweight women.21–23 There is growing evidence that such dysregulation may influence the physiologic changes during pregnancy leading to the development of many of the pathological conditions associated with obesity.17,24,34,35 For example, lipid levels differ across BMI strata and altered lipid homeostasis during pregnancy may contribute to poor placental development and disrupted endothelial function.35,36 Accordingly, prior studies suggest that the relationship between fetal growth and maternal lipids differs between these groups.34 Our current study suggests that such metabolic dysregulation may alter the relationship between factors, such as leptin and sFlt1, that may be associated with placental angiogenesis in obese/overweight compared to normal-weight women.
The prospective cohort design is a significant strength of the current study. We were able to obtain multiple measures of maternal serum leptin and sFlt1 concentrations on each participant starting in early first trimester and at multiple time points across pregnancy. Despite the modest sample size, this study is one of the largest longitudinal studies of either sFlt1 or leptin levels in the current literature, with 5 time points for data collection in pregnancy for nearly 300 women. Our analytic approach used linear mixed models that accounted for the longitudinal data structure. As a result, we were able to analyze the trajectory of maternal serum sFlt1 concentrations in different BMI groups sampled from a population-based cohort of pregnant women. Our study design and analytic plan also provided substantial statistical power to analyze the relation between leptin and sFlt1 and to analyze how factors such as maternal overweight/obesity could influence this relation.
The longitudinal design inherently limits study participation to a group of women who present for early prenatal care and are able to attend multiple study visits. Women without prenatal care or with late, interrupted, or sporadic care are less likely to have been included. As a result, maternal sociodemographic covariates, chronic disease, and pregnancy complications showed limited variation and may limit the generalizability of our study. This limitation is most evident with regard to race; more than 80% of our sample is white. Factors related to maternal weight and weight gain may have different effects in different racial strata. For example, the influence of the rate of maternal weight gain on infant birth weight occurs at different times in gestation in different racial strata.37 Such results suggest that the racial, cultural, and sociodemographic context of a pregnancy may influence pregnancy outcomes related to maternal weight and BMI status. While our sample is too small to address these issues, it will be very important to conduct these analyses in larger, multiethnic cohorts.
Nevertheless, while our sample is homogeneous with regard to measures of socioeconomic status, race, and illness, there is considerable variation in prepregnancy BMI. Our sample was equally distributed into 2 BMI subgroups. Because of this homogeneity, many potential confounding factors were accounted for by our sampling, allowing us to focus primarily on the effects of maternal overweight/obesity.
Prior work suggests that maternal obesity may be associated with an increased risk of placental dysfunction and adverse pregnancy outcomes.38 Our analysis is a step toward defining how factors associated with maternal overweight and obesity may influence pregnancy outcomes through their effects on factors that may influence placental function and development. Specifically, the relationships between maternal sFlt1, leptin, and obesity status begin to suggest that potential mechanisms by which excess adiposity may relate to placental angiogenesis. Future studies should focus on how the pathogenic effects of obesity may be associated with metabolic dysregulation and disruption of the balance among various angiogenic factors during pregnancy. Larger studies will be needed to address how these effects may, ultimately, contribute to poor pregnancy outcomes.
Acknowledgments
We gratefully acknowledge the infrastructure and personnel support of the Michigan Clinical Research Unit and the laboratory support of the Chemistry Core of the Michigan Diabetes Research and Training Center as well as the Michigan Molecular Genetics Laboratory.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: VKM was supported by a Doris Duke Clinical Scientist Development Award (Grant 2007092); and a NIH Mentored Scientist Award (K08-HD045609). The Michigan Clinical Research Unit is supported by a Clinical and Translational Science Award (UL1RR024986) from the National Institutes of Health. This work utilized the resources of the Chemistry Core of the Michigan Diabetes Research and Training Center funded by the National Institute of Diabetes and Digestive and Kidney Diseases (NIH5P60 DK020572). JKS was supported by a postdoctoral award from Wayne State University.
References
- 1. Regnault TR, Galan HL, Parker TA, Anthony RV. Placental development in normal and compromised pregnancies—a review. Placenta. 2002;23(suppl A):S119–S129. [DOI] [PubMed] [Google Scholar]
- 2. Smith GC, Wear H. The perinatal implications of angiogenic factors. Curr Opin Obstet Gynecol. 2009;21(2):111–116. [DOI] [PubMed] [Google Scholar]
- 3. Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Annu Rev Med. 2008;59(1):61–78. [DOI] [PubMed] [Google Scholar]
- 4. Straughen J, Kumar P, Misra VK. The effect of maternal soluble FMS-like tyrosine kinase 1 during pregnancy on risk of preterm delivery [published online March 27, 2012]. J Matern Fetal Neonatal Med. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest. 2007;117(9):2362–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–770. [DOI] [PubMed] [Google Scholar]
- 7. Park HY, Kwon HM, Lim HJ, et al. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001;33(2):95–102. [DOI] [PubMed] [Google Scholar]
- 8. Sierra-Honigmann MR, Nath AK, Murakami C, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281(5383):1683–1686. [DOI] [PubMed] [Google Scholar]
- 9. Anagnostoulis S, Karayiannakis AJ, Lambropoulou M, Efthimiadou A, Polychronidis A, Simopoulos C. Human leptin induces angiogenesis in vivo. Cytokine. 2008;42(3):353–357. [DOI] [PubMed] [Google Scholar]
- 10. Bouloumie A, Drexler HC, Lafontan M, Busse R. Leptin, the product of Ob gene, promotes angiogenesis. Circ Res. 1998;83(10):1059–1066. [DOI] [PubMed] [Google Scholar]
- 11. Highman TJ, Friedman JE, Huston LP, Wong WW, Catalano PM. Longitudinal changes in maternal serum leptin concentrations, body composition, and resting metabolic rate in pregnancy. Am J Obstet Gynecol. 1998;178(5):1010–1015. [DOI] [PubMed] [Google Scholar]
- 12. Masuzaki H, Ogawa Y, Sagawa N, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3(9):1029–1033. [DOI] [PubMed] [Google Scholar]
- 13. Hardie L, Trayhurn P, Abramovich D, Fowler P. Circulating leptin in women: a longitudinal study in the menstrual cycle and during pregnancy. Clin Endocrinol (Oxf). 1997;47(1):101–106. [DOI] [PubMed] [Google Scholar]
- 14. Hauguel-de Mouzon S, Lepercq J, Catalano P. The known and unknown of leptin in pregnancy. Am J Obstet Gynecol. 2006;194(6):1537–1545. [DOI] [PubMed] [Google Scholar]
- 15. Butte NF, Hopkinson JM, Nicolson MA. Leptin in human reproduction: serum leptin levels in pregnant and lactating women. J Clin Endocrinol Metab. 1997;82(2):585–589. [DOI] [PubMed] [Google Scholar]
- 16. Sagawa N, Yura S, Itoh H, et al. Possible role of placental leptin in pregnancy: a review. Endocrine. 2002;19(1):65–71. [DOI] [PubMed] [Google Scholar]
- 17. Misra VK, Trudeau S. The influence of overweight and obesity on longitudinal trends in maternal serum leptin levels during pregnancy. Obesity (Silver Spring). 2011;19(2):416–421. [DOI] [PubMed] [Google Scholar]
- 18. WHO. Obesity: preventing and managing the global epidemic. Report of a WHO Consultation. Geneva: World Health Organization; 2000. [PubMed] [Google Scholar]
- 19. Selvin S, Abrams B. Analysing the relationship between maternal weight gain and birthweight: exploration of four statistical issues. Paediatr Perinat Epidemiol. 1996;10(2):220–234. [DOI] [PubMed] [Google Scholar]
- 20. Hadlock FP, Harrist RB, Sharman RS, Deter RL, Park SK. Estimation of fetal weight with the use of head, body, and femur measurements—a prospective study. Am J Obstet Gynecol. 1985;151(3):333–337. [DOI] [PubMed] [Google Scholar]
- 21. Bluher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes. 2009;117(6):241–250. [DOI] [PubMed] [Google Scholar]
- 22. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314(1):1–16. [DOI] [PubMed] [Google Scholar]
- 24. Stuebe AM, McElrath TF, Thadhani R, Ecker JL. Second trimester insulin resistance, early pregnancy body mass index and gestational weight gain. Matern Child Health J. 2010;14(2):254–260. [DOI] [PubMed] [Google Scholar]
- 25. Ouyang Y, Chen H, Chen H. Reduced plasma adiponectin and elevated leptin in pre-eclampsia. Int J Gynaecol Obstet. 2007;98(2):110–114. [DOI] [PubMed] [Google Scholar]
- 26. Acromite M, Ziotopoulou M, Orlova C, Mantzoros C. Increased leptin levels in preeclampsia: associations with BMI, estrogen and SHBG levels. Hormones (Athens). 2004;3(1):46–52. [DOI] [PubMed] [Google Scholar]
- 27. Samolis S, Papastefanou I, Panagopoulos P, Galazios G, Kouskoukis A, Maroulis G. Relation between first trimester maternal serum leptin levels and body mass index in normotensive and pre-eclamptic pregnancies—role of leptin as a marker of pre-eclampsia: a prospective case-control study. Gynecol Endocrinol. 2010;26(5):338–343. [DOI] [PubMed] [Google Scholar]
- 28. Baksu A, Ozkan A, Goker N, Baksu B, Uluocak A. Serum leptin levels in preeclamptic pregnant women: relationship to thyroid-stimulating hormone, body mass index, and proteinuria. Am J Perinatol. 2005;22(3):161–164. [DOI] [PubMed] [Google Scholar]
- 29. Laml T, Preyer O, Hartmann BW, Ruecklinger E, Soeregi G, Wagenbichler P. Decreased maternal serum leptin in pregnancies complicated by preeclampsia. J Soc Gynecol Investig. 2001;8(2):89–93. [PubMed] [Google Scholar]
- 30. Kafulafula GE, Moodley J, Ojwang PJ, Kagoro H. Leptin and pre-eclampsia in black African parturients. Bjog. 2002;109(11):1256–1261. [DOI] [PubMed] [Google Scholar]
- 31. Lepercq J, Guerre-Millo M, Andre J, Cauzac M, Hauguel-de Mouzon S. Leptin: a potential marker of placental insufficiency. Gynecol Obstet Invest. 2003;55(3):151–155. [DOI] [PubMed] [Google Scholar]
- 32. Williams MA, Havel PJ, Schwartz MW, et al. Pre-eclampsia disrupts the normal relationship between serum leptin concentrations and adiposity in pregnant women. Paediatr Perinat Epidemiol. 1999;13(2):190–204. [DOI] [PubMed] [Google Scholar]
- 33. Nakatsukasa H, Masuyama H, Takamoto N, Hiramatsu Y. Circulating leptin and angiogenic factors in preeclampsia patients. Endocr J. 2008;55(3):565–573. [DOI] [PubMed] [Google Scholar]
- 34. Misra VK, Trudeau S, Perni U. Maternal serum lipids during pregnancy and infant birth weight: the influence of prepregnancy BMI. Obesity (Silver Spring). 2011;19(7):1476–1481. [DOI] [PubMed] [Google Scholar]
- 35. Vahratian A, Misra VK, Trudeau S, Misra DP. Prepregnancy body mass index and gestational age-dependent changes in lipid levels during pregnancy. Obstet Gynecol. 2010;116(1):107–113. [DOI] [PubMed] [Google Scholar]
- 36. Uzun H, Benian A, Madazli R, Topcuoglu MA, Aydin S, Albayrak M. Circulating oxidized low-density lipoprotein and paraoxonase activity in preeclampsia. Gynecol Obstet Invest. 2005;60(4):195–200. [DOI] [PubMed] [Google Scholar]
- 37. Misra VK, Hobel CJ, Sing CF. The effects of maternal weight gain patterns on term birth weight in African-American women. J Matern Fetal Neonatal Med. 2010;23(8):842–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cedergren MI. Maternal morbid obesity and the risk of adverse pregnancy outcome. Obstet Gynecol. 2004;103(2):219–224. [DOI] [PubMed] [Google Scholar]