

Abstract
For bottom-up proteomics there are a wide variety of database searching algorithms in use for matching peptide sequences to tandem MS spectra. Likewise, there are numerous strategies being employed to produce a confident list of peptide identifications from the different search algorithm outputs. Here we introduce a grid search approach for determining optimal database filtering criteria in shotgun proteomics data analyses that is easily adaptable to any search. Systematic Trial and Error Parameter Selection – referred to as STEPS – utilizes user-defined parameter ranges to test a wide array of parameter combinations to arrive at an optimal “parameter set” for data filtering, thus maximizing confident identifications. The benefits of this approach in terms of numbers of true positive identifications are demonstrated using datasets derived from immunoaffinity-depleted blood serum and a bacterial cell lysate, two common proteomics sample types.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) shotgun proteomics has become a widely applied technology for characterization of complex protein samples [1-3]. A critical step in the bottom-up proteomics data analysis pipeline is matching MS/MS spectra to theoretical fragmentation patterns of peptides derived from a protein sequence database. A number of search algorithms, such as SEQUEST [4], X!Tandem [5], and Mascot [6] are available for this purpose. Output from these algorithms is a list of peptide-to-spectrum matches (PSMs) along with metrics that reflect the quality of the match. Although robust, the process is prone to high rates of false positive and false negative peptide identifications. The challenge is to remove the false positives, while retaining the maximum number of true positives [7-11].
All of the database search engines provide PSM scoring metrics that aid in differentiating between true and false matches. Additional information such as mass measurement accuracy, charge state of the ion, number of tryptic termini, and search engine independent scoring algorithms, e.g., MS-Generating Function (MS-GF) [12] can be used to further refine research results. Mass measurement accuracy in particular is helpful for filtering high mass resolution data, and even more so when systematic error correction is employed [13-14]. Common approaches for increasing confident peptide identifications include combining scoring metrics, and partitioning PSMs based on charge state and number of tryptic termini prior to the generation of filtering criteria [10, 15]. The most commonly accepted metric for validating filtering criteria utilizes a decoy database search strategy to estimate the false discovery rate (FDR) [7, 16].
We developed STEPS (Systematic Trial and Error for Parameter Selection) as a means of optimizing filtering criteria to improve confidence in peptide identifications. The STEPS methodology (Figure 1) utilizes a freely available automated processing engine (APE; available at http://omics.pnl.gov/software/APE.php) that allows a user to leverage all information available to them for a particular MS/MS dataset to generate optimal filtering criteria for that dataset. The user specifies the parameter(s) to be used, the range over which to search them, and an increment. The integrated ranger tool uses this information to create an iteration table that consists of all possible combinations of the user-defined criteria. This table is read by APE, which generates the appropriate SQL statement to test each line of criteria one row at a time, quickly testing of thousands of potential parameter sets. Each iteration result is recorded in a table that is queried to retrieve the criteria set that produces the largest number of true positives below a user-specified false discovery rate (FDR). APE then uses these criteria to generate a final results table. The FDR is estimated using the reverse database method on non-redundant datasets (unique peptide identifications), which prevents frequently observed peptides from skewing the error estimate [16].
Figure 1.
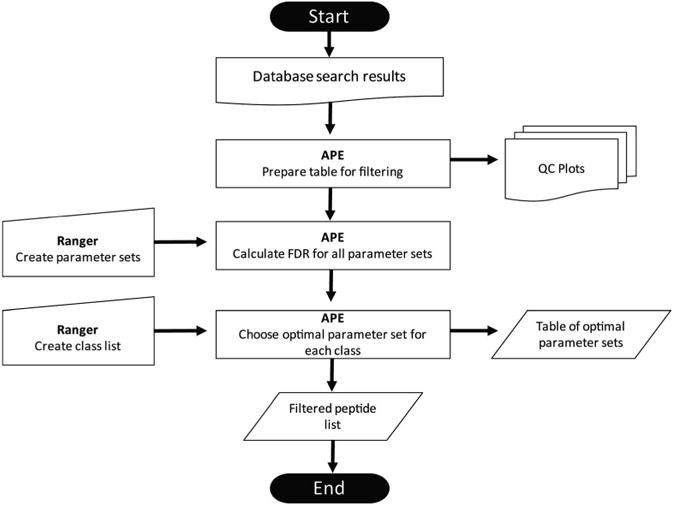
Schematic overview of the STEPS methodology. Data can be brought into APE as a flat text file or SQLite database file. Parameter tables are generated within APE using the ranger functionality. The first table defines the parameters to be used and the second table is used to select criteria that correspond to the user-defined FDR, charge state, and NTT. STEPS workflow will output a table of confident peptide identifications, a table of optimal parameters, a crosstab of spectral counting results, and selected QC plots.
In addition to the grid search capability, APE has a number of useful functionalities that make it a powerful data analysis tool, including integrated plotting capabilities. As a result, the STEPS workflow can be configured to generate metric histograms and scatter plots for quality control purposes and to generate optimal filtering parameters (Supplemental Figure 1). Additionally, APE can be configured to produce and export custom tables to facilitate further analysis or for publication. Additional plots and tables can be easily added or subtracted from the workflow. Using SQL Scripting to build workflows provides much faster analyses, elimination of human errors, and gives the ability to work with very large datasets (> 1 million data rows) easily. Moreover, APE maintains a record of all analyses in a time-date stamp fashion that is easily recalled. All analysis steps are visible to the analyst and added comments explain the processing performed for each workflow step (Supplemental Figure 2), which facilitates transparency in the data analysis process. APE effectively creates a detailed lab notebook for every data analysis. The workflow steps are readily modifiable to allow for custom workflows that can be exchanged among researchers. APE also allows for the integration of independent validation metrics, and provides a facile platform for investigating the effects of changes in workflow parameters. Thus, STEPS workflows can be used for “push button” processing, as well as for intensive data processing investigations by expert analysts.
To illustrate its usefulness, we applied the STEPS approach to datasets obtained for a Shewanella oneidensis cell lysate and immunoaffinity-depleted human blood serum [17-18]. Various combinations of filtering parameters were examined to determine discriminative power and to demonstrate the value of exploiting additional information. Spectra for both sample sets had been obtained previously using a custom built four-column high-pressure capillary LC system coupled on-line to an LTQ-Orbitrap mass spectrometer (Thermo Scientific, San Jose CA) via a nanoelectrospray interface manufactured in-house [19]. DeconMSn was utilized to generate .dta files [20], and SEQUEST (Version 27, Revision 12) was used to search all MS/MS data against either an in-house S. oneidensis FASTA protein sequence for MR-1 strain or the human protein database from SwissProt (20,276 entries, released 05/05/2010). FDR was estimated for non-redundant datasets, using a combined forward /reverse database search and the equation (2*Decoy)/(Decoy + Target).
We evaluated the effectiveness of SEQUEST parameters Xcorr, DeltaCn2, and MassDiff, as well as all combinations of these parameters, in terms of numbers of confident peptide identifications. (See Supplemental Table 1 for a summary of ranges and step sizes). Xcorr and DeltaCn2 represent SEQUEST scoring metrics commonly employed for filtering PSMs, and MassDiff is a value that represents the difference between the measured mass and the exact mass of the peptide from the database, which is useful for filtering high mass accuracy data [21]. For this evaluation, we utilized an FDR of 5%. PSMs were partitioned into classes based on their number of tryptic termini (NTT = 0, 1, 2) and charge state (1+, 2+, 3+, 4+, and 5+), which resulted in 15 unique peptide classes (unique combination of charge and NTT) for which an optimal parameter set was generated by STEPS. We also evaluated the effectiveness of incorporating MS-GF [12] values in conjunction with the SEQUEST parameters.
Figure 2 summarizes results obtained for the depleted serum and S. oneidensis datasets with (black bars) and without (grey bars) partitioning, using SEQUEST only parameters (Figure 2A and B) and SEQUEST parameters plus the MS-GF [12] value (Figure 2C and D). Note that for all parameter combinations in Figure 2A and B, partitioning by charge state and NTT provides substantially better differentiation, which agrees with findings in the literature [22]. In some cases without partitioning, single parameter filtering was unable to obtain a 5% FDR. For both datasets, the use of two-metric parameter sets outperformed single-metric filtering, and the utilization of all thee metrics represented the optimum. It is also worthwhile to note that different parameter combinations represent the optimal data model for the different datasets. In Figure 2C and D, incorporation of the MS-GF score provided further increases in the number of peptide identifications at a constant FDR. Interestingly, partitioning by class confers only modest improvements when incorporating the MS-GF score. Additionally, smaller gains are observed when a third parameter is introduced into the model.
Figure 2.
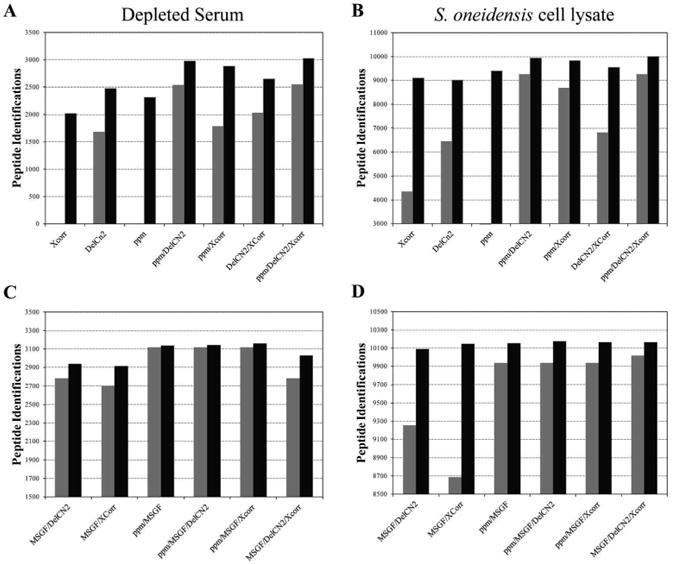
Bar plots depicting the number of confident peptide identifications obtained from A) depleted serum dataset using parameters specified by STEPS optimization, B) S. oneidensis dataset using parameters specified by STEPS optimization, C) serum dataset using STEPS parameters plus MS-GF scoring, and D) S. oneidensis dataset using STEPS parameters plus MS-GF scoring. Results obtained without partitioning peptides into classes are shown in grey and with partitioning are shown in black.
To benchmark performance, we compared results obtained using STEPS with results obtained using PeptideProphet [10], a widely-used PSM filtering algorithm. The comparison was performed using the number of unique peptide identifications at 5% FDR. PeptideProphet probability was used for filtering datasets analyzed with PeptideProphet. Utilization of the STEPS methodology yielded 10.7% increase in true identifications for the S. oneidensis dataset (Figure 3B) and 17.5% increase for the serum dataset (Figure 3A). Significant overlap (Figure 3C and D) was observed between the two filtering approaches. When optimizing parameters, it is always a concern that the parameters will have been over-fit to the specific dataset used to train the parameters. To test for this, the serum dataset (10 LC-MS runs) was randomly divided in 2 parts (5 + 5 LC-MS runs), using one part to train the parameters and the second half as a test set. This analysis was repeated for 4 different partitions of the dataset. On average, only a 2.5% decrease in confident peptide identifications is seen between using the parameters trained on the dataset and applying STEPS parameters to a complimentary dataset, Supplemental Figure 3. This difference is much smaller than the difference between STEPS and PeptideProphet, 17.5% for the serum dataset.
Figure 3.
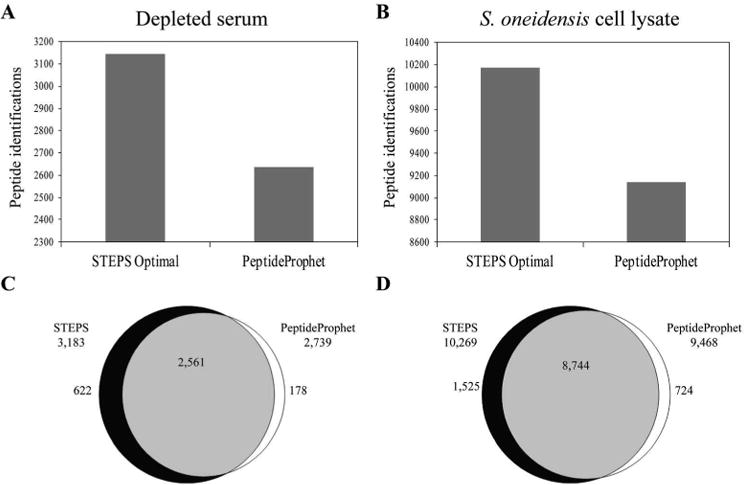
Comparison of confident peptide identifications obtained using STEPS and PeptideProphet at similar FDR. A) True identifications obtained for A) serum and B) S. oneidensis datasets. Overlap of peptide identifications between STEPS and PeptideProphet for C) depleted serum and D) S. oneidensis datasets.
Inspection of peptides that appeared in STEPS-optimized datasets only (data not shown) revealed peptides that had received low SEQUEST scores, but had high mass measurement accuracy. The majority of these identifications also have confident MS-GF scores, indicating they are legitimate identifications. Tables showing the optimal parameter sets obtained for the depleted serum and cell lysate datasets can be seen in Supplemental Tables 2 and 3, respectively.
To conclude, STEPS methodology represents a conceptually-simple, flexible, and highly discriminative approach to PSM filtering. Implemented using the automated processing engine APE, all data manipulations are visible and modifiable, which allows the analyst to create custom data models. As such, the STEPS methodology can be readily adapted to datasets from any sample type, mass spectrometer, and search engine. By leveraging more of the information available to the analyst, STEPS allows for greater retention of true positive matches from proteomics datasets. This becomes especially significant when filtering PSM's from very complex samples, as in the study of microbial communities[23]. Because of its inherent flexibility and simplicity, STEPS-based methodologies should have widespread applicability in the field.
Supplementary Material
Supplemental Figure 1: Example mass error histogram plot output by APE.
Supplemental Figure 2: Screenshot from APE showing the workflow layout for a STEPS process.
Supplemental Figure 3: Cross-Validation of serum dataset filtering with STEPS. For all comparisons, 5 datasets were used to train filtering parameters (grey bars) and these parameters were then applied to the remaining 5 datasets (black bars). An average of 2.5% reduction in confident peptide identifications was seen when applying the parameter sets developed to the test datasets when compared with using the dataset to train the parameters.
Supplemental Table 1: Ranges and step sizes used to generate iteration tables for Figure 2 comparisons.
Supplemental Table 2: Optimal filtering parameter table for depleted serum datasets generated by STEPS.
Supplemental Table 3: Optimal filtering parameter table for cell lysate datasets generated by STEPS.
Acknowledgments
This work was supported by NIH grant P01 DA026134 (to RDS). The software and STEPS methodology were developed in the NIH NCRR P41 Biomedical Technology Research Center for Proteomics (RR018522 to RDS). Portions of this research were supported by the U.S. Department of Energy's (DOE) Office of Biological and Environmental Research (OBER) Pan-omics program. This research was performed at the W.R. Wiley Environmental Molecular Science Laboratory (a national scientific user facility sponsored by the U.S. Department of Energy's Office of Biological and Environmental Research and located at PNNL). Pacific Northwest National Laboratory is a multiprogram national laboratory operated by Battelle for the DOE under Contract DE-AC05-76RL01830. The authors gratefully acknowledge Nancy Colton and Dr. Samuel Payne for critical reading of the manuscript.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson PL, Smith RD. Proteome analysis by mass spectrometry. Annu Rev Biophys Biomolec Struct. 2003;32:399–424. doi: 10.1146/annurev.biophys.32.110601.141854. [DOI] [PubMed] [Google Scholar]
- 3.Cravatt BF, Simon GM, Yates JR. The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 4.Yates JR, Eng JK, McCormack AL, Schieltz D. Method To Correlate Tandem Mass-Spectra Of Modified Peptides To Amino-Acid-Sequences In The Protein Database. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 5.Fenyo D, Beavis RC. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal Chem. 2003;75:768–774. doi: 10.1021/ac0258709. [DOI] [PubMed] [Google Scholar]
- 6.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Kall L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. Journal of Proteome Research. 2008;7:29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. Journal of Proteome Research. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 11.Qian WJ, Liu T, Monroe ME, Strittmatter EF, et al. Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: The human proteome. Journal of Proteome Research. 2005;4:53–62. doi: 10.1021/pr0498638. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Gupta N, Pevzner PA. Spectral probabilities and generating functions of tandem mass spectra: A strike against decoy databases. Journal of Proteome Research. 2008;7:3354–3363. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petyuk VA, Mayampurath AM, Monroe ME, Polpitiya AD, et al. DtaRefinery, a Software Tool for Elimination of Systematic Errors from Parent Ion Mass Measurements in Tandem Mass Spectra Data Sets. Mol Cell Proteomics. 2010;9:486–496. doi: 10.1074/mcp.M900217-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 15.Tabb DL. What's driving false discovery rates? Journal of Proteome Research. 2008;7:45–46. doi: 10.1021/pr700728t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 17.Pieper R, Su Q, Gatlin CL, Huang ST, et al. Multi-component immunoaffinity subtraction chromatography: An innovative step towards a comprehensive survey of the human plasma proteome. Proteomics. 2003;3:422–432. doi: 10.1002/pmic.200390057. [DOI] [PubMed] [Google Scholar]
- 18.Liu T, Qian WJ, Mottaz HM, Gritsenko MA, et al. Evaluation of multiprotein immunoaffinity subtraction for plasma proteomics and candidate biomarker discovery using mass spectrometry. Mol Cell Proteomics. 2006;5:2167–2174. doi: 10.1074/mcp.T600039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livesay EA, Tang KQ, Taylor BK, Buschbach MA, et al. Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal Chem. 2008;80:294–302. doi: 10.1021/ac701727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayampurath AM, Jaitly N, Purvine SO, Monroe ME, et al. DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics. 2008;24:1021–1023. doi: 10.1093/bioinformatics/btn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zubarev R, Mann M. On the proper use of mass accuracy in proteomics. Mol Cell Proteomics. 2007;6:377–381. doi: 10.1074/mcp.M600380-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Ma ZQ, Dasari S, Chambers MC, Litton MD, et al. IDPicker 2.0: Improved Protein Assembly with High Discrimination Peptide Identification Filtering. Journal of Proteome Research. 2009;8:3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aylward FO, Burnum KE, Scott JJ, Suen G, et al. Metagenomic and metaproteomic insights into bacterial communities in leaf-cutter ant fungus gardens. The ISME journal. 2012:1–14. doi: 10.1038/ismej.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Example mass error histogram plot output by APE.
Supplemental Figure 2: Screenshot from APE showing the workflow layout for a STEPS process.
Supplemental Figure 3: Cross-Validation of serum dataset filtering with STEPS. For all comparisons, 5 datasets were used to train filtering parameters (grey bars) and these parameters were then applied to the remaining 5 datasets (black bars). An average of 2.5% reduction in confident peptide identifications was seen when applying the parameter sets developed to the test datasets when compared with using the dataset to train the parameters.
Supplemental Table 1: Ranges and step sizes used to generate iteration tables for Figure 2 comparisons.
Supplemental Table 2: Optimal filtering parameter table for depleted serum datasets generated by STEPS.
Supplemental Table 3: Optimal filtering parameter table for cell lysate datasets generated by STEPS.