

Abstract
Our efforts in constructing the ABD-ring of phomactin A through an intramolecular oxa-[3 + 3] annulation strategy is described. This struggle entailed finding a practical and efficient preparation of annulation precursor, and a realization of the unexpected competing regioisomeric pathway. The success entailed accessing the A-ring through Diels-Alder cycloaddition of Rawal’s diene. Furthermore, the discovery that the regioisomers from the annulation existed as atropisomers with respect to the D-ring olefin and that they could be equilibrated to the desired ABD-tricycle, allowing large quantities of tricycle to be accessed.
Keywords: Phomactin A, ABD-tricycle, intramolecular oxa-[3 + 3] annulation, atropisomers, Rawal’s Diels-Alder Cycloaddition
1. Introduction
In 1991, as part of a program to isolate and identify new platelet activating factor [PAF]1 antagonists from marine sources, Sugano and Sato et al. reported the isolation and structure of (+)-phomactin A [Figure 1].2 (+)-Phomactin A was extracted and purified from the culture filtrate of a marine fungus, Phoma sp. [SANK 11486], a parasite which was collected from the shell of a crab, Chionoecetes opilio that was found off the coast of the Fukui prefecture in Japan. It was determined to have a moderate PAF aggregation inhibitory ability [IC50 = 10 μM]. The rough structure of (+)-phomactin A was determined using 1H and 13C NMR methods as well as IR and HRMS techniques. The authors were unable to assign the complete structure, and were forced to resort to crystallographic methods. Since (+)-phomactin A was reported as an oil, the C3-para-bromobenzoate derivative was prepared and a suitable crystal was obtained [Figure 1]. The crystal structure is of low quality, but clearly showed the unusual ABCD-tetracyclic topology of (+)-phomactin A, and also determined the absolute stereochemistry. Subsequently, nine additional phomactins were isolated from various fungal sources with many of them displaying anti-PAF activity:3–5 B,3 B1,3 B2,3 C3 [or Sch 479186], D,3 E,4 F,4 G,4 and finally, H5 [Figure 2].
Figure 1.

(+)-Phomactin A and Its C3-Para-Bromobenzoyl Ester.
Figure 2.

The Phomactin Family and the Sch Structures.
Intriguingly, Chu et al. at Schering-Plough independently reported the isolation of Sch 47918 in 1992, which is identical to (+)-phomactin C, although the opposite antipode is shown in the paper.6 In 1993, Chu et al. also reported structures of (+)-Sch 49026, (+)-Sch 49027, and (+)-Sch 49028.7 However, based on Pattenden’s work,8 Sch 49028 was mis-assigned, and in fact, it is now believed that it should actually be phomactin A. In addition, when Oikawa9 isolated phomactatriene [or Sch 49026: see Figure 3], the source from which all phomactin members are believed to originate biosynthetically, the C5a stereochemistry of (+)-Sch 49026 was corrected as shown, as it should be (S) and not (R). As a family, the phomactins posses an unusual and highly oxygenated diterpenoid molecular architecture, and including phomactin A, they all consist of a novel bicyclo[9.3.1]pentadecane core [red in (+)-Sch 49026]. Notably, Oikawa and co-workers used an elegant 13C-labeling study that involved incubation of Phoma sp. with 1-13C and 1,2-13C acetate to reveal the biosynthesis of phomactins with phomactatriene being a key biosynthetic intermediate commencing from geranylgeranyl diphosphate (GGDP) cyclization.9
Figure 3.

Challenging Features in (+)-Phomactin A.
Although not the most potent member of the family, phomactin A is the most challenging one structurally, as it has several key hurdles built into its architecture. The most challenging fragments are highlighted in Figure 3. In Box-A, the highly sensitive hydrated furan is prone to dehydration under acidic or basic conditions, and any total synthesis almost certainly must save introduction of this fragment until the end-game. Box-B relates to the strained, and somewhat twisted electron-rich double bond. This trisubstituted olefin is extremely reactive towards electrophilic oxidants. Additionally, we believe that the olefin is not free to rotate, and rotamer issues may arise in an attempted synthesis.
In Box-C, the cis-methyl groups at C6 and C7 are highlighted with C6 being the hindered quaternary center. This center must be installed at an early time, and will obstruct attempts to functionalize centers nearby. It is noteworthy that many conformations of phomactin A will always have one methyl group assuming an axial position, severely hindering any reagent approach to the A-ring from the top face, while the belt [we will refer to the C1′–C6′ portion as the belt – in blue] effectively blocks the bottom face. Lastly, Box-D showcases the unusual 12-membered [D-ring] bridged/fused ring system, which only serves to make the system more challenging; that fact would be thoroughly demonstrated through the work of other groups, as well as our own.
In the last fifteen years, the challenging structure of the phomactins, in particular phomactin A, coupled with the interesting biological activity10–15 has elicited an impressive amount of attention from the synthetic community.16–18 The most active member (+)-phomactin D was first synthesized by Yamada in 1996,19 and in 2007, Wulff20 reported the synthesis of (±)-phomactin B2. However, (+)-phomactin A seems to be the most popular target, presumably due to the additional stereochemical complexity and strain introduced by the presence of the 1-oxadecalin. To date, monumental total syntheses have been accomplished in racemic form by the Pattenden group21 at University of Nottingham beginning in the early 1990’s and enantioselectively by the Halcomb group22 at University of Colorado at Boulder in 2002 and 2003. Although details will not be described here,16,17 certain aspects of these two successful syntheses of phomactin A as well as many of the approaches have mimicked Yamada’s synthesis of D, thereby underscoring the remarkable influence of Yamada’s earlier work on the phomactin chemistry. After completing their synthesis of (±)-phomactin A, Pattenden23 completed a total synthesis of (±)-phomactin G via a modified route used for A. We wish to report here details of our efforts in constructing the ABD-tricycle of phomactin A.
2. Results and Discussions
2.1. Retrosynthetic Analysis
More than nine years ago, we commenced our own approach toward (±)-phomactin A with the simple intent to feature an oxa-[3 + 3] annulation strategy24–28 that was developed in our lab,29–32 and in particular, an intramolecular version of this annulation. While oxa-[3 + 3] annulations or related reaction manifolds33,34 are very well known and can be traced back more than six decades,35 an intramolecular variant of this reaction was not known.24–28 There were no applications of intramolecular oxa-[3 + 3] annulations in natural product synthesis27,28,36 until our approach toward phomactin A was disclosed. Subsequently, an account toward (±)-likonide B was reported.36i
More specifically, as shown in Scheme 1, we envisioned that phomactin A could be elaborated through C5a-homologation and C-ring closure of 1, which could be attained via a sequence of oxidative and reductive transformations involving the key ABD-tricycle 2. From the onset, we recognized that 2 possesses a unique structural topology. A Spartan™ model reveals a highly strained caged motif with the plane of the AB-ring being in close proximity with the D-ring olefin at C3′. Yet, our retrosynthetic assessments appeared to be very reasonable with the exception of the C5a-homologation, which would involve addition to a quite hindered C5a-carbonyl group. Finally, we used an intramolecular oxa-[3 + 3] annulation of an in situ generated vinyl iminium salt 3 to access ABD-tricycle 2.
Scheme 1.

The Hsung Group Approach to Phomactin A.
Mechanistically, the intermolecular oxa-[3 + 3] annulation of diketone 437 with α,β–unsaturated iminium ion 5 involves a tandem cascade38 of Knoevenagel condensation–6π–electron electrocyclic ring-closure of 1-oxatrienes 639–41 to give pyranyl heterocycles 7 [Scheme 2].42 This tandem sequence can be considered as a stepwise formal [3 + 3] cycloaddition43–45 in which two σ bonds are formed in addition to a new stereogenic center adjacent to the heteroatom, thereby representing a biomimetic strategy in natural product synthesis,36,40a and revealing the power of oxa-6π pericyclic ring-closures in organic synthesis that can find its precedent six decades ago.35 The challenge in this and other related annulations is the regiochemical control,24–28 head-to-head versus head-to-tail [see the box], which can be unpredictable and lead to complex mixtures.46 However, the use of α,β–unsaturated iminium salts in our annulations has led to head-to-head regioselectivity almost exclusively.24–28
Scheme 2.

Regiochemical Issues in Our Synthetic Approach.
The intramolecular version of this annulation could entail a different sequence consisting of O-1,4-addition followed by C-1,2-addition/β–elimination to give the desired ABD-tricycle 2. Like the intermolecular annulation, the intramolecular variant also represents an attractive sequential or domino pathway.34 In the context of phomactin A synthesis, approaching the D-ring construction through the usage of this intramolecular annulation strategy would further accentuate the synthetic prowess of the oxa-[3 + 3] annulation. In particular, upon evaluating all the synthetic approaches reported,16,17 the macrocyclic D-ring was constructed: (1) through ring-closing metathesis [RCM], sulfone alkylation, acetylenic addition, Julia olefination, Nozaki-Hiyama-Kishi [NHK], Stille, or Suzuki cross coupling, which is different from our strategy; and (2) at late stages of the synthesis. Thus, our approach would be unique, as the D-ring would be assembled at an early stage of the synthesis. However, we did overlook the equally plausible regiochemical orientation in the oxa-[3 + 3] annulation as shown in 3B, which could lead to disaster; and it was without recognizing such a possibility in our initial retrosynthetic assessment, that we commenced our endeavor.47–50
2.2. Initial Approaches to the Key Diketo-Enal 8
In our initial routes, we exert ed much effort to develop a direct approach toward the key diketo-enal 8 through 1,4-conjugate-addition/trapping with iodide 9 as shown in Scheme 3. However, these efforts did not lead to a fruitful outcome in terms of a concise and stereoselective synthesis of 8.51 Ultimately, in our third generation synthesis, instead of the belt being appended to the cyclohexanyl A-ring portion in a single operation as in the earlier approaches [first and second generations51], it would be stitched together at the C4′-C5′ bond using a B-alkyl Suzuki coupling of vinyl iodide 11 and borane 12. The former would come from 13 derived from a methyl-cuprate conjugate addition followed by trapping with allyl iodide. After these two pieces were joined, we could then investigate oxygenation of the cyclohexanone A-ring in ketone 10 to generate the required diketone motif in 8. These details have been described in our preliminary communication.47,51 While the entire endeavor took 22 steps from 2-methyl-cyclohexanone, it allowed us to examine carefully the key oxa-[3 + 3] annulation reaction.
Scheme 3.

Initial Approaches to the Key Diketo-Enal 8.
2.3. Annulation, Atropisomerism, and Equilibration
The key intramolecular oxa-[3 + 3] annulation reaction of diketo-enal 8 was attempted under high dilution conditions simply at room temperature using piperidinium acetate salt in THF. We were delighted to observe the formation of the desired annulation product 2, along with what we believed to be regioisomeric products 13a and 13b* arriving from annulation of 3B [Scheme 4].
Scheme 4.

Intramolecular Oxa-[3 + 3] Annulation of Diketo-Enal 8.
The undesired regioisomers 13a and 13b* were formed as an inseparable 4:1 mixture, which we were initially unable to assign. The overall ratio for 2:[13a + 13b*] ranged between 1:2.2 and 1:3.0, thereby implying that the desired ABD-tricycle in this annulation is actually the minor product. Although the overall yield of the reaction was 76%, we were disappointed by the poor ratio of desired cycloadduct to the isomeric products and the overall yield of the annulation varied with the low end being 35%.
The three annulation products, 2, 13a and 13b* would presumably be derived from the three respective transition states shown in Figure 4. However, when we obtained relative energies of 2, 13a and 13b* employing HF6-31G* and B3LYP6-31G* calculations using Spartan™–2002, we found a disconcerting result. That is, while the relative energetics appeared to be in the right order, the proposed minor diastereomer 13b* with the allylic [C3′] Me group pointing inward possesses a very high energy. This promoted us to very carefully examine the assignment of isomers 13a and 13b*, as we recognized the significant implication of being correct in these assignments to our ultimate total synthesis efforts.
Figure 4.

Three TS for the Key Oxa-[3 + 3] Annulation.
Toward this end, epoxidation of a mixture of 13a and 13b* mixture with m-CPBA proceeded in quantitative yield to give the mixture of epoxides 14a and 14b, which could be painstakingly separated via silica gel column chromatography to afford pure 14a and 14b, as white solids [Scheme 5]. Interestingly, 14b displayed strong fluorescence under a shortwave UV lamp; to our knowledge, no other compound of this type has ever exhibited this fluorescence.
Scheme 5.

Epoxidation and Reassignment of Regioisomers.
Attempts to assign the major epoxide 14a using an nOe experiment failed, as the 1H NMR of 14a suffered from a conformational exchange in solution, which caused line broadening. When the sample of 14a was heated to 80 °C in toluene d8, a suitable 1H NMR spectrum was obtained, but even at this temperature, the 13C lines were too broad to be observed. Fortunately, we were able to obtain crystals of 14a; which were suitable for X-Ray analysis [see the box, Scheme 5].52 On the other hand, the structure of minor epoxide 14b could be readily assigned using nOe experiment. Key nOe’s between H3a and H4′ in 14b unambiguously revealed that the proposed assignment of 13b* was likely incorrect. Epoxides 14a and 14b have the same stereochemistry at C2 but differ in the relative stereochemistry of the epoxide, thereby suggesting that different olefinic faces were exposed to epoxidation. This implies that diastereomeric tricycles 13a and 13b are atropisomeric in nature at the trisubstituted belt olefin [the D-ring], and that these tight belts are conformationally locked and do not equilibrate or freely rotate. The correct assignment should be 13b and not 13b*.
While the discovery of this novel D-ring atropisomerism reveals the unique and intricate architecture of these tricycles, it alerted us to the actual alignment of the C3′ allyl methyl group in the desired ABD-tri cycle 2, and more importantly, the vigor of its assignment. Thus, we turned our attention to the desired ABD-tri cycle 2 and the initial assignment of 2, which was not a trivial matter. The desired ABD-tricycle 2 existed as a single belt olefin rotamer and was observed to have a large Rf difference [less polar] from the mixture of undesired 13a and 13b. This can be explained by the presence of the quaternary center adjacent to the most Lewis basic site [the carbonyl] in the case of 2. While no crystal structure could be attained at this stage, interesting correlations using nOe on the single isomer 2 would strongly support our structural assignment and its respective transition state TS-2 for the annulation [Figure 5]. In addition, we observed a long range 1H-1H COSY interaction between the C3a vinyl proton and the C8 allylic protons; an interaction that is impossible in regioisomers 13a and 13b, due to the position of the quaternary center.
Figure 5.

Assignment of 2 and Energetics of Possible Atropisomers.
Further HF6-31G* and B3LYP6-31G* calculations would suggest that the C3′ Me group of 2 is also pointing in as in the assignment of 13a and 13b. The atropisomer of 2 [see model 2′ in Figure 5] is 6.61 kcal mol−1 less stable. Additionally, the C3′ atropisomer of the originally assigned 13b* possesses an even higher energy [11.08 kcal mol−1!!], the actual 13b, atropisomeric with 13a, is only 2.53 kcal mol−1. Consequently, we were pleased with the knowledge that we had obtained the three most stable isomers in the annulation according to our earlier calculations. Whether these other atropisomers were ever formed, or were simply too costly or unstable to form at room temperature, we did not screen for their existence.
Lastly and most importantly, when the undesired isomers 13a/b were stirred with roughly 1.0 equiv of piperidinium acetate in CH2Cl2 overnight, and subsequently, treat ed with excess Ac2O, we were able to obtain a mixture of 2 and 13a/b in the 1.7:4:1 ratio with an efficient 87% isolated yield. Mechanistically, we believe the equilibration proceeds through pericyclic ring-opening41 of 13a and 13b to give 1-oxatriene 15B, and the overall isomerization from 15B to 1-oxatriene 15A would require a sequence of 1,4-addition of piperidine, bond roatation, and β–elimination. This equilibration discovery helped to provide us with precious annulation product 2, which was much needed for the next major investigation leading to total synthesis of phomactin A.
2.4. A Short and Concise Route to Diketo Enal 8
Having definitively established the feasibility of this intramolecular oxa-[3 + 3] annulation, we recognized that the 22-step synthetic route to diketo-enal 8 [the third generation synthesis: see section 2.2] was too lengthy, and that another route would be required if we were to succeed in a total synthesis of phomactin A. This third generation route faltered after the side-chain was attached because oxidation of the cyclohexane moiety was impractical using the methods we attempted. Thus, a synthesis of the A-ring that incorporated as much oxygenation as possible was desirable, so as to avoid the difficult process of installing it at a later time. In addition, we felt that it would be best to form the diketone moiety in an already protected fashion and unmask it at the end. Consequently, our fourth generation approach to diketo-enal 8 began with the use of a Diels-Alder reaction to form the A ring with the added possibility of achieving an asymmetric manifold.
The revised retrosynthesis is shown in Scheme 7. While the A-ring core 19 would be constructed through a Diels-Alder cycloaddition of tiglic aldehyde and diene 21, the C2′-C3′ bond would be connected through a α-alkyl Suzuki-Miyaura reaction similar to the intramolecular one adopt ed in Halcomb’s successful total synthesis.22 We were quite excited by the convergence of this approach, and the amount of oxygenation that could potentially be brought into the Suzuki coupling.
Scheme 7.

The Fourth Approach to Diketo-Enal 8.
We quickly found that Diels-Alder cycloaddition of tiglic aldehyde with Danishefsky’s diene proceeded only at high temperature, and after workup, a mixture of products was seen including β–methoxyketone 22, which proved to be somewhat difficult to eliminate to the desired enone 20, and 4-hydroxy-acetophenone 23 via dimerization of the diene. The potential use of Totah’s diene18g would have been very attractive because of the diketone oxidation state resulting from the cycloaddition. However, we were concerned that this functionality might not survive installment of the side chain. Instead, we turned to Rawal’s diene.53
With Rawal’s diene, the cycloaddition occurred with remarkable ease at 50 °C in THF, and the reaction was observed to slowly occur even at low temperature [−10 °C]. This high level of reactivity towards a rather unreactive and/or hindered dienophile is presumably due to the dimethylamino group being more electron donating [significantly raising its HOMO level] relative to Danishefsky’s diene.54–56 In this fashion, high yields of the desired aminocyclohexene 25 were obtained. This key reaction sets the stereochemistry and provides us with extensive A-ring oxygenation. It was also observed that 25 was reasonably stable to moisture and could be manipulated to a certain extent, but it was unstable to chromatography and underwent clean elimination to give the enone under acidic conditions. We were pleased to find that Wittig olefination of the crude cycloadduct 25 proceeded with ease to give an alkene that was not isolated but exposed to HF to cleanly afford enone 20 in 72% yield from Rawal’s diene.
To introduce oxygenation, nucleophilic epoxidation using basic hydrogen peroxide cleanly afforded α–epoxide 26 as the only observed isomer [Scheme 9]. Subsequently, we began to investigate the epoxide opening and protecting group chemistry to access several possible candidates in anticipation of the Suzuki-Miyaura coupling. While direct reduction of 26 with LAH was unselective and afforded a mixture of 1,2 and 1,3-diols, NaBH4 reduction was also not selective and afforded a 1:1 mixture of α and β alcohols at C8a. Ultimately, Luche reduction of epoxide in 26 cleanly afforded β–alcohol 27. TES protection then afforded silyl ether 28. β–Alcohol 27 could also be protected as PMB ether 29 and reduced and protect ed as a TES ether to give 30.
Scheme 9.

Various Transformations of the Cycloadduct 20.
Finally, while the protection of an α–epoxy ketone [such as 26] as a ketal seems to be an under-investigated area, we found that the protection proceeded under standard conditions to give 31. Selective reduction of ketal 31 then represents a daunting proposition, as the epoxide is flanked on both sides with quaternary centers. However, upon exposure to LAH in refluxing THF, a very clean and selective reduction took place to afford the desired alcohol 32, which was transformed to TES ether 33. Presumably, the ketal oxygen atoms provided a chelation opportunity, which serves to direct the reduction at the observed position.
We then turned our attention to the required coupling partner vinyl bromide 18. A number of approaches were investigated here before we opted to use more standard chemistry. As shown in Scheme 10, bromination of trans-crotyl alcohol [~95% trans by 1H NMR] proceeded smoothly to afford a dibromide as nearly a single isomer [~95%] after Kugelrohr distillation. The LDA-induced elimination of the dibromide via 34-Li was reported by Corey57 to be highly selective for vinyl bromide E-35.58 The elimination proved to be somewhat problematic in our hands, giving slightly lower yield than was reported, but did reliably afford the desired vinyl bromide E-35 after extensive purification. The crude 1H NMR was typically very complex, with significant amounts of starting material always being present, along with varying amounts of product resulting from the undesired direction of elimination 36 along with the undesired vinyl bromide Z-35. After isolation, the crude material was typically filtered through SiO2, distilled using a Kugelrohr, distilled through a 20-cm vacuum-jacket ed column, and finally, carefully chromatographed to afford highly pure material. The method was very tedious but was capable of providing large quantities of the precious bromide E-35.
Scheme 10.

Synthesis of Vinyl Bromide 18.
We did some investigatory work into possibly improving the reaction in terms of selectivity as well as efficiency. The use of one equivalent of NaH, followed by exposure to LDA produced almost exclusively the wrong product of elimination as 36. The use of LiH followed by LDA returned mostly starting material. Protection of the al cohol as a TMS ether followed by exposure to LDA, and subsequent hydrolysis, returned only starting dibromide. Mechanistically, we believe the E2 elimination is simply controlled stereoelectronically through the required conformation as shown in 34-Li. Deprotonation of the allylic alcohol renders the distal hydrogen [in red] more acidic, and the presence of HMPA discourages chelation controlled or directed deprotonation, leading to E-35 as the major product.
To extend the chain, we were pleased that bromination using a slight excess of PBr3 [0.37 equiv] to a solution of the alcohol at 0 °C in dry ether provided a high yield of dibromide 37, which possess lachrymatory properties. To extend the chain, we wished to use acetone enolate to displace the allylic bromide. In practice, this failed, giving no alkylated product when the lithium enolate of acetone was reacted with the bromide. We then examined the alkylation using the dianion of ethylacetoacetate, which worked very well in our case, giving β–ketoester 38 in 32% yield. Treatment of β–ketoester 38 with excess KOH in 95% EtOH at 80 °C induced decarboxylation to afford methyl ketone 39 in good overall yield. Subsequent Horner-Wadsworth-Emmons olefination of 39 afforded a 3.5:1 [E:Z] mixture of α,β–unsaturated esters 40, which at this point could be separated for purposes of characterization. Upon scale-up, separation was not performed, as the olefin geometry was not important to us. DIBAL-H reduction of the ester then afforded allylic alcohol that was protected with TBDPSCl to give vinyl bromide 18, which was quite stable and could be stored for months in the freezer.
Again, we turned our attention to the B-alkyl Suzuki-Miyaura coupling, albeit this time the A-ring has more oxygenation and its coupling partner is more structurally advanced. As shown in Scheme 11, coupling of vinyl bromide 18 was very successful with alkene 33 to give the desired cross-coupled product 41. Suzuki-Miyaura coupling of 18 with alkenes 28, 30, and 31 also worked well but we elected to go with product 41 for its potential in providing a concise synthesis of diketo-enal 8.
Scheme 11.
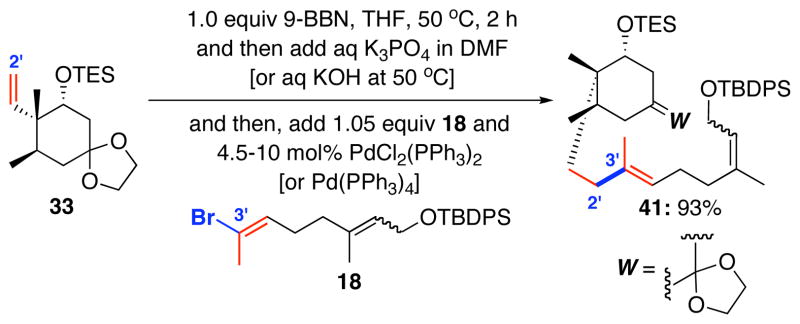
Suzuki-Miyaura Coupling of Vinyl Bromide 18.
Subsequently, the TES ether in 41 could be removed with moderate success and the corresponding alcohol oxidized to ketone 42 [Scheme 12]. We had hoped that we could eliminate the ketal and trap the resulting alkoxide as a stable vinylogous ester. In practice, elimination with KHMDS followed by exposure to MeI afforded a modest yield of the desired enone 43 along with what appeared to be the non-methylated byproduct. The TBDPS protected alcohol could then be cleaved with TBAF and oxidized to enal 44. Acidic hydrolysis then afforded the desired diketo-enal 8. We felt that this route was clever, but the additional length and the problematic elimination step led us to investigate other systems.
Scheme 12.

Completion of a 12-Step Synthesis of Diketo-Enal 8.
Ultimately, TBAF removal of both silicon ethers in 42 afforded diol 45, and an ensuing double Dess-Martin oxidation gave the enal intermediate. Finally, treatment of this enal with 1 N H2SO4 in acetone at 50 °C removed the ketal moiety and afforded the desired diketo-enal 8 as a mixture of enal isomers. The hydrolysis reaction was slow at room temperature, and when heated, the reaction was usually stopped with some starting material still present, due to the formation of an unknown by-product. Overall, at 12 linear steps, this route to diketo-enal 8 was much shorter than the third generation synthesis [22 steps], and was easily capable of supplying 8 in ≥10 gram quantities.
3. Conclusion
We have described here details of our struggle and ultimate success in constructing the ABD-ring of phomactin A through the use of an intramolecular oxa-[3 + 3] annulation strategy. The struggle entailed finding a practical and efficient preparation of annulation precursor, a definitive assessment of the annulation, and a realization of the unexpected competing regioisomeric pathway that almost terminated the total synthesis efforts. The success entailed (1) the construction of the A-ring through Diels-Alder cycloaddition of Rawal’s diene; and (2) the discovery that the regioisomers from the oxa-[3 + 3] annulation existed as atropisomers with respect to the D-ring olefin and could be equilibrated to the desired ABD-tricycle, allowing access to large quantity of the ABD-tri cycle practical for an eventual total synthesis endeavor.59 This work represents the first approach in which the 12-membered D-ring of phomactin A was constructed simultaneously with the 1-oxadecalin at an early stage; one that is also amenable for an enantioselective total synthesis49 through the use of Rawal’s asymmetric Diels-Alder cycloaddition.54–56
4. Experimental Section
All reactions were performed in flame-dried glassware under a nitrogen atmosphere. Solvents were distilled prior to use. Reagents we re used as purchased (Sigma-Aldrich, Acros), except where noted. Chromatographic separations were performed using Bodman 60 Å SiO2. 1H and 13C NMR spectra were obtained on Varian VI-300, VI-400, and VI-500 spectrometers using CDCl3 (except where noted) with TMS or residual CHCl3 in the solvent as standard. Melting points were determined using a Laboratory Devices MELTEMP and are uncorrected/calibrated. Infrared spectra were obtained using NaCl plates on a Bruker Equinox 55/S FT–IR Spectrophotometer, and relative intensities are expressed qualitatively as s (strong), m (medium), and w (weak).
TLC analysis was performed using Aldrich 254 nm polyester-backed plates (60 Å, 250 μm) and visualized using UV and a suitable chemical stain. Low- resolution mass spectra were obtained using an Agilent-1100-HPLC/MSD and can be either APCI or ESI, or an IonSpec HiRes-MALDI FT-Mass Spectrometer. High-resolution mass spectral analyses were performed at University of Wisconsin Mass Spectrometry Laboratories. All spectral data obtained for new compounds are reported.
4.1. Annulation, Atropisomerism, and Equilibration
4.1.1. The Intramolecular Oxa-[3 + 3] Annulation Procedure
Enal 8 (39.1 mg, 0.128 mmol) was taken up in THF (200 mL) and piperidinium acetate (37.2 mg, 0.256 mmol) was added. The solution was stirred for 18 h at room temperature. The following morning the THF was removed in vacuo and the residue subjected to flash chromatography (5– 15% EtOAc/hexanes) to afford the desired isomer 2 (8.4 mg, 23%) and a 4:1 mixture of isomers 13a and 13b (19.4 mg, 53%), which could only be unambiguously characterized through epoxides 14a and 14b.
Chromenone 2
Rf = 0.41 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) δ 1.02 (d, 3 H, J = 7.0 Hz ), 1.07 (s, 3 H), 1.38 (brm, 3 H), 1.47 (s, 3 H), 1.70 (ddd, 1 H, J = 3.0, 6.0, 15.0 Hz), 1.80 (td, 1 H, J = 3.5, 14.5 Hz ), 1.89 (ddd, 1 H, J = 3.5, 12.5, 15.0 Hz), 1.94 (ddd, 1 H, J = 2.0, 4.0, 14.5 Hz), 1.94 (dt, 1 H, J = 3.5, 14.0 Hz), 2.06 (ddd, 1 H, J = 1.5, 7.5, 14.5 Hz), 2.10 (d, 1 H, J = 19.5 Hz), 2.12(m, 1 H), 2.40–2.29 (m, 2 H), 2.87 (dd, 1 H, J = 6.5, 20.0 Hz), 4.99 (d, 1 H, J = 10.5 Hz), 5.15 (brd, 1 H, J = 10.5 Hz), 6.45 (d, 1 H, J = 10.5 Hz); 13C NMR (75 MHz, CDCl3) δ (ppm) 198.6, 169.1, 134.6, 125.3, 119.8, 118.0, 116.0, 82.9, 47.1, 46.1, 39.5, 37.0, 35.7, 34.5, 31.3, 25.2, 24.6, 20.0, 16.1; IR (ne at) cm−1 2976 (m), 2929 (m), 2901 (m), 2835 (w), 1640 (vs), 1607 (s), 1449 1449 (m), 1416 (s); ESI-MS (m/z) 309.2 (M+Na )+ (100), 197.0 (13); HRMS calcd for C19H26O2Na (M+Na )+ 309.1830; found 309.1818.
4.1.2. Characterization of Epoxides 14a and 14b to Assign Regioisomers 13a and 13b
The mixture of atropisomeric cycloadducts 13a and 13b (regioisomeric products of the oxa-[3 + 3] cycloaddition) (133.1 mg, 0.465 mmol) were taken up in CH2Cl2 (2.0 mL) and cooled to 0 °C with an ice bath. To the solution was added solid m-CPBA (77%, 112.1 mg, 0.50 mmol) was added in a single portion. TLC indicated consumption of starting material within five minutes, at which time dimethyl sulfide (100 μL) was added. It was noted that the product mixture seemed to form two separate spots in the TLC; the upper spot was observed to be highly fluorescent under the short wave UV lamp used for UV indication; the lower spot did not exhibit fluorescence whatsoever. The solution was diluted with CH2Cl2 (15 mL) and washed successively with 1 M NaOH (2.0 mL), water (5 mL), and brine (5 mL). The organic phase was dried with Na2SO4 and the crude 1H NMR indicated clean and quantitative conversion of both isomers to the corresponding epoxides. The mixture of isomers was partially separated using chromatography to afford analytical samples of 14a and 14b as white solids; the yields of which were not recorded.
Epoxide 14a
m.p. = 110–114 °C; Rf = 0.39 (60% EtOAc in hexanes); 1H NMR (300 MHz, toluene d8, 80 °C) δ 0.66 (d, 3H, J = 6.9 Hz), 0.81 (s, 3H), 1.06 (s, 3H), 1.10 (s, 3H), 1.20–1.41 (m, 5H), 1.50–1.60 (m, 3H), 1.61–1.72 (m, 2H), 1.76–1.86 (m, 2H), 2.04 (d, 1H, J = 17.1 Hz ), 2.37 (dd, 1H, J = 4.8, 17.1 Hz), 2.71 (d, 1H, J = 9.9 Hz), 4.61 (d, 1H, J = 10.2 Hz), 6.64 (d, 1H, J = 10.2 Hz); LRESIMS 627.3 (2M + Na)+ (35), 325.2 (M + Na) + (100), 303.2 (M + H)+ (93); HRESIMS calcd for C19H26NaO3 + 325.1774; found 325.1781, δ = 2.15 ppm.
Epoxide 14b
m.p. = 85–88 °C; Rf = 0.41 (60% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.91 (d, 3H, J = 6.5 Hz), 1.07 (dd, 1H, J = 10.5, 14.5 Hz), 1.08 (s, 3H), 1.36 (s, 3H), 1.39 (m, 1H), 1.39 (s, 3H), 1.70 (m, 2H), 1.93 (m, 3H), 2.20 (m, 2H), 2.28 (s, 1H), 2.29 (d, 1H, J = 2.0 Hz ), 2.73 (dd, 1H, J = 6.0, 9.0 Hz), 5.18 (d, 1H, J = 10.0 Hz ), 6.51 (d, 1H, J = 11.0 Hz); 13C NMR (75 MHz, CDCl3) δ 15.1, 17.3, 19.6, 26.1, 29.9, 30.5, 31.7, 33.2, 37.7, 41.4, 42.3, 60.3, 62.7, 84.1, 108.7, 117.3, 120.0, 175.1, 193.8; IR (film, cm−1) 2967 (m), 1653 (vs), 1584 (s), 1454 (m), 1413 (m); LRESIMS m/e (% rel. intensity) 929.5 (3M + Na)+ (30), 627.3 (2M + Na) + (100), 325.2 (M + Na)+ (22), 303.2 (M + H)+ (82); HRESIMS calcd for C19H26NaO3 + 325.1774; found 325.1780, δ = 1.85 ppm.
4.1.3. Equilibration Procedure
To a solution of 13a and 13b (3.55 g, 12.33 mmol) in CH2Cl2 was added piperidine (1.25 g, 14.77 mmol). This reaction mixture was stirred for 10 min at which point AcOH (0.90 g, 15.02 mmol) was added at rt, and allowed to stir for 16 h. The following day, Ac2O (7.57 g, 74.22 mmol) and piperidinium acetate salt (2.13 g, 14.69 mmol) were added to the reaction mixture. Once the reaction was deemed complete by TLC analysis, the mixture was quenched with H2O (50 mL) and most of the THF was removed by rotary evaporation. The resultant solution was diluted with EtOAc (300 mL) then washed with brine (100 mL), saturated aqueous NaHCO3 (100 mL), and again with brine (100 mL). The organic layer was dried with MgSO4 and concentrated to a crude residue that was purified by flash chromatography (5–15% EtOAc/hexanes) to afford the desired isomer 2 (0.97 g, 27%) and a mixture of isomers 13a and 13b (2.12 g, 60%).
4.2. A Short and Concise Route to Diketo Enal 8
4.2.1. Transformations of Cycloadducts
Enone 20
Rawal’s diene (49.97 g, 0.220 mol) was placed in a flame dried flask with the aid of ca. 25 mL of THF (for rinsing the diene’s container) along with tiglic aldehyde (Lancaster, 23.37 mL, 0.242 mol) and a magnetic stirbar. The flask was sealed with a rubber septem, evacuated, and filled with nitrogen. The flask was then lowered into a 50 °C oil bath and stirred at this temperature overnight. The following morning, a drop of the reaction mixture was taken for 1H NMR analysis. The analysis showed that the starting diene had been completely consumed (occasionally there was small amounts of diene remaining, in which case the reaction was heat ed for a few additional hours and re-checked). The stirbar was removed and toluene (25 mL) was added. The flask was placed on a hi-vacuum rotary evaporator (ca. 6 mmHg) and heated to approximately 40 °C while all volatiles were removed (mainly concerned about excess aldehyde). After 10 minutes, the flask was then exposed to high vacuum (ca. 0.20 mmHg) for 2 h.
While the aldehyde was under vacuum, a solution of methyl Wittig ylide was prepared by the addition of methyltriphenylphosphonium bromide (92.88g, 0.260 mol) to a flame dried 2 L 3-necked round bottom flask (joints: 2 × 24/40, 1 × 45/50) equipped with a nitrogen inlet, a vacuum line, a thermometer, and a large stirbar. Anhydrous THF (780 mL, quickly measured into a graduated cylinder under air) was added. The headspace was then flushed with nitrogen by periodically turning on the vacuum line. Stirring was begun, and the flask was immersed in an ice water bath and cooled. When the temperature dropped below 5 °C, n-butyllithium (2.55 M in hexane, 100.0 mL, 0.255 mol) was slowly added, maintaining the temperature below 5 °C, from an all-glass syringe. The mixture quickly turned bright yellow, and the solid slowly dissolved until only a small amount of salt remained. The color at this point was a bright, golden yellow. The ylide solution was stirred an additional 30 minutes and the temperature approached 0 °C.
The crude aldehyde 25 from above was sealed under nitrogen and THF (130 mL) was added. The flask was swirled to ensure complete salvation of the aldehyde and the solution was slowly transferred to the ylide solution via cannula. Occasionally, a thick white precipitate formed immediately upon the introduction of the aldehyde, but in this case no such precipitate formed immediately. When all of the aldehyde solution had been added, the flask was rinsed with additional THF (25 mL) and this was added to the ylide solution. The flask was removed from the ice bath, and allowed to warm to ambient temperature. After 1 h, a 0.7 mL aliquot was taken and placed in an NMR tube. 1H NMR analysis of the mixture indicated complete consumption of the aldehyde. (Occasionally, the aldehyde was not entirely gone; in this case, a solution of additional ylide was prepared and added to the reaction mixture.) Excess Wittig ylide was destroyed by addition of acetone (ca. 10 mL); this addition induced the formation of a massive amount of a thick, gooey, orange precipitate. The reaction mixture was then filtered through a coarse fritted funnel, and the volume reduced to 300 mL by concentration in vacuo. The remaining thick slurry was recombined with the filtered precipitate and placed in a 4-L beaker with hexanes (1 L) and water (1 L). The mixture was stirred and mashed with a large glass rod for 10–15 minutes as the solid material gradually became less gooey and more crystalline. Additional precipitate formed as well, this material being in the form of a powder. The entire mixture was then filtered again and the solid was placed in a triphenylphoshine oxide waste container. The water was drained and the organic layer was washed again with H2O (500 mL) and then brine (250 mL). The organic layer was dried with Na2SO4, and some additional precipitate was observed. Filtration and removal of the solvent afforded a crude alkene.
The above alkene was taken up in THF (220 mL) and cooled with an ice bath. A ca. 4 M solution of HF in THF (132 mL, 0.528 mol) was slowly added, the cooling bath removed, and the reaction stirred overnight. The following day, saturated NaHCO3 (100 mL) was slowly added and the mixture was stirred and transferred into a separatory funnel. MTBE (500 mL) was added, and the mixture was cautiously shaken with frequent venting. The aqueous layer was discarded, and the organic layer was washed with H2O (100 mL), brine (100 mL) and dried with Na2SO4. Concentration afforded a thick orange syrup which contained primarily a mixture of phosphine waste and the desired enone. The enone was purified by Kugelrohr bulb-to-bulb distillation (65 °C oven temperature, ca. 0.22 mmHg). A small amount of fore-run was noted, and the bulb was changed. This material consisted mostly of desired enone 20 contaminated with unknown impurities. The main fraction was taken, and the bath temperature was increased to 100 °C, causing an additional small amount of enone to distill. The enone was transferred to a tared flask with the aid of a minimal amount of CH2Cl2 and the solvent was removed in vacuo to afford pure enone 20 (23.80 g, 72% from Rawal’s diene) as a clear, light-yellow oil with a pleasant sweet/flowery odor. The distillation residue occasionally contained additional enone which could be obtained by filtration through silica gel with 20% EtOAc in hexanes. Rf = 0.23 (10% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.94 (d, 3H, J = 6.5 Hz), 1.11 (s, 3H), 2.12 (dqd, 1H, J = 4.0, 7.0, 11.0 Hz), 2.24 (dd, 1H, J = 11.0, 16.5 Hz), 2.42 (dd, 1H, J = 3.5, 16.5 Hz), 5.08 (dd, 1H, J = 1.0, 17.5 Hz), 5.15 (dd, 1H, J = 1.0, 11.0 Hz), 5.80 (dd, 1H, J = 11.0, 17.5 Hz), 5.94 (dd, 1H, J = 1.0, 10.5 Hz), 6.59 (d, 1H, J = 10.5 Hz); 13C NMR (125 MHz, CDCl3) δ 15.9, 18.0, 37.8, 42.2, 67.3, 114.6, 127.8, 144.6, 157.2, 200.0; IR (neat, cm−1) 2968 (s), 2880 (m), 1685 (vs), 1639 (m), 1457 (m), 1415 (m), 1387 (m).
Epoxide 26
Enone 20 (12.99g, 86.47 mmol) was taken up in MeOH (90.0 mL) and hydrogen peroxide (30% solution, 16.2 ml, 0.130 mol) was added. The solution was cooled to 0 °C, and powdered K2CO3 (1.20 g, 8.65 mmol) was added. The solution was allowed to warm to ambient temperature and after 4 h, UV activity on TLC had ceased. The solution was poured into H2O (200 mL) and extracted with MTBE (4 × 75 mL). The combined extracts were washed with 10% Na2S2O3 (100 mL), brine (100 mL), and dried with Na2SO4. Concentration and chromatography (10% EtOAc in hexanes) afforded epoxide 26 (12.79 g, 76.96 mmol, 89%) as a clear, colorless oil. Rf = 0.24 (10% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.81 (d, 3H, J = 7.0 Hz), 1.00 (s, 3H), 1.95 (dd, 1H, J = 11.5, 19.0 Hz), 2.25 (qd, 1H, J = 7.0, 14.0 Hz), 2.44 (dd, 1H, J= 6.0, 19.5 Hz), 3.25 (d, 1H, J = 4.0 Hz), 3.26 (d, 1H, J = 4.0 Hz), 5.24 (dd, 1H, J = 1.0, 18.0 Hz ), 5.26 (dd, 1H, J = 1.0, 10.5 Hz ), 5.97 (dd, 1H, J = 11.0, 17.5 Hz ); 13C NMR (125 MHz, CDCl3) δ 13.6, 14.4, 15.4, 29.5, 40.1, 41.5, 55.4, 63.6, 115.1, 143.4, 205.5; IR (neat, cm−1) 3085 (w), 2970 (s), 2882 (m), 1716 (vs), 1638 (m); LRESIMS m/e (% relative intensity) 301.2 (18), 256.1 (100), 233.1 (88), 221.1 (M + Na + MeOH)+ (90), 189.1 (M + Na)+ (11); HRESIMS calcd for C10H14NaO2 + 189.0886; found 189.0877, δ = 4.76 ppm.
β-Alcohol 27
Epoxide 26 (32.1 mg, 0.193 mmol) was taken up in MeOH (1.0 mL), CeCl3•7H2O (36.0 mg, 96.9 μmol) was added and the mixture was stirred until the cerium had dissolved completely. The solution was cooled to −78 °C and NaBH4 (14.6 mg, 0.386 mmol) was added in one portion. A white precipitate formed, and the mixture was stirred for 30 min, at which time TLC indicated complete consumption of starting material. The mixture was then warmed to ambient temperature and pipetted into a separatory funnel containing water (5 mL) and MTBE (10 mL). The mixture was shaken and the organic layer was then washed with brine and dried over Na2SO4. The crude 1H NMR indicated the presence of only one alcohol. The crude material was purified by chromatography to afford alcohol 27 (25.6 mg, 0.164 mmol, 85%) as a clear, colorless oil. Rf = 0.25 (40% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.77 (d, 3H, J = 7.0 Hz), 1.03 (s, 3H), 1.23 (ddd, 1H, J = 10.5, 12.5, 13.5 Hz), 1.56 (dqd, 1H, J = 2.5, 6.5, 13.5 Hz), 1.72 (dddd, 1H, J = 1.5, 2.5, 7.5, 13.5 Hz), 2.01 (br, 1H), 2.88 (d, 1H, J = 3.5 Hz), 3.11 (dd, 1H, J = 1.5, 3.5 Hz), 4.03 (dd, 1H, J = 7.0, 10.0 Hz), 5.16 (dd, 1H, J = 1.0, 18.0 Hz), 5.17 (dd, 1H, J = 1.0, 10.0 Hz), 5.87 (dd, 1H, J = 11.0, 17.5 Hz); 13C NMR (125 MHz, CDCl3) δ 13.9, 15.5, 28.5, 35.3, 39.9, 57.6, 62.7, 66.4, 114.3, 144.1; IR (neat, cm−1) 3414 (br), 3084 (w), 2968 (s), 2935 (s), 2879 (m), 1638 (m), 1463 (m), 1417 (m), 1381 (m), 1031 (s), LRESIMS m/e (% relative intensity) 391.2 (28), 207.1 (M + K)+ (7), 191.1 (M + Na )+ (100), 151.1 (M + H − H2O) + (8); HRESIMS calcd for C10H16NaO2 + 191.1042; found 191.1041, δ = 0.52 ppm.
Silyl Ether 28
β-alcohol 27 (42.1 mg, 0.25 mmol) was taken up in CH2Cl2 (1.0 mL) and imidazole (0.51 g, 0.75 mmol) was added. When the imidazole had dissolved, chlorotriethylsilane (62.9 μL, 0.38 mmol) was added and a white precipitate formed. After 18 h, the mixture was diluted with hexanes (10 mL) and washed with water (2 × 5 mL), brine (5 mL), and dried with Na2SO4. Concentration and chromatography (5% MTBE in hexanes) afforded silyl ether 28 (64.3 mg, 0.23 mmol, 91%) as a clear, colorless oil. Rf = 0.35 (5% MTBE in hexanes); 1H NMR δ 0.64 (q, 6H, J = 7.5 Hz ), 0.74 (d, 3H, J = 6.5 Hz ), 0.98 (t, 9H, J = 8.0 Hz), 1.02 (s, 3H), 1.28 (dt, 1H, J = 10.5, 13.0 Hz), 1.51 (dqd, 1H, J = 2.5, 7.0, 14.0 Hz), 1.57 (dddd, 1H, J = 1.5, 2.5, 6.5, 12.5 Hz ), 2.84 (d, 1H, J = 3.5 Hz), 3.05 (dd, 1H, J = 1.5, 3.5 Hz), 3.94 (dd, 1H, J = 6.5, 10.0 Hz), 5.14 (dd, 1H, J = 1.5, 18.0 Hz), 5.15 (dd, 1H, J = 1.0, 11.5 Hz), 5.87 (dd, 1H, J = 11.5, 18.5 Hz); 13C NMR (125 MHz, CDCl3) δ 4.9 (3C), 7.0 (3C), 13.9, 15.5, 28.7, 36.1, 40.0, 58.4, 62.8, 67.0, 114.0, 144.3; IR (neat, cm−1) 3085 (w), 2958 (s), 2912 (s), 2878 (s), 1638(w), 1460 (m), 1097 (s); LRESIMS m/e (% relative intensity) 321.2 (M + K) + (8), 305.2 (M + Na)+ (100), 256.1 (30), 233.1 (24); HRESIMS calcd for C16H30NaSiO2 + 305.1907; found 305.1910, δ = 0.98 ppm.
Silyl Ether 30
Alcohol 27 (1.26 g, 7.50 mmol) was placed in a 50 mL flask with a magnetic stir bar and dissolved in DMF (15 mL). Sodium hydride (60% in mineral oil, 0.45 g, 11.3 mmol) was added in one portion. Vigorous foaming ensued, and the solution turned cloudy. After 1 h, the flask was swirled by hand to ensure complete incorporation of reagents, and PMBCl (1.53 mL, 11.3 mmol) was added. The solution became very hot within 2 min and the reaction was completed within 0.5 h. The solution was poured into water (200 mL) and extracted with hexanes (4 × 35 mL). The combined organic extracts were washed with water (50 mL), brine (50 mL), and dried over Na2SO4. Evaporation of solvent followed by flash chromatography (gradient elution 5 to 10% EtOAc in hexanes) afforded epoxide 29 (1.79 g, 83%) as a clear, colorless oil.
Lithium aluminum hydride (195 mg, 5.14 mmol) was cautiously added through a plastic funnel into a 100 mL round bottom flask containing anhydrous THF (10 mL) at 0 °C. Special care was taken to avoid spilling any LAH into the ice bath. The funnel was rinsed with THF (25 mL) and the flask was fitted with a rubber septum. A 25 mL point-tipped flask was charged with epoxide 29 (0.99 g, 3.43 mmol) in THF (14 mL). This solution was slowly added to the 0 °C suspension of LAH via cannula; after the addition the flask was rinsed with an additional 2 mL of THF which was added to the LAH solution. A water-cooled condenser was fitted to the flask and the reaction was connected to a nitrogen line. The ice bath was replaced with an oil bath and the reaction was heated to reflux (ca. 75 °C bath temperature) overnight. The following morning, the reaction was cooled to 0 °C and removed from the condenser. Sulfuric a cid (3 M, 25 mL) and ice chunks (25 mL) were placed in a 500 mL beaker which in turn was placed in a bucket of ice water. The THF solution was then slowly poured into the beaker with constant stirring using a glass rod.
The quench was quite vigorous and the mixture grew warm (at which point more ice was added). When all of the THF solution had been added, the flask was rinsed with acid and EtOAc (5 mL each). The mixture in the beaker was stirred and more acid was added in 1 mL portions until all solids had dissolved. The mixture was then transferred to a 250 mL separatory funnel and the lower layer was drained and set aside. The top layer was added to a round bottom flask and concentrated until bumping became violent. The resulting biphasic syrup was transferred into the separatory funnel with the aid of EtOAc (25 mL). The aqueous layer was reintroduced and the mixture extracted with EtOAc (5 × 15 mL). The combined extracts were washed with water (1 × 15 mL), brine (2 × 15 mL), and dried with Na2SO4. The crude 1H NMR indicated that the material was sufficiently pure to proceed without purification, giving an intermediate alcohol as a clear, colorless oil. The material was quickly chromatographed through a short silica gel loaded column (20– 30% EtOAc in hexanes, gradient elution) to afford an intermediate alcohol.
The intermediate alcohol (0.88 g, 3.03 mmol) was taken up in CH2Cl2 (6 mL) and imidazole (0.41 g, 6.06 mmol) was added. When the imidazole had dissolved, chlorotriethylsilane (0.51 mL, 3.03 mmol) was added and a white precipitate formed. After 18 h, the mixture was diluted with hexanes (10 mL) and washed with water (2 × 5 mL), brine (5 mL), and dried with Na2SO4. Concentration and chromatography (5% MTBE in hexanes) afforded triethylsilyl ether 30 (1.10 g, 89%) as a clear, colorless oil.
Ketal 31
Epoxy-ketone 26 (19.77 g, 0.119 mol) was taken up in benzene (240 mL) and ethylene glycol (33.16 mL, 0.595 mol) was added, along with PPTS (2.99 g, 11.90 mmol) and the reaction was fitted to a Dean-Stark trap equipped with a water-cooled condenser. A heating mantle was fitted to the apparatus, and heating begun. Aluminum foil was wrapped around the Dean-Stark trap to help distillation of benzene/water azeotrope. The solution was vigorously refluxed for 18 hours. The following morning, the reaction was cooled and poured into water (100 mL). MTBE (240 mL) was added and, after shaking, the aqueous phase was removed. The organic phase was washed with water two additional times (100 mL). The organic layer was dried with brine (100 mL) and Na2SO4. Removal of the solvents and chromatography (15–25% EtOAc in hexanes) afforded ketal 31 (19.77g, 94.01 mmol, 79%) as a clear, colorless oil, along with recovered ketone 26 (1.78 g, 10.71 mmol, 9%). 31: Rf = 0.24 (20% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.72 (d, 3H, J = 7.0 Hz ), 0.97 (s, 3H), 1.51–1.57 (m, 2H), 1.77 (sextet, 1H, J = 7.0 Hz), 2.89 (d, 1H, J = 4.0 Hz), 3.02 (d, 1H, J = 3.5 Hz ), 3.93–4.01 (m, 2H), 4.03–4.06 (m, 1H), 4.10–4.13 (m, 1H), 5.15 (d, 1H, J = 16.5 Hz), 5.15 (d, 1H, J = 11.5 Hz), 5.88 (dd, 1H, J = 16.5 Hz); 13C NMR (125 MHz, CDCl3) δ 13.3, 15.3, 28.3, 37.8, 39.9, 55.6, 61.9, 64.8, 65.1, 104.6, 114.4, 143.9; IR (neat, cm−1) 3084 (m), 2968 (vs), 2883 (s), 1638 (m), 1460 (m), 1439 (m), 1421 (m); LRESIMS m/e (% relative intensity) 443.3 (2M + Na)+ (13), 233.1 (M + Na)+ (100); HRESIMS calcd for C12H18NaO3 + 233.1148; found 233.1141, δ = 3.00 ppm.
Alcohol 32
Lithium aluminum hydride (5.72 g, 150.87 mmol) was gradually and cautiously added through a plastic funnel into a 500 mL round bottom flask containing anhydrous THF (200 mL) at 0 °C. Special ca re was taken to avoid spilling any LAH into the ice bath. The funnel was rinsed with THF (25 mL) and the flask was fitted with a rubber septum. A 100 mL point-tipped flask was charged with ketal 31 (21.15 g, 100.58 mmol) in THF (50 mL). This solution was slowly added to the 0 °C suspension of LAH via cannula; after the addition the flask was rinsed with an additional 15 mL of THF which was added to the LAH solution. A water-cooled condenser was fitted to the flask and the reaction was connected to a nitrogen line. The ice bath was replaced with an oil bath and the reaction was heated to reflux (ca. 75 °C bath temperature) overnight.
The following morning, the reaction was cooled to 0 °C and removed from the condenser. Sulfuric acid (3 M, 200 mL) and ice chunks (200 mL) were placed in a 1 L beaker which in turn was placed in a bucket of ice water. The THF solution was then slowly poured into the beaker with constant stirring using a glass rod. The quench was quite vigorous and the mixture grew warm ( at which point more ice was added). When all of the THF solution had been added, the flask was rinsed with acid and EtOAc (50 mL each). The mixture in the beaker was stirred and more acid was added in 5 mL portions until all solids had dissolved. The mixture was then transferred to a 1 L separatory funnel and the lower layer was drained and set aside. The top layer was added to a round bottom flask and concentrated until bumping became violent.
The resulting biphasic syrup was transferred into the separatory funnel with the aid of EtOAc (100 mL). The aqueous layer was reintroduced and the mixture extracted with EtOAc (5 × 75 mL). The combined extracts were washed with water (1 × 50 mL), brine (2 × 50 mL), and dried (Na2SO4). On several occasions, the crude 1H NMR indicated that the material was sufficiently pure to proceed without purification, giving 32 (20.71 g, 97.56 mmol, 97%) as a clear, colorless oil. When impurities were noted, the material could be chromatographed (20–40% EtOAc in hexanes, gradient elution) to afford the pure alcohol 32. Rf = 0.12 (20% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.79 (d, 3H, J = 7.0 Hz), 0.90 (s, 3H), 1.85 (td, 1H, J = 3.0, 14.5 Hz), 1.96 (dd, 1H, J = 3.5, 14.5 Hz ), 2.08 (dqd, 1H, J = 4.0, 7.0, 12.5 Hz), 3.51 (td, 1H, J = 3.5, 9.5 Hz), 3.60 (d, 1H, J = 9.5 Hz), 3.92– 4.04 (m, 4H), 5.03 (dd, 1H, J = 1.5, 18.0 Hz), 5.14 (dd, 1H, J = 1.5, 11.0 Hz), 6.08 (d, 1H, J = 10.5, 17.5 Hz); 13C NMR (125 MHz, CDCl3) δ 14.4 16.2, 30.7, 36.2, 39.0, 43.6, 64.1, 64.7, 77.0, 109.5, 113.2, 145.9; IR (ne at, cm−1) 3523 (br), 3080 (m), 2965 (vs), 2890 (vs), 1637 (m), 1459 (s), 1416 (s), 1363 (s); LRESIMS m/e (% relative intensity) 447.3 (2M + Na )+ (14), 349.2 (19), 235.1 (M + Na )+ (100); HRESIMS calcd for C12H20NaO3 + 235.1305; found 235.1302, δ = 1.28 ppm.
Silyl Ether 33
Alcohol 32 (7.51 g, 35.38 mmol) was taken up in CH2Cl2 (70.0 mL) and imidazole (6.02 g, 88.45 mmol) was added. When the imidazole had completely dissolved, chlorotriethyl silane (7.13 mL, 42.46 mmol) was added over a one minute period. A white precipitate formed immediately, and the mixture was stirred at ambient temperature overnight. The mixture was poured into hexanes (300 mL) and washed with water (3 × 100 mL), brine (50 mL), and dried with Na2SO4. Purification via chromatography afforded silyl ether 33 (10.40 g, 31.84 mmol, 90%) as a clear, colorless oil. Rf = 0.39 (10% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.58 (q, 6H, J = 8.0 Hz), 0.83 (d, 3H, J = 7.5 Hz ), 0.93 (s, 3H), 0.96 (t, 9H, J = 8.0 Hz), 1.47 (ddd, 1H, J = 1.0, 9.5, 12.5 Hz), 1.73 (ddd, 1H, J = 1.5, 5.5, 13.5 Hz), 1.76 (ddd, 1H, J = 2.0, 4.5, 15.0 Hz), 1.83 (ddd, 1H, J = 1.0, 4.0, 13.5 Hz), 2.15 (dqd, 1H, J = 4.5, 7.0, 10.0 Hz), 3.67 (dd, 1H, J = 3.5, 6.0 Hz ), 3.87– 3.95 (m, 4H), 5.01 (dd, 1H, J = 1.5, 17.5 Hz), 5.05 (dd, 1H, J = 1.5, 11.0 Hz), 6.08 (dd, 1H, J = 10.5, 17.5 Hz); 13C NMR (125 MHz, CDCl3) δ 5.0 (3C), 7.0 (3C), 16.2, 17.3, 31.8, 38.4, 39.3, 43.6, 63.8, 64.3, 75.1, 109.2, 112.2, 145.7; IR (neat, cm−1) 3080 (w), 2964 (vs), 2879 (vs), 1637 (w), 1459 (s), 1416 (s), 1364 (s), 1302 (m); LRESIMS m/e (% relative intensity) 349.2 (M + Na)+ (100), 307.2 (11), 172.1 (32); HRESIMS calcd for C18H34NaSiO3 + 349.2169; found 349.2169, δ = 0.00 ppm.
4.2.2 Synthesis of Vinyl Bromide
Ketone 39
Crotyl alcohol (48.78 g, 0.676 mol) was placed in a 1 L 3-necked flask equipped with a thermometer and taken up in CH2Cl2 (670 mL). The solution was cooled to between − 40 and −30 °C using a dry ice/acetone bath and maintained at this temperature during the addition. Bromine (~35 mL) was added slowly until the orange color became persistent, at which time bromine addition was ceased and the solution was allowed to warm. A large amount of precipitate typically formed upon the completion of the addition, as the product crashed out. The solution was washed with thiosulfate until colorless, washed with brine, dried with Na2SO4, and concentrated. Kugelrohr distillation (air bath temperature 80– 85 °C, 0.20 mmHg) afforded the dibromide (137.47 g, 0.593 mol, 88%) as an off-white semi-solid. Rf = 0.39 (30% EtOAc in hexanes); 1H NMR (300 MHz, CDCl3) δ 1.91 (d, 3H, J = 6.6 Hz), 2.18 (s, 1H), 4.09 (br, 2H), 4.25 (td, 1H, J = 4.2, 8.1 Hz), 4.38 (qd, 1H, J = 6.6, 9.3 Hz).
A 5 L 3-necked flask was dried in an oven overnight and fitted with a thermometer, a vacuum/nitrogen inlet, and a dropping funnel. A solution of LDA was prepared in the flask by addition of n-butyllithium (2.15 M, 600 mL, 1.29 mol) to diisopropyl amine (191.0 mL, 1.37 mol) in THF (1.8 L). The temperature of the solution during the BuLi addition was maintained below 5 °C by means of a dry ice/acetone bath. After the last of the BuLi had been added, the solution was stirred at approximately 0 °C for 0.5 h. After 30 minutes, HMPA (52.0 mL, 0.297 mol) was added, and the solution cooled to below -70 °C. A solution of the dibromide (137.47 g, 0.593 mmol) in THF (1 L) was then slowly added, while the temperature of the solution was maintained below −70 °C. The addition took about 90 minutes. When the addition was complete, the solution was stirred and additional 2 hours and then MeOH (50 mL) was added, and the solution warmed.
Water (100 mL) was added, and the volume of the solution was reduced by 75% in vacuo. The mixture was then diluted with MTBE (1 L) and washed with 3 M HCl (200 mL portions) until the aqueous layer remained acidic. The solution was washed with brine and dried with Na2SO4. Concentration afforded a black oil which was filtered through a bed of silica gel, eluting with 40% EtOAc in hexanes. Concentration afforded an orange oil which was distilled in a Kugelrohr apparatus (bath temp 110 °C, 15 mmHg). This material was then distilled on a 20 cm vacuum jacketed column at 15 mmHg and the forerun was discarded. Finally the material was carefully chromatographed (25–45% EtOAc in hexanes) to afford pure bromide E-35 (34.99 g, 0.232 mol, 39%) as a colorless oil. Rf = 0.20 (30% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 1.47 (t, 1H, J = 5.5 Hz), 2.37 (s, 3H), 4.20 (t, 2H, J = 6.0 Hz), 6.18 (t, 1H, J = 7.5 Hz).
Bromide E-35 (34.99 g, 0.232 mol) was taken up in anhydrous ether (230 mL) and cooled to 0 °C. Phosphorus tribromide (8.14 mL, 85.84 mmol) was slowly added over a 5 minute period. After 15 minutes, TLC indicated complete consumption of the starting material, and the mixture was partially concentrated on a rotary evaporator (caution, stench) to a light yellow syrup.
The syrup was taken up in 10% ether in pentane and filtered through a 5-inch bed of silica with the ether pentane solvent mixture. The top layer of the silica turned dark orange, but the filtrate was colorless. Concentration afforded dibromide 37 (45.68 g, 0.213 mol, 92%) as a colorless oil with a potent, irritating, and persistent odor. Rf = 0.39 (hexanes); 1H NMR (500 MHz, CDCl3) δ 2.32 (d, 3H, J = 1.5 Hz ), 3.90 (d, 3H, J = 9.0 Hz), 6.18 (qt, 1H, J = 1.5, 8.5 Hz); 13 C NMR (125 MHz, CDCl3) 23.1, 27.0, 127.0, 127.7; IR (neat, cm −1) 1641 (vs), 1204 (vs).
Ethyl acetoacetate (0.77 mL, 6.09 mmol) was slowly added to a suspension of sodium hydride (60% in mineral oil, 0.26 g, 6.55 mmol) in THF (19 mL) at 0 °C. After foaming had subsided, the solution was cooled to −78 °C, and BuLi (2.41 M, 2.52 mL, 6.08 mmol) was added. After 30 minutes, dibromide 37 (1.00 g, 4.68 mmol) in THF (5 mL) was added. The solution was stirred for 30 minutes, and warmed. The solution was diluted with EtOAc (50 mL) and washed with 1 M HCl (25 mL). The organic phase was dried with brine and concentrated to a crude syrup which consisted primarily of β-ketoester 38 [Rf = 0.29 (20% EtOAc in hexanes)] and ethyl acetoacetate, and was not purified further. Rf = 0.29 (20% EtOAc in hexanes).
The crude β-ketoester 38 (prepared from 1.00 g of 37) was taken up in 95% EtOH (15 mL) and KOH (1.54 g, 23.40 mmol) was added. The solution was heated to reflux for 2 h. Removal of most of the EtOH on a rotary evaporator, dilution with MTBE (50 mL), washing with water (4 × 25 mL), brine (25 mL), and drying with Na2SO4 afforded the crude ketone 39 (0.71 g, 79% for 2 steps), which was not purified further. An analytical sample was prepared by column chromatography (15% EtOAc in hexanes). 39: Rf = 0.34 (20% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 2.15 (s, 3H), 2.24 (d, 3H, J = 1 Hz), 2.27 (q, 2H, J = 7.5 Hz), 2.52 (t, 2H, J = 7.0 Hz), 5.79 (qt, 1H, J = 1.5, 7.5 Hz); 13C NMR (125 MHz, CDCl3) δ 23.8, 30.2, 42.6 (2C), 120.7, 130.5, 207.5; LRESIMS m/e (% rel. intensity) 213.1 (M + Na) + (23); 207.2 (100); HRESIMS calcd for C7H11BrNaO+ 212.9885, found 212.9898, δ = 6.10 ppm.
α, β-Unsaturated Ester 40
Sodium hydride (60% in mineral oil, 2.22 g, 55.59 mmol) was taken up in MTBE (130 mL) and cooled to 0 °C. Phosphonate (10.30 mL, 51.31 mmol) was added slowly, as hydrogen was evolved. When all of the phosphonate had been added, the solution was stirred an additional 15 minutes, after which time a solution of ketone 39 (8.17 g, 42.76 mmol) in MTBE (20 mL) was added via cannula. The resulting solution was stirred overnight at ambient temperature. The resulting gel was poured into water (150 mL) and additional MTBE (100 mL) was added. After shaking, and draining the lower layer, the organic phase was washed with an additional 100 mL portion of water, brine (100 mL), and dried with Na2SO4. Concentration and purification by chromatography (5–10% MTBE in hexanes, gradient elution) afforded the esters 40 (10.47 g, 40.19 mmol, 94%) as a 3.5: 1 E: Z mixture of isomers. Trans isomer: Rf = 0.19 (5% MTBE in hexanes); 1H NMR (500 MHz, CDCl3) δ 1.28 (t, 3H, J = 7.0 Hz), 2.16 (d, 3H, J = 1.5 Hz), 2.21 (m, 4H), 2.22 (d, 3H, J = 0.5 Hz), 4.15 (q, 2H, J = 7.5 Hz ), 5.67 (s, 1H), 5.80 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 14.5, 23.3 (2 C), 27.7, 40.0, 59.7, 116.4, 120.4, 130.6, 158.2, 166.8; IR (neat, cm−1) 2981 (m), 2937 (m), 1715 (s), 1649 (s), 1148 (s). LRESIMS m/e (% rel. intensity) 283.1 (M + Na)+ (100), 261.2 (M + H)+ (31); HRESIMS calcd for C11H17BrNaO2 283.0304, found 283.0307, δ = 1.10 ppm.
Vinyl Bromide 18
Under a nitrogen atmosphere, ester 40 (7.99 g, 29.04 mmol) was taken up in anhydrous toluene (29.0 mL) and cooled to 0 °C using an ice/water bath. DIBAL-H (1.0 M in toluene, 60.98 mL, 60.98 mmol) was then slowly added via syringe over a period of 15 minutes. 30 minutes after the addition had been completed, TLC indicated complete conversion. The solution was slowly transferred with the aid of 200 mL MTBE into a 1-L beaker containing 100 g of ice and 100 mL of 3 M H2SO4 (caution, a large exotherm as well as hydrogen evolution occurred, the beaker was also placed in a bucket of ice water) and rapidly stirred with a mechanical stirrer. Additional acid was added incrementally until the emulsion was destroyed. When the biphase was completely clear, the mixture was poured into a separatory funnel and the organic layer separated. The aqueous layer was extracted with MTBE (5 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried with Na2SO4. Concentration afforded an intermediate alcohol (6.79 g, 29.04 mmol 100%, contained some toluene) which was used without further purification.
An analytical sample was prepared by chromatography (30% EtOAc in hexanes). Rf = 0.24 (30% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 1.54 (br, 1H), 1.67 (s, 3H), 2.08 (t, 2H, J = 7.5 Hz ), 2.15 (q, 2H, J = 7.5 Hz), 2.21 (s, 3H), 4.16 (d, 2H, J = 7.0 Hz), 5.42 (dt, 1H, J = 1.0, 7.0 Hz), 5.81 (dt, 1H, J = 1.5, 7.5 Hz); 13C NMR (125 MHz, CDCl3) δ 16.4, 23.3, 28.0, 38.7, 59.4, 119.7, 124.4, 131.6, 138.4; IR (neat, cm−1) 3351 (br), 2980 (vs), 2923 (vs), 2880 (vs), 1652 (s), 1433 (vs), 1381 (vs).
The above intermediate alcohol (1.19 g, 5.43 mmol) and imidazole (0.74 g, 10.86 mmol) were taken up in CH2Cl2 (11.0 mL) and TBDPSCl (1.41 mL, 5.43 mmoL) was added. After one hour, TLC indicated consumption of the starting material. The mixture was diluted with hexanes (50 mL) and washed with water (2 × 25 mL), brine (25 mL), and dried with Na2SO4. Concentration and filtration through silica (20% CH2Cl2 in hexanes) yielded vinyl bromide 18 (2.36 g, 5.16 mmol, 95%) as a colorless oil which yellowed upon storage. Rf = 0.33 (20% CH2Cl2 in hexanes); 1H NMR (500 MHz, CDCl3) δ 1.05 (s, 9H), 1.43 (s, 3H), 2.01 (t, 2H, J = 7.0 Hz), 2.09 (q, 2H, J = 7.0 Hz), 2.20 (s, 3H), 4.22 (d, 2H, J = 6.0 Hz), 5.38 (qd, 1H, J = 1.0, 6.0 Hz), 5.81 (dq, 1H, J = 1.5, 7.0 Hz), 7.36–7.69 (m, 6H), 7.69 (dd, 4H, J = 1.5, 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 16.5, 19.4, 23.4, 27.1 (3C), 28.1, 38.7, 61.3, 119.5, 125.1, 127.8 (4C), 129.7 (2C), 131.8, 134.2, 135.8 (4C), 136.0; IR (neat, cm−1) 3071 (m), 3049 (m), 2956 (s), 2931 (s), 2857 (s), 1652 (m), 1471 (m), 1429 (s), 1111 (vs), 1060 (vs); LRESIMS m/e (% relative intensity) 495.2 (M + K) + (27), 479.2 (M + Na)+ (82), 463.2 (28), 447.2 (87), 307.1 (33), 285.1 (25), 277.2 (100), 256.1 (61), 233.1 (23); HRESIMS calcd for C25H33BrNaOSi+ 479.1376; found 479.1389, δ = 2.71 ppm.
4.2.3. Synthesis of Diketo-Enal 8
Cross-Coupled Product 41
Ketal 33 (1.29 g, 3.95 mmol) was placed in a 50 mL round bottem flask and was dried azeotropically with benzene (10 mL). A magnetic stirbar was added and the flask was placed under an atmosphere of nitrogen. To the neat ketal was added 9-BBN ( 0.5 M in THF, 8.3 mL, 4.15 mmol). A yellow color formed (cause unknown, but the color formation was always observed), and the solution was placed in a 50 °C oil bath for two hours. During the hydroboration, vinyl bromide 18 (1.98 g, 4.35 mmol) was placed in a 25 mL point-tipped flask with a stirbar. The flask was evacuated and filled with nitrogen. THF (8.3 mL) was added, the septum briefly removed, and Pd(PPh3)4 (205.3 mg, 0.178 mmol, scrupulously stored under nitrogen at 0 °C in an foil wrapped bottle) was quickly added in a single portion. The headspace was flushed with nitrogen by inserting a second nitrogen line and turning one of the lines to vacuum for 10–15 seconds. The mixture was then stirred/swirled until the yellow palladium complex dissolved completely. When the hydroboration had been heated for two hours, the flask was removed from the oil bath and placed in a room-temperature water bath.
A freshly prepared KOH solution (based on 85% KOH by weight, 2.0 M, 4.35 mL, 8.70 mmol) was added to the borane solution. A small amount of bubbling was observed. After the KOH had been added, the palladium/bromide solution was rapidly added via cannula. The resulting yellow solution was vigorously stirred and gradually changed to a golden orange color. After 45 min, TLC indicated nearly complete consumption of the bromide and the reaction was deemed to be complete (the borane was never detected by TLC) and was poured into hexanes (150 mL) and water 50 mL and vigorously shaken. The color rapidly changed to brownish-black, and a dark brown precipitate formed. The entire mixture was filtered through a medium fritted funnel, and the water layer was then discarded. The organic layer was repeatedly washed with water and filtered as needed until further precipitate was not observed. The organic layer was washed with brine and then dried with Na2SO4. The dry solution was then filtered through a 3 cm bed of Celite® to remove a fine, cloudy precipitate. Concentration afforded a brown oil and the crude material was purified by chromatography (8–10% MTBE, gradient elution) to afford the coupled product 41 (2.56 g, 3.63 mmol, 93%) as a light yellow/brown oil (color was undoubtedly an impurity). Rf = 0.36 (10% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.61 (dq, 6H, J = 3.0, 8.0 H), 0.84 (s, 3H), 0.90 (d, 3H, J = 7.5 Hz), 0.97 (t, 9H, J = 8.0 Hz), 1.04 (s, 9H), 1.38 (dt, 1H, J = 4.5, 13.0 Hz), 1.44 (s, 3H), 1.47 (m, 1H), 1.59 (m, 1H), 1.61 (s, 3H), 1.71–2.00 (m, 7H), 2.06 (m, 2H), 3.80 (dd, 1H, J = 5.0, 7.0 Hz), 3.89–3.91 (m, 4H), 4.22 (d, 2H, J = 6.5 Hz), 5.12 (qt, 1H, J = 1.0, 7.0 Hz), 5.39 (qt, 1H, J = 1.0, 6.5 Hz), 7.35–7.43 (m, 6H), 7.70 (dd, 4H, J = 1.5, 8.0 Hz ); 13C NMR (125 MHz, CDCl3) δ 5.5 (3C), 7.3 (3C), 16.2, 16.5, 16.6, 19.4, 19.5, 26.7, 27.1 (3C), 32.4, 32.7, 33.7, 38.2, 39.3, 39.8 (2C), 61.4, 64.2, 64.2, 73.2, 109.6, 123.6, 124.1, 127.8 (4C), 129.7 (2C), 134.3 (2C), 135.8 (4C), 136.5, 137.4; IR (neat, cm−1) 3071 (m), 3049 (m), 2957 (vs), 2878 (vs), 1668 (w), 1590 (w), 1461 (s), 1428 (s); LRESIMS m/e (% relative intensity) 727.6 (M + Na)+ (100), 172.1 (11); HRESIMS calcd for C43H68NaSiO4 + 727.4548; found 727.4537, δ = 1.51 ppm.
Diketo-enal 8
Ketal 41 (69.7 mg, 0.12 mmol) was taken up in MeOH (3.0 mL) and a few crystals of PPTS were added. After 4 h, TLC indicated consumption of most of the starting material. The solution was diluted with MTBE (10 mL) and washed with water (5 mL). The organic phase was washed with brine and dried with Na2SO4. Concentration and chromatography (10–20% EtOAc in hexanes, gradient elution) afforded an intermediate alcohol as a colorless oil. Rf = 0.23 (20% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.75 (s, 3H), 0.85 (d, 3H, J = 6.5 Hz), 1.04 (s, 9H), 1.34 (dt, 1H, J = 4.5, 13.5), 1.43 (s, 3H), 1.49–1.55 (m, 2H), 1.61 (m, 1H), 1.62 (s, 3H), 1.80–1.93 (m, 4H), 1.98 (m, 2H), 2.04–2.14 (m, 3H), 3.47 (d, 1H, J = 9.5 Hz), 3.68 (td, 1H, J = 3.5, 9.5 Hz), 3.90– 3.99 (m, 4H), 4.22 (d, 2H, J = 6.0 Hz), 5.15 (t, 1H, J = 6.0 Hz), 5.38 (qt, 1H, J = 1.0, 6.0 Hz), 7.35–7.43 (m, 6H), 7.70 (dd, 1H, J = 1.5, 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 15.6, 16.1, 16.4, 16.6, 19.4, 26.7, 27.1 (3C), 33.1, 33.5, 36.0, 36.5, 39.3, 39.7, 39.7, 61.4, 64.1, 64.6, 73.3, 109.6, 123.6, 124.1, 127.8 (4C), 129.7 (2C), 134.3, 135.8 (4C), 136.6, 137.4; IR (neat, cm−1) 3531 (br), 2957 (s), 2933 (s), 2888 (m), 2857 (m), 1428 (m), 1111 (vs); LRESIMS m/e (% relative intensity) 613.4 (M + Na) + (100); HRESIMS calcd for C37H54NaSiO4 + 613.3684; found 613.3688, δ = 0.65 ppm.
This intermediate alcohol (471 mg, 0.797 mmol) was taken up in CH2Cl2 (3.2 mL) and Dess-Martin periodinane (440 mg, 1.03 mmol) was added in one portion. Almost instantly, TLC indicated consumption of the starting material. The mixture was directly loaded onto a short silica gel column packed in 10% EtOAc and filtered through the silica. Concentration afforded ketone 42 (443 mg, 0.752 mmol, 94%) as a colorless oil.
Ketone 42 (443 mg, 0.752 mmol) was taken up in THF (4.5 mL) and cooled to −78 °C. KHMDS (0.5 M in toluene, 1.96 mL) was dripped in the solution fairly rapidly and solution was stirred at −78 °C for 1 h and then removed from the bath for 30 min. The reaction mixture was cooled back down to −78 °C and MeI (0.14 mL, 2.28 mmol) was added and the mixture was maintained at this temperature for 5 min, upon which time the mixture was removed from the −78 ° C bath and stirred for 16 h. The next day, the solution was cooled back down to −78 °C and more MeI (0.14 mL, 2.28 mmol) was added to the reaction and allowed to stir another 16 h at room temperature. To the reaction mixture was added MTBE (25 mL) and 1 M HCl (25 mL). Separation of the organic layer followed by washing with brine (25 mL) and drying with Na2SO4 gave enone 43 (213 mg, 43%).
Enone 43 (121 mg, 0.20 mmol) was placed in a 25 ml round bottom flask and TBAF (1 M in THF, ca. 10 % water, 0.3 mL, 0.3 mmol) was added along with THF (1 mL). The solution was stirred at ambient temperature for 50 min. The solution was poured into water (0.5 mL) and extracted with EtOAc (4 × 5 mL). The organic extracts were washed with brine (1 mL), and dried with Na2SO4. Concentration afforded a crude syrup which was filtered through SiO2 using 1:1 THF: hexanes to afford the intermediate alcohol along with contamination from the removed silyl alcohol.
The crude intermediate alcohol from above was taken up in CH2Cl2 (1 mL) and Dess-Martin periodinane (102 mg, 0.24 mmol) was added in one portion. After 15 minutes, TLC indicated consumption of the starting material. The mixture was filtered through silica gel using 1:1 THF: hexanes. Concentration afforded a semi-solid consisting of an intermediate keto-enal 44 along with trace contamination from DMP (46.8 mg, 77% over 2 steps).
To a solution of crude intermediate keto-enal 44 from above in acetone (2 mL) was added 1.0 N aq H2SO4 (5 drops). The reaction mixture was stirred at 50–60 °C for 20 h, and monitored carefully using TLC analysis. After the reaction was completed, the mixture was partitioned between equal volume of EtOAc, and H2O. The organic layer was dried over Na2SO4, and filtered, concentrated in vacuo. The crude residue was purified using silica gel flash column chromatography to give the pure annulation precursor enal 8 (68%). Rf = 0.18 (50% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) mixture of isomers; major isomer δ 0.94–1.01 (m, 3H), 1.08 (d, 3H, J = 2.0 Hz), 1.58–1.66 (m, 4H), 1.84–1.95 (m, 2H), 2.17 (d, 3H, J = 1.0 Hz), 2.19–2.29 (m, 5H), 2.42 (dqd, 1H, J = 2.0, 3.5, 10.0 Hz), 2.59 (t, 1H, J = 7.5 Hz), 2.76–2.83 (m, 1H), 3.32 (ddd, 1H, J = 2.5, 4.0, 17.5 Hz), 3.50 (dd, 1H, J = 12.5, 18.0 Hz), 5.09– 5.13 (m, 1H), 5.88 (t, 1H, J = 8.0 Hz), 9.99 (d, 1H, J = 8.0 Hz); minor isomer δ 9.87 (d, 1H, J = 8.0 Hz); IR (neat) cm−1 2971brw, 2360m, 2342m, 1667brm, 1599brm; mass spectrum (APCI): m/e (% relative intensity) 305 (100) (M+H)+, 287 (40); m/e (EI) calcd for C19H28O3 304.2033, found 304.2032. [δ = 3.29 ppm].
Diol 45
Ketal 41 (11.79 g, 20.54 mmol) was placed in a 100 ml round bottom flask and TBAF (1 M in THF, ca. 10 % water, 45.2 mL, 45.2 mmol) was added. The solution was stirred at ambient temperature overnight. The following morning, the solution was poured into water (200 mL) and extracted with EtOAc (4 × 100 m). The organic extracts were washed with water (100 mL), brine (100 mL), and dried with Na2SO4. Concentration afforded a crude syrup which was purified by chromatography (50–100% EtOAc in hexanes, gradient elution) to afford diol 45 (6.95 g, 19.72 mmol, 96%) as a light yellow oil. Rf = 0.37 (80% EtOAc in hexanes; 1H NMR (500 MHz, CDCl3) δ 0.76 (s, 3H), 0.86 (d, 3H, J = 7.0 Hz ), 1.35 (dt, 1H, J = 4.0, 12.5 Hz), 1.50–1.60 (m, 4H), 1.62 (s, 3H), 1.67 (s, 3H), 1.80–1.92 (m, 4H), 2.02–2.13 (m, 4H), 3.50 (d, 1H, J = 10.0 Hz), 3.68 (td, 1H, J = 2.5, 10.0 Hz), 3.91–4.01 (m, 4H), 4.14 (d, 2H, J = 7.0 Hz), 5.14 (t, 1H, J = 7.0 Hz), 5.14 (qt, 1H, J = 1.0, 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 15.6, 16.1, 16.4, 26.4, 33.1, 33.4, 35.9, 36.4, 39.3, 39.6, 39.7, 59.5, 64.6, 73.3, 109.5, 123.3, 123.7, 136.8, 139.7; IR (neat, cm−1) 3470 (br), 2940 (s), 1667 (m); LRESIMS m/e (% relative intensity) 727.5 (2M + Na )+ (9), 391.2 (M + K) + (13), 375.2 (M + Na)+ (100); HRESIMS calcd for C21H36NaO4 + 375.2506; found 375.2505, δ = 0.27 ppm.
Diketo-Enal 8 from Diol 45
Diol 45 (3.31 g, 9.39 mmol) was taken up in CH2Cl2 (60 mL) and Dess-Martin periodinane (9.96 g, 23.47 mmol) was added in 3 portions over 5 minutes. The mixture became quite warm, and a fine white precipitate quickly formed. TLC indicated consumption of the starting material within 30 minutes. Excess oxidant was destroyed by addition of 2-propanol (1 mL) and the mixture was poured into 30% EtOAc in hexanes (300 mL) and allowed to stand for 30 minutes. The mixture was filtered though a medium fritted funnel and concentrated. The resulting oil was purified by chromatography (30–50 EtOAc in hexanes gradient elution) to afford an intermediate enal (2.81 g, 8.08 mmol, 86%) as a clear, colorless oil. Rf = 0.23 (30% EtOAc in hexanes); 1H NMR (500 MHz, CDCl3) δ 0.97 (d, 3H, J = 7.0 Hz ), 1.01 (s, 3H), 1.37 (m, 1H), 1.65 (s, 3H), 1.79–1.94 (m, 5H), 2.11 (dqd, 1H, J = 4.5, 7.0, 11.5 Hz), 2.17 (s, 3H), 2.18–2.26 (m, 4H), 2.50 (dd, 1H, J = 3.0, 15.0 Hz), 2.75 (d, 1H, J = 15.0 Hz), 3.95 (m, 4H), 5.10 (brt, 1H, J = 6.5 Hz), 5.88 (md, 1H, J = 7.5 Hz), 10.00 (d, 1H, J = 7.5 Hz); 13C NMR (125 MHz, CDCl3) δ 15.0, 16.3, 17.8, 19.1, 25.9, 31.4, 33.8, 34.3, 39.5, 40.8, 48.4, 50.7, 64.7, 64.8, 108.8, 122.4, 127.6, 137.3, 164.1, 191.5, 211.3; IR (neat, cm−1) 2969 (vs), 2886 (vs), 2771 (m), 1711 (vs), 1673 (vs); LRESIMS m/e (% relative intensity) 719.4 (2M + Na)+ (40), 387.2 (M + K)+ (21), 371.2 (M + Na )+ (100); HRESIMS calcd for C21H32NaO4 + 371.2193; found 371.2192, δ = 0.27 ppm.
To a solution of intermediate acetal enal (400.0 mg, 1.15 mmol) in acetone (9 mL) was added 1.0 N aq H2SO4 (2.30 mL). The reaction mixture was stirred at 50–60 °C for 4–5 h, and monitored carefully using TLC analysis. After the reaction was completed, the mixture was partitioned between equal volume of EtOAc, and H2O. The organic layer was dried over Na2SO4, and filtered, concentrated in vacuo. The crude residue was purified using silica gel flash column chromatography to give the pure annulation precursor enal 8 with yields varying between 59–88%. Rf = 0.18 (50% EtOAc/hexanes). Spectroscopic information for 8 was already disclosed (vide supra).
Scheme 6.

A Scucsessful Equilibration.
Scheme 8.

Choices of Diene for the Cycloaddition.
Acknowledgments
Authors thank the NIH [NS38049] for financial support. We thank Dr. Victor G. Young and Dr. William W. Brennessel of University of Minnesota for solving X-ray structures. GSB thanks the AFPE Fellowship sponsored by Procter and Gamble Company. KPC thanks ACS for an Organic Division Fellowship sponsored by Schering-Plough Research Institute. We are very grateful for all the invaluable discussions with Professor Viresh Rawal and Professor Yong Huang on the key Diels-Alder cycloaddition.
Footnotes
With the deepest respect and most heartfelt appreciation, this paper is dedicated to Professor Gilbert Stork on the very special occasion of his 90th birthday.
References
- 1.PAF is a proinflammatory phospholipid mediator found in many different cell types in the human body, referring to a class of compounds with the general structure of 1-O-alkyl/acyl-2-acetylglycero-3-phosphocholine.
- 2.Sugano M, Sato A, Iijima Y, Oshima T, Furuya K, Kuwano H, Hata T, Hanzawa H. J Am Chem Soc. 1991;113:5463. [Google Scholar]
- 3.Sugano M, Sato A, Iijima Y, Oshima T, Furuya K, Haruyama H, Yoda K, Hata T. J Org Chem. 1994;59:564. [Google Scholar]
- 4.Sugano M, Sato A, Iijima Y, Oshima T, Furuya K, Kuwano H, Hata T. J Antibiot. 1995;48:1188. doi: 10.7164/antibiotics.48.1188. [DOI] [PubMed] [Google Scholar]
- 5.Koyama K, Ishino M, Takatori K, Sugita T, Kinoshita K, Takahashi K. Tetrahedron Lett. 2004;45:6947. [Google Scholar]
- 6.Chu M, Truumees I, Gunnarsson I, Bishop WR, Kreutner W, Horan AC, Patel MG, Gullo VP, Puar MS. J Antibiot. 1993;46:554. doi: 10.7164/antibiotics.46.554. [DOI] [PubMed] [Google Scholar]
- 7.Chu M, Patel MG, Gullo VP, Truumees I, Puar MS, McPhail AT. J Org Chem. 1992;57:5817. [Google Scholar]
- 8.Cheing JWC, Goldring WPD, Pattenden G. Chem Commun. 2003:2788. doi: 10.1039/b310753a. [DOI] [PubMed] [Google Scholar]
- 9.Tokiwano T, Fukushi E, Endo T, Oikawa H. Chem Commun. 2004:1324. doi: 10.1039/b401377h. [DOI] [PubMed] [Google Scholar]
- 10.PAF mediates an incredibly diverse array of biological processes including wound healing, inflammation, bronchoconstriction, vascular permeability, apoptosis, angiogenesis, reproduction and long-term potentiation.11 PAF are also implicated in many inflammatory, respiratory related problems including asthma and pulmonary edema, and cardiovascular diseases.12–14 Only limited structure activity relationships have been reported by Sugano et al. focusing on derivatization of phomactin D, the most potent member of the family in inhibiting PAF aggregation [IC50 = 0.80 μM],12 and by Rawal et al. on some simple but interesting phomactin structural analogs.15.
- 11.Stafforinif DM, McIntyre TM, Zimmerman GA, Prescott SM. Crit Rev in Clin Lab Sciences. 2003;40:643. doi: 10.1080/714037693. [DOI] [PubMed] [Google Scholar]
- 12.Sugano M, Sato A, Saito K, Takaishi S, Matsushita Y, Iijima Y. J Med Chem. 1996;39:5281. doi: 10.1021/jm950640q. [DOI] [PubMed] [Google Scholar]
- 13.Braquet P, Touqui L, Shen TY, Varaftig BB. Pharm Rev. 1987;39:97–145. [PubMed] [Google Scholar]
- 14.Nagase T, Ishii S, Kume K, Uozumi N, Izumi T, Ouchi Y, Shimizu T. J Clin Investigations. 1999;104:1071. doi: 10.1172/JCI7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu X, Lambertino AT, Houghton TJ, McGilvra JD, Xu C, Rawal VH, Leff AR. Life Sciences. 2003;73:3005. doi: 10.1016/j.lfs.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 16.For an excellent review on synthetic efforts toward phomactins, see: Goldring WPD, Pattenden G. Acc Chem Res. 2006;39:354. doi: 10.1021/ar050186c.
- 17.Also see: Cole KP, Hsung RP. Chem Tracts. 2003;16:811.
- 18.For synthetic approaches, see: Foote KM, Hayes CJ, Pattenden G. Tetrahedron Lett. 1996;37:275.Chen D, Wang J, Totah NI. J Org Chem. 1999;64:1776. doi: 10.1021/jo982488g.Seth PP, Totah NI. J Org Chem. 1999;64:8750.Seth PP, Totah NI. Org Lett. 2000;2:2507. doi: 10.1021/ol0061788.Kallan NC, Halcomb RL. Org Lett. 2000;2:2687. doi: 10.1021/ol0062345.Chemler SR, Danishefsky SJ. Org Lett. 2000;2:2695. doi: 10.1021/ol0062547.Seth PP, Chen D, Wang J, Gao X, Totah NI. Org Lett. 2000;56:10185.Foote K, John M, Pattenden G. Synlett. 2001:365.Mi B, Maleczka R. Org Lett. 2001;3:1491. doi: 10.1021/ol015807q.Chemler SR, Iserloh U, Danishefsky SJ. Org Lett. 2001;3:2949. doi: 10.1021/ol0161357.Houghton T, Choi S, Rawal VH. Org Lett. 2001;3:3615. doi: 10.1021/ol0163833.Mohr PJ, Halcomb RL. Org Lett. 2002;4:2413. doi: 10.1021/ol026159t.DiBlasi CM, Hamblett CL, Leighton JL. Abstracts of Papers. 224th National Meeting of the American Chemical Society; Boston, MA. Fall. 2002. p. ORGN-364.DiBlasi CM. Thesis Advisor: Professor James L Leighton. Columbia University; 2003. Ph.D. Dissertation.Balnaves AS, McGowan G, Shapland PDP, Thomas EJ. Tetrahedron Lett. 2003;44:2713.Cheing JWC, Goldring WPD, Pattenden G. Chem Commun. 2003:2788. doi: 10.1039/b310753a.Ryu K, Cho YS, Jung SI, Cho CG. Org Lett. 2006;8:3343. doi: 10.1021/ol061231z.Huang J, Wang H, Wu C, Wulff WD. Org Lett. 2007;9:2799. doi: 10.1021/ol070904q.Seth PP, Chen D, Wang J, Gao X, Totah NI. Tetrahedron Lett. 2007;48:4605. doi: 10.1016/j.tetlet.2007.04.122.Peng W, Lee CS. Synlett. 2008;142Furuya Y, Hata K, Kuwano T, Antibiot HJ. 1995;48:1188. doi: 10.7164/antibiotics.48.1188.Ciesielski J, Canterbury DP, Frontier AJ. Org Lett. 2009;11:4374. doi: 10.1021/ol901721y.Shapland PDP, Thomas EJ. Tetrahedron. 2009;65:4201.Schwartz KD, White JD. Org Lett. 2011;13:248. doi: 10.1021/ol1026816.
- 19.For total synthesis of (+)-phomactin D, see: Miyaoka H, Saka Y, Miura S, Yamada Y. Tetrahedron Lett. 1996;37:7107.
- 20.For total synthesis of (±)-phomactin B2, see: Huang J, Wu C, Wulff WD. J Am Chem Soc. 2007;129:13366. doi: 10.1021/ja074275r.
- 21.For total synthesis of (±)-phomactin A, see: Goldring WPD, Pattenden G. Chem Commun. 2002:1736. doi: 10.1039/b206041h.For detailed full papers, see: Diaper CM, Goldring WPD, Pattenden G. Org Biomol Chem. 2003;1:3949. doi: 10.1039/b307986d.Foote KM, Hayes CJ, John MP, Pattenden G. Org Biomol Chem. 2003;1:3917. doi: 10.1039/b307985f.
- 22.For total synthesis of (+)-phomactin A, see: Mohr PJ, Halcomb RL. J Am Chem Soc. 2003;125:1712. doi: 10.1021/ja0296531.
- 23.For total synthesis of (±)-phomactin G, see: Goldring WPD, Pattenden G. Org Biomol Chem. 2004;2:466. doi: 10.1039/b314816e.
- 24.Harrity JPA, Provoost O. Org Biomol Chem. 2005;3:1349. doi: 10.1039/b502349c. [DOI] [PubMed] [Google Scholar]
- 25.Hsung RP, Kurdyumov AV, Sydorenko N. Eur J Org Chem. 2005;1:23. [Google Scholar]
- 26.Hsung RP, Wei L-L, Sklenicka HM, Shen HC, McLaughlin MJ, Zehnder LR. Trends in Heterocyclic Chem. 2001;7:1–24. [Google Scholar]
- 27.Hsung RP, Cole KP. In: Strategies and Tactics in Organic Synthesis. Harmata M, editor. Vol. 4. Elsevier Science, Pergamon Press; Oxford, UK: 2004. p. 41. [Google Scholar]
- 28.Buchanan GS, Feltenberger JB, Hsung RP. Curr Org Syn. 2010;7:363. doi: 10.2174/157017910791414490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsung RP, Shen HC, Douglas CJ, Morgan CD, Degen SJ, Yao LJ. J Org Chem. 1999;64:690. doi: 10.1021/jo982165k. [DOI] [PubMed] [Google Scholar]
- 30.Hsung RP, Wei LL, Sklenicka HM, Douglas CJ, McLaughlin MJ, Mulder JA, Yao L. Org Lett. 1999;1:509. [Google Scholar]
- 31.Shen HC, Wang J, Cole KP, McLaughlin MJ, Morgan CD, Douglas CJ, Hsung RP, Coverdale HA, Gerasyuto AI, Hahn JM, Liu J, Wei LL, Sklenicka HM, Zehnder LR, Zificsak CA. J Org Chem. 2003;68:1729. doi: 10.1021/jo020688t. [DOI] [PubMed] [Google Scholar]
- 32.Kurdyumov AV, Lin N, Hsung RP, Gullickson GC, Cole KP, Sydorenko N, Swidorski JJ. Org Lett. 2006;8:191. doi: 10.1021/ol0523042. [DOI] [PubMed] [Google Scholar]
- 33.For recent related recent studies, see: Sagar R, Park J, Koh M, Park SB. J Org Chem. 2009;74:2171. doi: 10.1021/jo8023889.Yamamoto Y, Itonaga K. Org Lett (c) doi: 10.1021/ol802800s.Zhong W, Lin F, Chen R, Su W. Synthesis. 2008:2561.Hubert C, Moreau J, Batany J, Duboc A, Hurvois JP, Renaud JL. Adv Synth Catal. 2008;350:40.Zhu M-K, Wei Q, Gong L-Z. Adv Synth Catal. 2008;350:1281.Brioche JCR, Goodenough KM, Whatrup DJ, Harrity JPA. J Org Chem. 2008;73:1946. doi: 10.1021/jo702620e.Yavari I, Sabbaghan M, Hossaini Z. Synlett. 2008:1153.Zhong W, Lin F, Chen R, Su W. Synthesis. 2008:2561.Brioche JCR, Goodenough KM, Whatrup DJ, Harrity JPA. Org Lett. 2007;9:3491. doi: 10.1021/ol701553j.Lee YR, Xia L. Synthesis. 2007:3240.Epstein OL, Rovis T. J Am Chem Soc. 2006;128:16480. doi: 10.1021/ja066794k.Sheibani H, Mosslemin MH, Behzadi S, Islami MR, Saidi K. Synthesis. 2006:435.
- 34.For earlier work, see: Appendino G, Cravotto G, Tagliapietra S, Nano GM, Palmisano G. Helv Chim Acta. 1990;73:1865.Schuda PF, Price WA. J Org Chem. 1987;52:1972.de March P, Moreno-Mañas M, Casado J, Pleixats R, Roca JL. J Heterocycl Chem. 1984;21:85.Tietze LF, v Kiedrowski G, Berger B. Synthesis. 1982:683.de Groot A, Jansen BJM. Tetrahedron Lett. 1975;16:3407.
- 35.For the earliest work in oxa-[3 + 3] annulations, see: Ikawa M, Stahmann MA, Link KP. J Am Chem Soc. 1944;66:902.
- 36.For applications of oxa-[3 + 3] annulations in natural product syntheses, see: Yamashita S, Iso K, Kitajima K, Himura M, Hirama M. J Org Chem. 2011;76:2408. doi: 10.1021/jo2002616.Seijiro Hosokawa S, Matsushita K, Tokimatsu S, Toriumi T, Suzuki Y, Tatsuta K. Tetrahedron Lett. 2010;51:5532.Jeso V, Nicolaou KC. Tetrahedron Lett. 2009;50:1161. doi: 10.1016/j.tetlet.2008.12.096.Yamashita S, Iso K, Hirama M. Org Lett. 2008;10:3413. doi: 10.1021/ol8012099.Brioche JCR, Goodenough KM, Whatrup DJ, Harrity JPA. Org Lett. 2007;9:3941. doi: 10.1021/ol701553j.Kurdyumov AV, Hsung RP. J Am Chem Soc. 2006;128:6272. doi: 10.1021/ja054872i.Liu H, Siegel DR, Danishefsky SJ. Org Lett. 2006;8:423. doi: 10.1021/ol052618p.Monda M, Puranik VG, Argade NP. J Org Chem. 2006;71:4992. doi: 10.1021/jo0606655.Olson BS, Trauner D. Synlett. 2005:700.Malerich JP, Maimone TJ, Elliott GI, Trauner D. J Am Chem Soc. 2005;127:6276. doi: 10.1021/ja050092y.Lee YR, Wang X. Bull Korean Chem Soc. 2005;26:1933.Sunazuka T, Handa M, Nagai K, Shirahata T, Harigaya Y, Otoguro K, Kuwajima I, Õmura S. Tetrahedron. 2004;60:7845.Hu H, Harrison TJ, Wilson PD. J Org Chem. 2004;69:3782. doi: 10.1021/jo049703f.Kurdyumov AV, Hsung RP, Ihlen K, Wang J. Org Lett. 2003;5:3935. doi: 10.1021/ol030100k.Hsung RP, Cole KP, Zehnder LR, Wang J, Wei LL, Yang XF, Coverdale HA. Tetrahedron. 2003;59:311.Malerich JP, Trauner D. J Am Chem Soc. 2003;125:9554. doi: 10.1021/ja036026i.Cole KP, Hsung RP. Tetrahedron Lett. 2002;43:8791.Sunazuka T, Handa M, Nagai K, Shirahata T, Harigaya Y, Otoguro K, Kuwajima I, Õmura S. Org Lett. 2002;4:367. doi: 10.1021/ol017046x.McLaughlin MJ, Hsung RP. J Org Chem. 2001;66:1049. doi: 10.1021/jo001368h.Zehnder LR, Hsung RP, Wang J, Golding GM. Angew Chem Int Ed. 2000;39:3876. doi: 10.1002/1521-3773(20001103)39:21<3876::AID-ANIE3876>3.0.CO;2-C.
- 37.For recent key reviews on chemistry of 1,3-dicarbonyls, see: Simon C, Constantieux T, Rodriguez J. Eur J Org Chem. 2004:4957.
- 38.For beautiful reviews and seminal accounts on tandem reactions, see: Nicolaou KC, Edmonds DJ, Bulger PG. Angew Chem Int Ed. 2006;45:7134. doi: 10.1002/anie.200601872.Nicolaou KC, Montagnona T, Snyder SA. Chem Commun. 2003:551. doi: 10.1039/b209440c.Tietze LF. Chem Rev. 1996;96:115. doi: 10.1021/cr950027e.Tietze LF, Beifuss U. Angew Chem, Int Ed. 1993;32:131.Tietze LF. J Heterocycl Chem. 1990;27:47.
- 39.For reviews for pericyclic ring-closures, see: Marvell EN. Thermal Electrocyclic Reactions. Academic Press; New York: 1980. Okamura WH, de Lera AR. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Paquette LA, editors. Vol. 5. Pergamon Press; New York: 1991. pp. 699–750.
- 40.For informative reviews on ring-closure in natural product synthesis, see: Beaudry CM, Malerich JP, Trauner D. Chem Rev. 2005;105:4757. doi: 10.1021/cr0406110.Pindur U, Schneider GH. Chem Soc Rev. 1994:409.
- 41.For leading references on electrocyclic ring-closures involving 1-oxatrienes, see: Shishido K, Shitara E, Fukumoto K. J Am Chem Soc. 1985;107:5810.Shishido K, Hiroya K, Fukumoto K, Kametani T. Tetrahedron Lett. 1986;27:971.
- 42.For a leading review, see: Tang Y, Oppenheimer J, Song Z, You L, Zhang X, Hsung RP. Tetrahedron. 2006;62:10785.
- 43.The term “formal [3 + 3]” was used to describe [3 + 3] carbo-cycloadditions. See: Seebach D, Missbach M, Calderari G, Eberle M. J Am Chem Soc. 1990;112:7625.
- 44.For earlier studies on [3 + 3] carbo-cycloadditions, see: Landesman HK, Stork G. J Am Chem Soc. 1956;78:5129.
- 45.For leading reviews on step-wise carbo-[3 + 3] formal cycloadditions, see: Filippini MH, Rodriguez J. Chem Rev. 1999;99:27. doi: 10.1021/cr970029u.Filippini M-H, Faure R, Rodriguez J. J Org Chem. 1995;60:6872.and see references 21–33 cited therein. Butkus E. Synlett. 2001:1827.Feist H, Langer P. Synthesis. 2007:327.
- 46.For a key seminal study, see: de March P, Moreno-Mañas M, Casado J, Pleixats R, Roca JL, Trius A. J Heterocyclic Chem. 1984;21:1369.
- 47.For an initial communication on the synthesis of diketo-enal 8, see: Cole KP, Hsung RP. Org Lett. 2003;5:4843. doi: 10.1021/ol030115i.
- 48.For an initial communication on recognition of the unique structural topology of ABD-tricycle 2, see: Cole KP, Hsung RP. Chem Commun. 2005:5784. doi: 10.1039/b511338e.
- 49.For an initial communication on revised and enantioselective approach to ABD-tricycle 2, see: You L, Hsung RP, Bedermann AA, Kurdyumov AK, Tang Y, Buchanan GS, Cole KP. Adv Syn Catal. 2008;350:2885.
- 50.For an initial communication on completion of our total synthesis of (±)-phomactin A, see: Tang Y, Cole KP, Buchanan GS, Li G, Hsung RP. Org Lett. 2009;11:1591. doi: 10.1021/ol900237e.
- 51.Details can be found in: Cole KP. PhD Dissertation. University of Minnesota; 2005.
- 52.CCDC for compound 14a is 28069. CCDC 28069 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request.cif.
- 53.Kozmin SA, He S, Rawal VH. Org Synth. 2000;78:152, 160. [Google Scholar]
- 54.(a) Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628. doi: 10.1021/ja017863s. [DOI] [PubMed] [Google Scholar]; (b) Kozmin SA, Green MT, Rawal VH. J Org Chem. 1999;64:8045. doi: 10.1021/jo981563k. [DOI] [PubMed] [Google Scholar]
- 55.Huang Y, Iwama T, Rawal VH. J Am Chem Soc. 2000;122:7843.Huang Yong. PhD thesis. Huang Y, Iwama T, Rawal VH. Org Lett. 2002;4:1163. doi: 10.1021/ol0255716.Also see: Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628. doi: 10.1021/ja017863s.Kozmin SA, Rawal VH. J Am Chem Soc. 1998;124:4628. doi: 10.1021/ja017863s.
- 56.Huang Y, Iwama T, Rawal VH. J Am Chem Soc. 2002;124:5950. doi: 10.1021/ja026088t. [DOI] [PubMed] [Google Scholar]
- 57.Corey EJ, Bock MG, Kozikowski AP, Rama Rao AV, Floyd D, Lipshutz B. Tetrahedron Lett. 1978;19:1051. [Google Scholar]
- 58.Schlosser M, Hammer E. Helv Chim Acta. 1974;57:2547. [Google Scholar]
- 59.Buchanan GS, Cole KP, Tang Y, Hsung RP. J Org Chem. 2011;76:7027. doi: 10.1021/jo200936r. [DOI] [PMC free article] [PubMed] [Google Scholar]