

Abstract
Patients with PMP22 deficiency present with focal sensory and motor deficits when peripheral nerves are stressed by mechanical force. It has been hypothesized that these focal deficits are due to mechanically induced conduction block (CB). To test this hypothesis, we induced 60–70% CB (defined by electrophysiological criteria) by nerve compression in an authentic mouse model of hereditary neuropathy with liability to pressure palsies (HNPP) with an inactivation of one of the two pmp22 alleles (pmp22+/−). Induction time for the CB was significantly shorter in pmp22+/− mice than that in pmp22+/+ mice. This shortened induction was also found in myelin-associated glycoprotein knock-out mice, but not in the mice with deficiency of myelin protein zero, a major structural protein of compact myelin. Pmp22+/− nerves showed intact tomacula with no segmental demyelination in both noncompressed and compressed conditions, normal molecular architecture, and normal concentration of voltage-gated sodium channels by [3H]-saxitoxin binding assay. However, focal constrictions were observed in the axonal segments enclosed by tomacula, a pathological hallmark of HNPP. The constricted axons increase axial resistance to action potential propagation, which may hasten the induction of CB in Pmp22 deficiency. Together, these results demonstrate that a function of Pmp22 is to protect the nerve from mechanical injury.
Introduction
Peripheral myelin protein-22 (PMP22) is primarily expressed in the compact myelin of peripheral nerves and is encoded by the PMP22 gene within the DNA segment of human chromosome 17p11.2 (Jetten and Suter, 2000). This gene is important clinically. Overexpression of PMP22 causes Charcot–Marie–Tooth disease type 1A (CMT1A), the most common heritable neuropathy afflicting ∼1:5000 people of all ethnic backgrounds. Alternatively, haploinsufficiency of PMP22 results in a different disorder, hereditary neuropathy with liability to pressure palsies (HNPP) (Chance et al., 1993; Li et al., 2007). Pathologically, CMT1A is characterized by dysmyelination/demyelination with frequent onion bulb formation (Dyck and Lambert, 1968; Robertson et al., 2002), whereas HNPP is characterized by the presence of frequent focal myelin folds known as tomacula (Madrid and Bradley, 1975; Yoshikawa and Dyck, 1991).
HNPP is characterized by focal episodes of weakness and sensory loss (Madrid and Bradley, 1975; Marazzi et al., 1988; Li et al., 2004). These focal deficits are usually triggered by physical activities, including stretching, repetitive motions of the affected limb, or compression (Li et al., 2004), suggesting that there is nerve vulnerability to mechanical force in PMP22 deficiency. Whether this is the case remains to be demonstrated and is an important issue that is not only clinically relevant but essential for understanding the biological function of PMP22, since HNPP is a “loss-of-function” model.
PMP22 has been suggested to play a role in proliferation, differentiation, and death of Schwann cells (Jetten and Suter, 2000; Sancho et al., 2001; Amici et al., 2007). These functions can be further evaluated in null models such as homozygous pmp22-deficient (pmp22−/−) mice (Sancho et al., 2001; Amici et al., 2007). However, it is unlikely that any of these three functions play a role in HNPP, a heterozygous disorder, since pmp22+/− mice, an authentic model of the human disease, have normal Schwann cell numbers and compact myelin (Adlkofer et al., 1997).
It has been hypothesized that the transient focal deficits in HNPP are caused by reversible conduction block (CB) (Lewis et al., 2000; Li et al., 2002, 2004), a failed propagation of the action potential along myelinated nerve fibers (Cornblath et al., 1991; Kaji, 2003). To investigate this possibility and the role of PMP22 in nerve resistance to mechanical stress, we have created a CB model in pmp22+/− mice by nerve compression. Our results demonstrated that CB can be mechanically induced in pmp22+/− nerves more rapidly than in normal nerves.
Materials and Methods
Pmp22-deficient mice, genotyping, and cross-breeding
The pmp22+/− mouse was generated by using homologous recombination technique to inactivate the pmp22 gene (Adlkofer et al., 1995). Peripheral nerves from this mouse showed typical pathological changes of HNPP, including tomacula (Adlkofer et al., 1995, 1997). Due to cytomegalovirus infection, this mouse had to be rederived embryonically to eliminate the viruses (assisted by Van Andel Rearch Institute, Grand Rapids, MI). The rederived mouse showed clinical phenotype and pathological changes identical to the mouse from original colony.
A breeding colony is maintained in Wayne State University animal facility. The Animal Investigational Committee in the institution has approved the use of animals for this study. For genotyping, DNA was isolated from clipped mouse tails and subjected to PCR analysis. Genotypes were determined as described in previous publications (Adlkofer et al., 1995, 1997). For the control mice with heterozygous deletion of myelin protein zero (MPZ) and homozygous knock-out of myelin-associated glycoprotein (mag), procedures for breeding and genotyping have been described in previous publications (Shy et al., 1997; Yin et al., 1998).
To study axonal constriction, we have cross-bred pmp22+/− mouse with YFP transgenic mouse (YFPtg+/+). The YFPtg mouse was transferred from Dr. Derron Bishop's laboratory (Indiana University School of Medicine, Muncie, IN) and carries a transgene expressing yellow fluorescent protein (YFP) specifically in neurons under the drive of Thy-1 promotor (Bishop et al., 2004). This gives clear and intense labeling of all axons.
Nerve conduction study
Mice were anesthetized with Avertin (250 mg/kg). Body temperature was allowed to equilibrate at room temperature. One pair of stimulating electrodes was positioned percutaneously at the sciatic notch. A second pair was inserted adjacent to the tibial nerve at the ankle. Compound muscle action potential (CMAP) was recorded from the intrinsic foot muscle using needle electrodes. CMAP amplitudes were measured from baseline to the peak of negative deflection. The ratio between CMAP amplitude from proximal (P) and distal (D) stimulation (P/D ratio) was calculated. This minimized the effect of changes in the absolute amplitude values due to variation in the electrode placement during different recordings in the same animals.
Induction of CB by compression
All mice in compression experiments were 2–3 months old. Anesthetized mice were placed on a Styrofoam board. Nerve conduction study (NCS) was done as described above. CMAP was recorded before compression. CB was induced at tibial and sciatic nerves by two different techniques as described below.
CB at the tibial nerve.
Mice were placed in prone position. The right leg rested on a metal plate with a width of 1 cm. Compression was delivered by a loop of nylon cord of 1 mm width with different weights attached (200–1000 g). The cord ran cross the dorsal side of the leg 3 mm above the ankle to compress the tibial nerve (Fig. 1A–C). Conduction velocities and P/D ratios were obtained every 15 min after the compression started. Toward the onset of CB, changes in conductions became accelerated. Thus, measurements were performed more frequently (every 5 min) until the ratio was reduced to 0.4 to 0.3 (60–70% CB). It has not been possible to make CB exactly at 60 or 70%; thus, all CBs were targeted between 60 and 70%.
Figure 1.
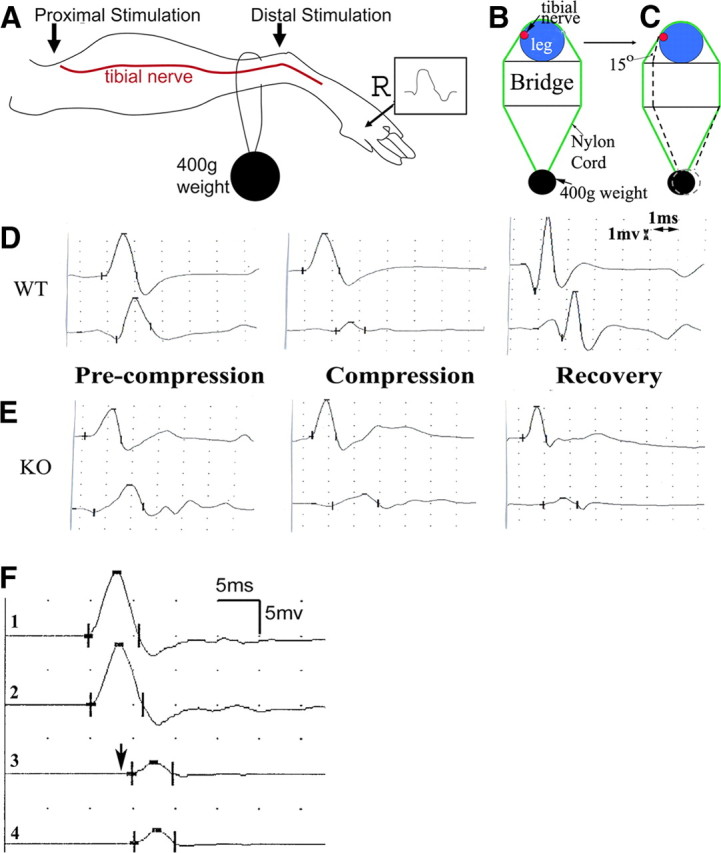
Experimental paradigm for nerve compression and mechanically induced CB in mice. A, This diagram shows the placement of stimulation (arrows) and recording (marked by R) electrodes for NCS. B, Compression was delivered by a nylon cord loop attached a 400 g weight. This diagram shows the cross-section at the site of nylon cord. The leg rests on a metal plate (marked as “bridge”) with a width of ∼1 cm. Tibial nerves usually run on the medial side of the ankle (red dot). C, In the second set of experiments, the angle between the nylon cord and the vertical line of leg is reduced by 15 degrees by shortening the length of the bridge (arrow). D, An example of NCS from a compression experiment on a wild-type mouse is shown. On the left, similar amplitudes of CMAP were evoked by stimulations at the distal and proximal sites. After compression was applied, CMAP amplitude from the proximal stimulation was reduced by >60% from the CMAP amplitude by the distal stimulation, called CB (middle). At this point, compression was removed. At day 5 after the compression, CB recovered (right). E, The same experiment was performed on a pmp22+/− mouse. CB did not recover at day 5 following compression (right). Sensitivity = 1 mV; speed = 1 ms. Notice that the onset of CMAP in mice may be obscured by an evolving positive deflection. Thus, peaks of CMAP were often used to calculate the latency and conduction velocity. Nevertheless, this issue does not affect the measurement of CMAP amplitudes or any of our conclusions. F, An original trace recorded at the hypothenar muscle of a patient with HNPP: stimulations on the ulnar nerve inched 1 cm/step across the elbow, a location subject to compression. A focal slowing (a long delay from trace 2 to 3 in contrast to a very short delay from 1 to 2 or from 3 to 4) across the elbow was identified within a 1 cm segment of the nerve, demonstrating the very focal nature of the slowing. CB was conspicuous in this case. The arrow indicates the third response, which had a >50% amplitude drop of motor response and was associated with weakness in muscles innervated by the ulnar nerve.
CB at the sciatic nerve.
An incision was made around the sciatic notch to expose the sciatic nerve. The nerve was wrapped with two layers of gauze that was soaked with saline. Compression was applied on the surface of gauze with a 20 g/mm2 vessel clamp (clamp width = 1.5 mm; catalog number Tks-1-20 g; Aros Surgical Instruments). Conduction velocity and P/D ratios were collected as described above.
Semithin section and EM
These techniques were described in our previous studies (Li et al., 2005; Zhang et al., 2008). In brief, mice were transcardially perfused with 4% paraformaldehyde and 3.5% glutaraldehyde. Sciatic or tibial nerves were dissected and postfixed for 24 h. Nerves were then osmicated for 1.5 h, dehydrated, and embedded in Epon. Tissue blocks were sectioned with 1 μm thickness and stained with methylene blue for light microscopic examination. The blocks were then trimmed and sectioned into ultrathin sections for EM examination (Zeiss EM 900).
For osmicated teased nerve fiber studies, nerves were dissected and fixed as described above, followed by dehydration and osmication in 1% osmium tetroxide before embedding in Epon. The nerve bundles were teased in fluid Epon under the dissection microscope by using fine forceps. The glass slides with teased nerve fibers were hardened in the 60°C oven, mounted with coverslip, and examined under the light microscope.
Teased nerve fiber immunohistochemistry
This technique has been described in our previous studies (Bai et al., 2006; Zhang et al., 2008). In brief, nerves were fixed in 4% paraformaldehyde for 30 min to 16 h (depending on which primary antibodies were to be used). Sciatic nerves were teased into individual fibers on glass slides. The slides were dried overnight, reacted with primary antibodies, stored at 4°C overnight, and then incubated for 1.5 h with secondary antibodies. The stained slides were examined under a Leica fluorescent or a Nikon confocal microscope. All antibodies used are listed in Table 1.
Table 1.
Primary antibodies
| Antibody | Source and catalog number | Species raised in | Specific antigen | Type | Reference |
|---|---|---|---|---|---|
| MAG | Zymed Laboratories 34-6200 | Rabbit | Shared C-terminal region of the small and large MAG | Polyclonal | Yamamoto et al., 1994 |
| Kv1.2 | Sigma P 8732 | Rabbit | GST fusion protein with aa 417-498 | Polyclonal | McKinnon, et al., 1989 |
| Pan-Nav | Sigma S 8809 | Mouse | Synthetic peptide CTEEQKKYYNAMKKLGSKK | Polyclonal IgG | Ulzheimer et al., 2004 |
| Caspr | Dr. E. Pellesa | Rabbit | Rat cytoplasmic domain | Polyclonal IgG | Einheber et al., 1997 |
| MBP | Chemicon MAB 386 | Rat | aa 82–87, bovine MBP | Monoclonal IgG | Li et al., 2005 |
aa, Amino acids.
aDepartment of Molecular Cell Biology, The Weizmann Institute of Science, Rehovot, Israel.
[3H]-saxitoxin binding assay
Peripheral nerves, including bilateral sciatic, tibial, peroneal, roots, and brachial plexus, were dissected from each mouse. Membrane protein extracts were prepared as previously described (Catterall et al., 1979). [3H]-saxitoxin ([3H]-STX) binding was performed as described previously, using rat brain membranes as a positive control (Isom et al., 1995; Chen et al., 2002). Briefly, membrane extracts were washed in binding buffer [containing the following (in mm): 50 HEPES-Tris, pH 7.5, 130 choline chloride, 5.4 KCL, 0.8 MgSO4 5.5 dextrose]. Membranes were then pelleted by centrifugation at 4°C and resuspended in ice-cold binding buffer. Two hundred microliters of each sample was aliquoted into polypropylene tubes on ice (in triplicate), containing 25 μl of a 50 nm [3H]-STX stock (5 nm final concentration in the assay), plus 25 μl of 100 μm unlabeled tetrodotoxin stock (10 μm final) or binding buffer to assess nonspecific binding. All samples were incubated on ice in a 4°C cold room for 1 h. Binding was terminated by rapid vacuum filtration over Whatman GF/C filters that have been presoaked in wash buffer [containing the following (in mm): 163 choline chloride, 5 HEPES-Tris, pH 7.5, 1.8 CaCl, 0.8 MgSO4) containing 1 mg/ml BSA, followed by three rapid washes with ice-cold wash buffer. [3H]-STX bound to the filters was assessed by scintillation counting. Specific binding is assessed by subtraction of nonspecific binding in the presence of tetrodotoxin. Specific 3H activity was normalized to protein concentration and finally expressed as femtomoles of sodium channels per milligram of protein, based on the established 1:1 stoichiometry of STX binding to neuronal sodium channels. For each experiment, two samples were collected from 11 to 15 pmp22+/+ mice and from 11 to 15 pmp22+/− mice.
Results
CB mechanically was induced more rapidly in pmp22+/− nerves, but not in MPZ-deficient nerves
To generate a model of CB in mice we performed a series of compressions on the tibial nerve using a nylon cord attached to different weights (200–1000 g) (Fig. 1A–C). We identified an ideal weight of 400 g that induces CB within 1.5 h. The 400 g weight was then used for the remaining experiments. Before compression, amplitudes of CMAP from proximal and distal stimulations were similar (Fig. 1D). Within 4–5 min of compression, CMAP amplitude from proximal stimulation began to decrease and continued to drop if the compression was not removed. Once the P/D ratio reached 0.4 – 0.3, a 60–70% CB was diagnosed and the compression was removed (Fig. 1D). There was a progressive prolongation of latencies with proximal stimulation before the CB while the distal motor latency remained unchanged, suggesting that there was focal slowing of conduction across the site of compression. Once 60–70% CB was achieved, the mice were returned to their cage. On waking from anesthesia (<1 h), the mice dragged their compressed leg for ∼12 h. The compression system used in our experiments was chosen to deliver a mild force and minimize the possibility of axonal transection.
To examine susceptibility to compression in Pmp22-deficient nerves, we performed the compression experiments in 19 pmp22+/− and 21 pmp22+/+ mice. The average time to induce CB in the pmp22+/− mice was 35 ± 13 min, which was significantly shorter than with wild-type mice (55 ± 38 min; p < 0.01).
Tibial nerves are located on the medial side of the ankle in mice as well as humans. To determine whether different contact angles between nylon cord and the vertical line of the mouse leg were affecting our results, we performed another set of compression experiments in which we altered the angle by 15 degrees (Fig. 1C, arrow). Similar results were observed. The mean duration for inducing CB between the pmp22+/− mice (n = 17) and wild-type (n = 17) was 31.3 ± 9.5 and 56.8 ± 14.8 min, which were again significantly different (p < 0.01).
Tibial nerves are cushioned by skin and subcutaneous tissues, which may vary from one mouse to another. In addition, the anatomical position of tibial nerves on the medial side of the ankle may also vary from mouse to mouse. To eliminate these potential factors, we surgically exposed sciatic nerves in the mice and compressed them directly using a vessel clamp (see technical details in Method section). Once again, the mean of duration for CB induction was shorter in pmp22+/− mice than that in pmp22+/+ mice (17.0 ± 4.4 vs 29.1 ± 5.8 min; n = 7 mice for each genotype; p < 0.001).
To ensure that the shortened induction of CB is not due to the reduced CMAP amplitudes in pmp22+/− mice, we reanalyzed our data from both sets of experiments using nylon cord compression (32 pmp22+/− or 35 pmp22+/+ mice). Mice were separated into low and high CMAP groups. No significant difference between these groups was found in the onset of CB (supplemental Table 1, available at www.jneurosci.org as supplemental material) (p > 0.05).
To determine whether the shortened induction of CB is specific to the deficiency of Pmp22, we performed similar studies with heterozygous mpz knock-out mice (mpz+/−). Like PMP22, MPZ is a major structural protein (>50% of all PNS myelin proteins) specific to compact myelin in the PNS (Shy et al., 2001). Deficiency of MPZ would be expected to negatively affect myelin stability. Indeed, homozygous deletion of mpz impairs myelin compaction (Martini et al., 1995), but abnormalities of myelin and axons occur even during embryonic stage and make the homozygotes unsuitable for control experiments. However, heterozygous null mpz (mpz+/−) mice do not develop significant electrophysiological and pathological abnormalities until the age of 6–12 months (Shy et al., 1997). Thus, before the age of 6 months old, they are an excellent animal model for controls. The induction time of CB on the vessel-clamp compressed sciatic nerves was not significantly different between the mpz+/+ and mpz+/− mice (25.3 ± 5.6 vs 25.3 ± 6.9 min; n = 6 mice for each genotypic group; p > 0.05). In addition, teased nerve fiber studies on the sciatic nerves showed no segmental demyelination or tomacula in mpz+/− mice. Together, these data demonstrate that a function of Pmp22 is to protect the nerve from mechanical injury, which is not necessarily related to myelin stability.
CB mechanically induced more rapidly in mag+/− nerves
Next, we performed the nerve compression in mag−/− mice using the vessel clamp technique. We chose these animals because homozygous mag null mice also develop tomacula. Tomacula have been detected in nerves of these mice at 1 month of age and affect 50% of paranodes in 3-month-old mag−/− mice (Yin et al., 1998). This prevalence of tomacula is comparable to that in 6-month-old pmp22+/− mice. Heterozygous mag-deficient mice (mag+/−), unlike heterozygous pmp22+/− animals, have no tomacula and no pathological phenotype (Yin et al., 1998). We therefore performed the compression experiments in mag−/− mice at 2 months of age. Surprisingly, teased nerve fiber examination showed no tomacula/axonal constrictions in mag−/− nerves. Nevertheless, CB was induced more rapidly in mag−/− mice than that in mag+/+ animals (9.0 ± 2.5 vs 20.9 ± 9.4 min; n = 7 mice for each genotypic group; p < 0.01). Since there were no tomacula in these homozygous Mag-deficient mice, this increased susceptibility to develop CB has to be caused by a different mechanism(s) than in the pmp22+/− mice. These findings suggest that vulnerability to mechanically induced CB in the peripheral nerves is not specific to Pmp22 deficiency, although the mechanisms for the CB may differ for different molecules.
Recovery of CB delayed in Pmp22-deficient nerves
NCS were repeated 3 or 5 d after the induction of CB in the first compression model, in which a nylon cord, attached to a weight, compressed the tibial nerve. CB completely recovered in 3 of 5 wild-type mice by the third day after compression and in 6 of the 7 wild-type mice by the fifth day (seventh with a partial recovery) (Table 2, Fig. 1D). In contrast, CB showed no complete recovery in any of 11 pmp22+/− mice at day 3 or day 5, and partial recovery in only half of the mice by day 5 (Table 2, Fig. 1E). These findings suggest that deficiency of Pmp22 results in slowed recovery from CB in the PNS.
Table 2.
Recovery of CB in pmp22+/− mice
| Genotype | No recovery |
Recovery |
||
|---|---|---|---|---|
| 3 d | 5 d | 3 d | 5 d | |
| Wild type | 2a/5b (40%) | 1/7 (14%) | 3c/5d (0pe) | 6/7 (1p) |
| pmp22+/− | 4/4 (100%) | 4/7 (57%) | 0/4 (0p) | 3/7 (2p) |
aAnimals with no recovery.
bTotal animals in this group.
cAnimals with recovery.
dTotal animals in this group.
e0p, No animals with partial recovery; 1p, one animal with partial recovery; 2p, two animals with partial recovery.
To evaluate the pathological changes in the compressed nerves, we next obtained semithin sections at transverse 2–3 mm distal to the compression site at both days 3 and 5 postcompression. Axons with some signs of acute Wallerian degeneration were found in 4 of 11 pmp22+/− mice (n = 12), but not in any of the wild-type mice (n = 12). However, only 2 of 12 pmp22+/− mice had severe acute axonal degeneration, as defined by over two-thirds of myelinated nerve fibers showing swelling and collapsed myelin (Fig. 2).
Figure 2.
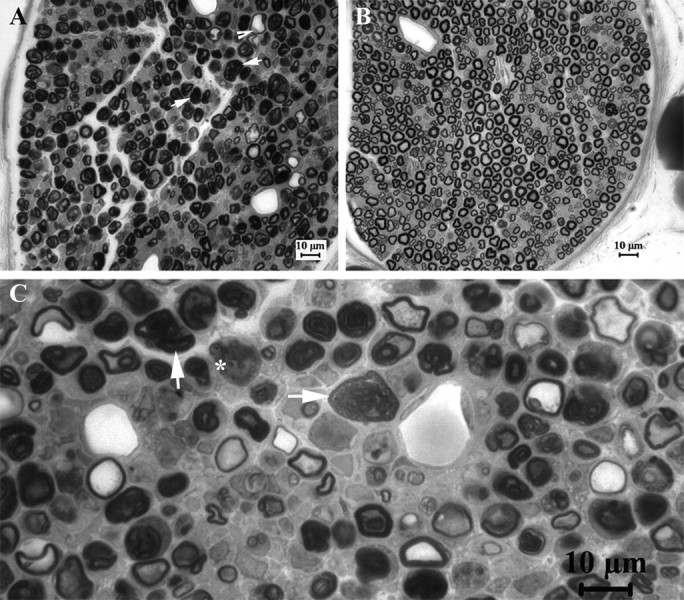
Axonal injuries in compressed PMP22-insufficient nerves. At postcompression days 3 and 5 we obtained semithin sections at transverse 2–3 mm distal to the compression site and examined them under light microscopy. Axons with signs of acute Wallerian degeneration, including extensively collapsed myelin (arrows in A and C), degenerated axons (asterisk in C), and swollen nerve fibers (arrowhead in A), were found in 4 of 11 pmp22+/− mice (n = 11) but not in wild-type mice (n = 12) (B). Two of these four pmp22+/− mice were severe (A). These results suggest that Pmp22 insufficiency may make nerve susceptible to axonal injuries in a small portion of animals.
We also evaluated the recovery over multiple time points, using the second compression model in which the sciatic nerve was compressed by a vessel clamp. The compressed nerves were harvested at day 0 (immediately after the compression), and at days 5, 7, 14, 21, and 30 (n = 3 mice for each time point). The reduced CMAP amplitudes induced by the proximal stimulations were completely recovered by day 7 in the wild-type mice (distal CMAP = 6.9 ± 2.6 mV vs proximal CMAP = 6.4 ± 2.5 mV; p > 0.05), but the recovery was delayed until day 14 in pmp22+/− mice (At day 7, distal CMAP = 2.4 ± 0.6 mV vs proximal CMAP = 1.9 ± 0.5 mV; At day 14, 2.7 ± 0.5 vs 2.6 ± 0.4, p > 0.05).
To determine whether axonal size of mutant or wild-type axons recovered differently following compression we evaluated axons at various time points after inducing CB. The nerves in both pmp22+/+ and pmp22+/− mice appeared stretched with smaller diameters, losing their normal undulated, “wave-like” appearance under light microscopy (Fig. 3). This undulated pattern and smaller diameters partially recovered at day 21, and returned to their normal state by day 30 postcompression in both pmp22+/+ and pmp22+/− nerves (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, persistent diffusely smaller diameters of axons could not explain the prolonged recovery from CB in the mutant animals. We could not determine whether focal constrictions of mutant nerves under tomacula changed following compression. The compressed nerves in all mice were fragile during mechanical manipulations such that any attempt at teasing nerve fibers introduced unacceptable artifact in the focally constricted region.
Figure 3.

Molecular architecture and septate junctions in naive and compressed nerves. A1–A3, Wild-type mice at 2–3 months old were perfused. Noncompressed sciatic nerves were dissected and teased into individual nerve fibers. Slides were stained with antibodies against MBP (blue) and Caspr (green). The former revealed internodal myelin (blue in A1). Caspr stained paranodes (arrows) that flanked the node of Ranvier. Caspr was also expressed at the Schmidt–Lanterman incisures (arrowhead in A2) and along the inner mesaxon as it spirals around the axon (arrow in A3). Voltage-gated potassium channels (Kv1.2; red in A3) were found in the juxtaparanodes (arrows in A3). Nav were concentrated in the node of Ranvier (arrowhead in A3) and appeared as a narrow band. B–E, From noncompressed pmp22+/− nerves. Nav, Kv1.2, Caspr, and MBP were all localized in their proper regions. In addition, MAG was also correctly localized in the paranodes (D, arrows) and incisures (arrowhead in D) similar to what was observed in the wild-type myelinated nerve fibers. Tomacula were found mainly in the paranodal regions (B, E) and almost always extended beyond the paranodes and into juxtaparanodes and internodes (between arrows in B and E). F, EM was performed on the longitudinal section of a noncompressed pmp22+/− sciatic nerve. Normal paranodal septate junctions (arrowhead array) were observed in the region with tomacula. G, H, Compressed sciatic nerves from the second compression model (surgically exposed and clamped sciatic nerve) (G) and noncompressed sciatic nerves (H) were sectioned into 10 μm thickness and stained with antibodies against Caspr. The localization of Caspr in the paranodes (arrowheads in G and insets and also in supplemental Fig. 1, available at www.jneurosci.org as supplemental material) was normal in compressed nerves. So was that of Kv1.2. However, axons revealed by YFP had smaller diameters and appeared stretched (G). Supporting this notion, we observed the typical undulating “wave” appearance of axons in noncompressed nerve (asterisk array in H). However, these waves disappeared in the compressed nerve (G), suggesting that the compressed nerve was physically stretched during the compression. Notice that the intensity of axonal YFP was much weaker in the compressed nerves. This change has been very helpful for precisely defining the region of compression (please see supplemental Fig. 1, available at www.jneurosci.org as supplemental material, for details). Within the compressed region, there were small islands of axons with strong intensity (arrows in G and in supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), which likely were spared from compression forces. Insets, Caspr localization in paranodes was visualized under high power.
Morphological examination at each time point demonstrated no or, at most, minimal damage in occasional axons in the compressed nerves of wild-type mice (Fig. 2; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, axonal damages were found in all compressed sciatic nerves of pmp22+/− mice, but only limited to a small portion of nerve fibers (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These degenerating axons were gradually diminishing during the recovery and were rarely detectable by day 30. In addition, on semithin sections, the prevalence of tomacula in the postcompressed nerves appeared similar to that in the nerves from the noncompressed side.
These results suggest that axonal injury was not contributing to CB in wild-type and played a minor role in Pmp22-deficient mice. However, the fact that a small portion of axons in Pmp22-deficient animals had some evidence of axonal damages suggested that Pmp22 insufficiency may make axons more susceptible to injury, which may contribute to the delayed recovery.
Compression does not cause axonal invagination, tomacula damage, or segmental demyelination in pmp22+/− nerves
Nodal invagination has been considered to be the basis of mechanically induced CB in a series of elegant studies by Gilliatt and his colleagues (Ochoa et al., 1971, 1972; Ochoa, 1972; Gilliatt et al., 1974; Rudge et al., 1974). In these studies, either a tourniquet or a nylon cord with an attached weight was applied to baboon's limb, and reversible CB was induced without significant axonal damage. The results suggested that the early phase of CB was caused by displacement of the nodes at the edge of compression, so that the nodes invaginated into the adjacent paranodal area. In addition, paranodal myelin swelling and rupture were also noticed. However, in our osmicated teased nerve fibers, these changes in nodes/paranodes were not found unless we used a much stronger vessel clamp (60 g/mm2) to induce CB. Otherwise CB developed in the absence of nodal invagination in our model when milder compression was used.
Tomacula have been considered to be an unstable pathological structure that degenerates as animals age (Adlkofer et al., 1997). We hypothesized that Pmp22-deficient myelin may also be unstable and could break down during compression, causing CB. To address this issue, we examined the compressed sciatic nerves from our second compression model, CB induced by clamping exposed sciatic nerve. In this model the compressed nerve segment was wider and much easier to be identified. Osmicated teased nerve fiber studies were performed in three pmp22+/− and three pmp22+/+ mice. None of the >100 randomly selected myelinated nerve fibers from either pmp22+/− or pmp22+/+ mice showed disruption of myelin. All tomacula examined were intact. Thus, stability of tomacula and myelin did not explain predisposition to CB in our pmp22+/− mice.
Molecular architecture and septate junctions of myelinated nerve fibers were normal in pmp22+/− mice
We next hypothesized that alterations of the molecular organization of pmp22+/− nerves might be responsible for the predisposition to develop CB, since these alterations are known to affect action potential propagation. Along these lines, recent investigations of myelinated axons and their nodes of Ranvier have demonstrated a surprising structural complexity, but very organized architecture, which is thought to be critical for the physiological functions of myelinated nerves (Scherer and Arroyo, 2002; Devaux and Scherer, 2005). To evaluate this architecture in our animals we performed teased nerve fiber immunohistochemistry (IHC) on noncompressed sciatic nerves from both pmp22+/− and pmp22+/+ mice. Proteins specific to each domain of myelinated nerve fibers, including node, paranode, juxtaparanode, and internode, appeared well expressed and organized identically in mutant and wild-type mice. These included voltage-gated sodium channels (Nav) in the nodes of Ranvier, Caspr, and MAG in the paranodes, Kv1.2 in juxtaparanode, and myelin basic protein (MBP) in internode (Fig. 3). Therefore, alterations in the localization of these molecules could not explain the predisposition to develop CB in pmp22+/− mice.
We next investigated septate junctions, the structures that connect paranodal loops of myelinating Schwann cell membrane onto the paranodal axolemma. Thus, they seal the inner mesaxon space and separate the juxtaparanodes from nodes. Disrupting septate junctions would allow outward current from juxtaparanodal potassium channels to counteract the inward current from nodal Nav, thereby raise the threshold of action potential production and decrease the safety factor for action potential propagation, resulting in CB (Lafontaine et al., 1982; Boyle et al., 2001). However, septate junctions in the paranodes with tomacula appeared normal under EM, in longitudinal ultrathin sections of noncompressed sciatic nerves (Fig. 3F, arrowheads).
Although we had demonstrated a normal molecular architecture in noncompressed Pmp22-deficient nerves we recognize that Pmp22 deficiency may predispose changes in the molecular architecture during compression. We dissected sciatic nerves from mutant and wild-type mice after CB was reached. IHC was performed with antibodies against Kv1.2 and Caspr. As with noncompressed nerves, no change of expression pattern of these proteins was found in the compressed sciatic nerves from either pmp22+/− or pmp22+/+ mice. However, axons appeared to have smaller diameters in the compressed nerves (Fig. 3G,H; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Concentration of Nav was normal in pmp22+/− mice
Although the localization of Nav appeared normal, we recognized that a decrease of Nav concentration may also predispose CB by reducing the safety factor for action potential production (Kearney et al., 2002). We therefore performed a [3H]-STX binding assay to determine Nav concentration in noncompressed peripheral nerves. Due to the overall low concentration of Nav in the peripheral nerves (Lombet et al., 1985), we had to collect nerves from 11 to 15 mice of each genotype and pooled membrane proteins together for each experiment. Three experiments were done. The mean of Nav concentration was 537 ± 153 fmol/mg in pmp22+/+ mice and 603 ± 132 fmol/mg in pmp22+/− mice; these differences were not significant (p > 0.05). Thus the total quantity of Nav could not explain the predisposition to CB in the mutant mice.
Axons encased by tomacula were constricted, a potential mechanism for shortened induction-time of CB in PMP22 insufficiency
Because there was no evidence of any myelin or molecular architecture change to explain the predisposition to CB in the preceding experiments, we next examined potential differences of axons between mutant and wild-type animals. We cross-bred pmp22+/− mice with YFPtg+/+ mice. Axons in all offspring were labeled by YFP, so that axonal structures can be visualized with great clarity.
We found focal constrictions in the axonal segments enclosed by tomacula in noncompressed pmp22+/− myelinated nerve fibers (Fig. 4B,C,F). We defined tomacula as a focal enlargement of myelin by 1/3 of internodal diameter of the myelinated fiber. Axonal constriction was defined as segments with diameters reduced by 1/3 of the internodal axonal diameter. Axonal diameters decrease naturally in the nodal and paranodal regions of a normal myelinated nerve fiber (Fig. 4A). Tomacula caused the axonal constriction to extend well beyond the paranodes. In some cases, tomacula caused a greater reduction in axonal caliber than seen at nodes and paranodes (Fig. 4B,C). To quantify this observation we randomly selected 20 paranodes with tomacula/axonal constrictions from 5 pmp22+/− mice and 20 paranodes without tomacula from five wild-type mice. Lengths of constricted axonal segments were measured in each paranode. The mean length of the constricted segments was 19.4 ± 4.9 μm for paranodes with tomacula/constrictions and 4.2 ± 0.2 μm for paranodes without tomacula (p < 0.0001) (Fig. 4G).
Figure 4.

Axonal constrictions in tomacula. All data in this figure were derived from noncompressed nerves. pmp22+/− mice were cross-bred with YFPtg+/+ mice, so that all axons were labeled by YFP expressed from the YFP transgene. Sciatic nerves were dissected, fixed, and teased into individual fibers, which were then stained with antibodies against Caspr. A1–A3, Three nodes from three myelinated nerve fibers were flanked by the Caspr-stained paranodes. Some Caspr spirals were also visible (arrowheads in A1). In the nodal/paranodal regions, there was a natural decrease of axonal diameter (between arrows in A2). There were no tomacula in these regions under phase-contrast imaging (overlay in A3). B1–B3, Another myelinated nerve fiber was examined and showed tomacula in both paranodes that flanked the node of Ranvier (arrowheads in B3). Axonal segments in the tomacula were constricted and the narrowed axonal segments extended far beyond the paranodal regions (between arrows in B3). C1–C3, A pmp22+/− myelinated nerve fiber had an asymmetry in its paranodes. The left paranode was affected by a tomacula with constricted axon (between arrows in C3). In contrast, the right paranode with no tomacula showed a normal axonal diameter. D, In some tomacula, axons may become enlarged (between arrows in D). E, The appearance of the enlarged axon in D is consistent with the EM finding in E. Axonal membranes occasionally became convoluted along the folding of myelin in tomacula (arrowhead array). This membrane would appear enlarged when it is labeled by YFP and viewed under light microscopy. F, A constricted axon on transverse section. G, The lengths of normal paranodal axons and constricted axonal segments were measured in randomly selected fibers (diameters of these selected fibers were comparable between the groups), and showed in this histogram (4.2 ± 0.2 μm for normal paranodal axons vs 19.4 ± 4.9 μm for constricted axonal segments; p < 0.0001).
The prevalence of tomacula and axonal constrictions was manually assessed in 50 randomly selected nodes from each 6-month-old pmp22+/− or pmp22+/+ mouse (n = 5 mice for each genotype) (Table 3). For most selected fibers, only one node in each myelinated fiber was evaluated. Regions with Schwann cell nucleus were avoided since there is a natural reduction of axonal diameter around the nucleus. Nearly half of the myelinated nerve fibers (48.4 ± 12.3%) contained tomacula in 6-month-old pmp22+/− mice. Tomacula were mainly found in the paranodal regions of large myelinated fibers (>5 μm) although they occasionally occurred in the internodes. Because only one node was examined in each selected myelinated fiber, the actual prevalence of tomacula may even have been higher. Focal constrictions were developed by 15.9 ± 4.8% of all evaluated axons. These constrictions were always within tomacula. Interestingly, a few axons in the tomacula (4.0 ± 2.5%) were enlarged by at least 1/3 of internodal axonal diameter (Fig. 4D). These enlargements presumably reflected axons that extended into myelin folds (Fig. 4E; supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material). Other axonal deformities within tomacula were also noticed (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Overall, these pathological changes (tomacula, axonal constriction, and enlargements) were present but slightly less (12% of evaluated axons) in 3-month-old pmp22+/− mice (Table 3).
Table 3.
Prevalence of tomacula and axonal constrictions in pmp22+/− mice
| Age | Tomacula (%) | Axonal constrictions (%) | Axonal enlargement (%) |
|---|---|---|---|
| 3 months (n = 6 mice) | 45.3 ± 0.4%a | 26.7 ± 7.5b or 12.1 ± 3.4a | 5.3 ± 3.7b |
| 6 months (n = 5 mice) | 48.4 ± 12.3%a | 32.0 ± 9.9b or 15.9 ± 4.8a | 4.0 ± 2.5b |
aPercentage of all counted myelinated nerve fibers.
bPercentage of myelinated fibers with tomacula.
Discussion
Our results demonstrate that mechanically induced CB occurs more rapidly and lasts longer in Pmp22-deficient nerves than in wild-type nerves. This finding was replicated in three sets of experiments by two different compression techniques. These findings confirm a hypothesis that has long been held concerning HNPP; namely that 50% of the normal PMP22 levels are inadequate to fully protect myelinated nerve fibers from compressive injury. Therefore one biological function of PMP22 must be protection from nerve injury.
The ease to develop CB cannot be explained simply because of reduced CMAP amplitudes in the mutant nerves (Fig. 1E). CB was determined by using P/D ratio, not the absolute value of CMAP amplitudes. Moreover, when induction time of CB was compared between two groups of mice with low and high CMAP, no significant difference was found (supplemental Table 1, available at www.jneurosci.org as supplemental material). Therefore, we believe that CB is directly related to Pmp22 deficiency.
We have identified focal axonal constrictions within tomacula as a novel potential cause of increased susceptibility to CB in Pmp22-deficient mice. Axonal constrictions were identified only within segments enclosed by tomacula and were triple/quadruple the length of constricted segments normally found in nodes/paranodes. Reduced axonal diameter rapidly raises axial resistance to action potential propagation since axial electric resistance of axons is inversely proportion to the square of axonal diameter (Hartline and Colman, 2007). Once the resistance increases to a critical level, CB occurs. We hypothesize that these multiple constricted axons within tomacula predispose the axons to CB. Furthermore, when the nerve is compressed, this causes even further stretching and thinning of axons (Fig. 3G; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). We postulate that this is the basis for the predisposition to CB observed in HNPP. Supporting our findings, reduced axonal diameters within tomacula have been identified in EM sections of sural nerve biopsies from patients with HNPP, although these findings were not quantified (Madrid and Bradley, 1975).
A potential concern for our hypothesis is whether the quantity of axonal constrictions we observed would be sufficient to predispose CB. Internodal length in mouse myelinated nerve fibers is 561 ± 136 μm (Perrot et al., 2007). A vessel clamp with a width of 1.5 mm (1500 μm) covers 2–3 nodes. When one node per myelinated nerve fiber was examined in 3-month-old pmp22+/− mice, 12% of fibers showed focal axonal constrictions (Table 3). Thus, up to 36% of fibers, and most of the largest diameter fibers, have constrictions in nerve segments that are covered by the vessel clamp, which also means that 36% of fibers under the compression of a 1.5-mm-width clamp are predisposed to CB. More importantly, it is the large myelinated fibers that are responsible for the major portion of CMAP, which is reduced in CB. For instance, with stronger and longer compression to achieve 60–100% CB in previous studies, pathological changes were still observed mainly in large myelinated fibers (Ochoa et al., 1972), and small fibers were spared (Ochoa et al., 1972; Fowler and Ochoa, 1975). In fact, early occurrence of slowing across the compression site in our experiments and previous compression studies (Rudge et al., 1974) also indicated that the large myelinated fibers were preferentially affected since conduction velocity measures the conduction of largest myelinated fibers (Gasser HS and Grundfest H, 1939). Finally, other pathological alterations in pmp22+/− nerves, such as the enlargements of paranodal axons in the tomacula (Fig. 4D,E), may also contribute to the rapid induction of CB. These convoluted axolemma may increase the total areas of paranodal axolemma and raise the capacitance, leading to a reduction of safety factor for action potential propagation (Joyner et al., 1980).
Our data do not claim that axonal constrictions in tomacula are the only cause for the CB, and the causal role of axonal deformities in the CB remains to be established. For example, wild-type mice also developed a decline in CMAP amplitudes after 5 min of compression and CB within 30 min. It simply occurs more rapidly in the mutant mice for a given level of compression. We do not know what other factors cause CB in wild-type animals. However the classical studies of Gilliatt and colleagues implicating nodal invagination cannot explain CB in our models since even wild-type mice did not demonstrate nodal invagination associated with their block unless stronger compression was applied.
How tomacula and axonal constrictions are formed remains a mystery. Tomacula are not unique to PMP22 insufficiency. They have been found in other neuropathies and animal models, including anti-MAG neuropathy, CMT1B, chronic inflammatory demyelinating neuropathy, Tangier's disease, as well as several animal models of peripheral nerve disorders (Sander et al., 2000; Cai et al., 2002, 2006). It has been hypothesized that tomacula result from excessive myelin folding collapsed on constricted axons in MAG knock-out mice (Yin et al., 1998). However, these findings have been challenged after focal hypermyelination was found in mag−/− mice at young age when axonal diameters were still normal (Cai et al., 2002). Thus, these findings suggest that tomacula are formed first and subsequently induce focal axonal constrictions by unknown mechanisms in MAG deficiency, such as abnormal Schwann cell-axon interaction. Whether this is also the case in PMP22 deficiency remains to be clarified. Whether there is an abnormal signaling through MAG in PMP22 deficiency is also unknown. Nevertheless, our data suggest that normal dosage of PMP22 is required to prevent formation of axonal constriction, which may be the mechanism of the protection it provides from nerve injury.
Susceptibility to mechanically induced CB was not restricted to PMP22-deficient nerves. It was also found in our mag−/− mice in the absence of tomacula. A recent study suggests that MAG deficiency destabilizes axons through a signaling pathway independent of Nogo protein complex.(Nguyen et al., 2009). Thus, results from both pmp22+/− and mag−/− nerves suggest that this susceptibility to CB is likely due to effects on the axon, rather than from myelin instability alone since this vulnerability was not found in Mpz-deficient nerves.
Finally, we found axonal damage in a small portion of pmp22+/− nerves that were compressed. These alterations disrupted axons between the proximal and distal stimulation sites and produce what might initially appear as CB (pseudo-CB). However, evidence of Wallerian degeneration was not seen in the vast majority of axons and is not a viable explanation for CB for our results. We do not know what other factors contributed to the delayed recovery from CB in the mutant mice. Our results did not show a delayed recovery from nerve stretching or an increased number of tomacula in the postcompressed nerve fibers. It is possible that a delayed recovery of focally constricted axons, under tomacula, is a contributing factor.
Although acute axonal damage occurred only rarely in pmp22+/− mice, it does have important clinical significance in humans. In one case report, a 20-year-old woman with HNPP developed massive axonal degeneration in her arms after strenuous physical activities, including running with a 34 kg backpack and performing 150 push-ups a day (Horowitz et al., 2004). We also identified a similar case. This 48-year-old man with HNPP developed arm weakness after heavy weight lifting. Needle EMG showed significant denervation in his arms, demonstrating that he had undergone acute Wallerian degeneration. Together, mechanical challenges during long or extraneous physical activities may be safe for healthy people, but could be dangerous to patients with HNPP.
Footnotes
This research is supported by grants from the National Institute of Neurological Disorders and Stroke (K08 NS048204 to J.L.), the Muscular Dystrophy Association (MDA 4029 to J.L.), the Veterans Administration (B6243R to J.L.), the Swiss National Science Foundation and the National Center of Competence and Research Neural Plasticity and Repair (to U.S.), the National Multiple Sclerosis Society (RG2882 and RG3771A4/3 to L.L.I.), the University of Michigan Training Program in Cellular and Molecular Biology (National Institutes of Health General Medicine GM007315-31 to H.O.), and University of Michigan Rackham Predoctoral Fellowship to H.O. We thank Ms. Yanmei Yuan for her technical assistance.
References
- Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- Adlkofer K, Frei R, Neuberg DH, Zielasek J, Toyka KV, Suter U. Heterozygous peripheral myelin protein 22-deficient mice are affected by a progressive demyelinating tomaculous neuropathy. J Neurosci. 1997;17:4662–4671. doi: 10.1523/JNEUROSCI.17-12-04662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amici SA, Dunn WA, Jr, Notterpek L. Developmental abnormalities in the nerves of peripheral myelin protein 22-deficient mice. J Neurosci Res. 2007;85:238–249. doi: 10.1002/jnr.21118. [DOI] [PubMed] [Google Scholar]
- Bai Y, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, Ceuterick-de Groote C, Mazanec R, Seeman P, Shy ME, Li J. R69C mutation in P0 gene alters myelination and ion channel subtypes. Arch Neurol. 2006;63:1787–1794. doi: 10.1001/archneur.63.12.1787. [DOI] [PubMed] [Google Scholar]
- Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. doi: 10.1016/j.neuron.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Cai Z, Sutton-Smith P, Swift J, Cash K, Finnie J, Turnley A, Thompson PD, Blumbergs PC. Tomacula in MAG-deficient mice. J Peripher Nerv Syst. 2002;7:181–189. doi: 10.1046/j.1529-8027.2002.02023.x. [DOI] [PubMed] [Google Scholar]
- Cai Z, Blumbergs PC, Cash K, Rice PJ, Manavis J, Swift J, Ghabriel MN, Thompson PD. Paranodal pathology in Tangier disease with remitting-relapsing multifocal neuropathy. J Clin Neurosci. 2006;13:492–497. doi: 10.1016/j.jocn.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Morrow CS, Hartshorne RP. Neurotoxin binding to receptor sites associated with voltage-sensitive sodium channels in intact, lysed, and detergent-solubilized brain membranes. J Biol Chem. 1979;254:11379–11387. [PubMed] [Google Scholar]
- Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- Chen C, Bharucha V, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ, 3rd, Kazen-Gillespie KA, Kazarinova-Noyes K, Shrager P, Saunders TL, Macdonald RL, Ransom BR, Scheuer T, Catterall WA, Isom LL. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci U S A. 2002;99:17072–17077. doi: 10.1073/pnas.212638099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornblath DR, Sumner AJ, Daube J, Gilliat RW, Brown WF, Parry GJ, Albers JW, Miller RG, Petajan J. Conduction block in clinical practice. Muscle Nerve. 1991;14:869–871. doi: 10.1002/mus.880140913. discussion 867–868. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Scherer SS. Altered ion channels in an animal model of Charcot–Marie–Tooth disease type IA. J Neurosci. 2005;25:1470–1480. doi: 10.1523/JNEUROSCI.3328-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol. 1968;18:603–618. doi: 10.1001/archneur.1968.00470360025002. [DOI] [PubMed] [Google Scholar]
- Einheber S, Zanazzi G, Ching W, Scherer S, Milner TA, Peles E, Salzer JL. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J Cell Biol. 1997;139:1495–1506. doi: 10.1083/jcb.139.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler TJ, Ochoa J. Unmyelinated fibers in normal and compressed peripheral nerves of the baboon: a quantitative electron microscopic study. Neuropathol Appl Neurobiol. 1975;1:247–265. [Google Scholar]
- Gasser HS, Grundfest H. Axon diameter in relation to the spike dimentions and conduction velocity in mammalian A fibers. Am J Physiol. 1939;127:393–414. [Google Scholar]
- Gilliatt RW, McDonald WI, Rudge P. Proceeding: the site of conduction block in peripheral nerves compressed by a pneumatic tourniquet. J Physiol. 1974;238:31P–32P. [PubMed] [Google Scholar]
- Hartline DK, Colman DR. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr Biol. 2007;17:R29–R35. doi: 10.1016/j.cub.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Horowitz SH, Spollen LE, Yu W. Hereditary neuropathy with liability to pressure palsy: fulminant development with axonal loss during military training. J Neurol Neurosurg Psychiatry. 2004;75:1629–1631. doi: 10.1136/jnnp.2003.029314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA. Functional co-expression of the beta 1 and type IIA alpha subunits of sodium channels in a mammalian cell line. J Biol Chem. 1995;270:3306–3312. doi: 10.1074/jbc.270.7.3306. [DOI] [PubMed] [Google Scholar]
- Jetten AM, Suter U. The peripheral myelin protein 22 and epithelial membrane protein family. Prog Nucleic Acid Res Mol Biol. 2000;64:97–129. doi: 10.1016/s0079-6603(00)64003-5. [DOI] [PubMed] [Google Scholar]
- Joyner RW, Westerfield M, Moore JW. Effects of cellular geometry on current flow during a propagated action potential. Biophys J. 1980;31:183–194. doi: 10.1016/S0006-3495(80)85049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji R. Physiology of conduction block in multifocal motor neuropathy and other demyelinating neuropathies. Muscle Nerve. 2003;27:285–296. doi: 10.1002/mus.10273. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Buchner DA, De Haan G, Adamska M, Levin SI, Furay AR, Albin RL, Jones JM, Montal M, Stevens MJ, Sprunger LK, Meisler MH. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6) Hum Mol Genet. 2002;11:2765–2775. doi: 10.1093/hmg/11.22.2765. [DOI] [PubMed] [Google Scholar]
- Lafontaine S, Rasminsky M, Saida T, Sumner AJ. Conduction block in rat myelinated fibres following acute exposure to anti-galactocerebroside serum. J Physiol. 1982;323:287–306. doi: 10.1113/jphysiol.1982.sp014073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, Sumner AJ, Shy ME. Electrophysiological features of inherited demyelinating neuropathies: a reappraisal in the era of molecular diagnosis. Muscle Nerve. 2000;23:1472–1487. doi: 10.1002/1097-4598(200010)23:10<1472::aid-mus3>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Li J, Krajewski K, Shy ME, Lewis RA. Hereditary neuropathy with liability to pressure palsy: the electrophysiology fits the name. Neurology. 2002;58:1769–1773. doi: 10.1212/wnl.58.12.1769. [DOI] [PubMed] [Google Scholar]
- Li J, Krajewski K, Lewis RA, Shy ME. Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2004;29:205–210. doi: 10.1002/mus.10521. [DOI] [PubMed] [Google Scholar]
- Li J, Bai Y, Ghandour K, Qin P, Grandis M, Trostinskaia A, Ianakova E, Wu X, Schenone A, Vallat JM, Kupsky WJ, Hatfield J, Shy ME. Skin biopsies in myelin-related neuropathies: bringing molecular pathology to the bedside. Brain. 2005;128:1168–1177. doi: 10.1093/brain/awh483. [DOI] [PubMed] [Google Scholar]
- Li J, Ghandour K, Radovanovic D, Radovanovic D, Shy RR, Krajewski KM, Shy ME, Nicholson GA. Stoichiometric alteration of PMP22 protein determines the phenotype of HNPP. Arch Neurol. 2007;64:974–978. doi: 10.1001/archneur.64.7.974. [DOI] [PubMed] [Google Scholar]
- Lombet A, Laduron P, Mourre C, Jacomet Y, Lazdunski M. Axonal transport of the voltage-dependent Na+ channel protein identified by its tetrodotoxin binding site in rat sciatic nerves. Brain Res. 1985;345:153–158. doi: 10.1016/0006-8993(85)90846-7. [DOI] [PubMed] [Google Scholar]
- Madrid R, Bradley G. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): studies on the formation of the abnormal myelin sheath. J Neurol Sci. 1975;25:415–448. [Google Scholar]
- Marazzi R, Pareyson D, Scaioli V, Corbo M, Boiardi A, Chiodelli G, Sghirlanzoni A. Recurrent familial neuropathy due to liability to pressure palsies. Ital J Neurol Sci. 1988;9:355–363. doi: 10.1007/BF02333999. [DOI] [PubMed] [Google Scholar]
- Martini R, Mohajeri MH, Kasper S, Giese KP, Schachner M. Mice doubly deficient in the genes for P0 and myelin basic protein show that both proteins contribute to the formation of the major dense line in peripheral nerve myelin. J Neurosci. 1995;15:4488–4495. doi: 10.1523/JNEUROSCI.15-06-04488.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon D. Isolation of a cDNA clone coding for a putative second potassium channel indicate the existence of a gene family. J Biol Chem. 1989;264:8230–8236. [PubMed] [Google Scholar]
- Nguyen T, Mehta NR, Conant K, Kim KJ, Jones M, Calabresi PA, Melli G, Hoke A, Schnaar RL, Ming GL, Song H, Keswani SC, Griffin JW. Axonal protective effects of the myelin-associated glycoprotein. J Neurosci. 2009;29:630–637. doi: 10.1523/JNEUROSCI.5204-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa J. Ultrathin longitudinal sections of single myelinated fibres for electron microscopy. J Neurol Sci. 1972;17:103–106. doi: 10.1016/0022-510x(72)90026-3. [DOI] [PubMed] [Google Scholar]
- Ochoa J, Danta G, Fowler TJ, Gilliatt RW. Nature of the nerve lesion caused by a pneumatic tourniquet. Nature. 1971;233:265–266. doi: 10.1038/233265a0. [DOI] [PubMed] [Google Scholar]
- Ochoa J, Fowler TJ, Gilliatt RW. Anatomical changes in peripheral nerves compressed by a pneumatic tourniquet. J Anat. 1972;113:433–455. [PMC free article] [PubMed] [Google Scholar]
- Perrot R, Lonchampt P, Peterson AC, Eyer J. Axonal neurofilaments control multiple fiber properties but do not influence structure or spacing of nodes of Ranvier. J Neurosci. 2007;27:9573–9584. doi: 10.1523/JNEUROSCI.1224-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AM, Perea J, McGuigan A, King RH, Muddle JR, Gabreëls-Festen AA, Thomas PK, Huxley C. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J Anat. 2002;200:377–390. doi: 10.1046/j.1469-7580.2002.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudge P, Ochoa J, Gilliatt RW. Acute peripheral nerve compression in the baboon. J Neurol Sci. 1974;23:403–420. doi: 10.1016/0022-510x(74)90158-0. [DOI] [PubMed] [Google Scholar]
- Sancho S, Young P, Suter U. Regulation of Schwann cell proliferation and apoptosis in PMP22-deficient mice and mouse models of Charcot-Marie-Tooth disease type 1A. Brain. 2001;124:2177–2187. doi: 10.1093/brain/124.11.2177. [DOI] [PubMed] [Google Scholar]
- Sander S, Ouvrier RA, McLeod JG, Nicholson GA, Pollard JD. Clinical syndromes associated with tomacula or myelin swellings in sural nerve biopsies. J Neurol Neurosurg Psychiatry. 2000;68:483–488. doi: 10.1136/jnnp.68.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Arroyo EJ. Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst. 2002;7:1–12. doi: 10.1046/j.1529-8027.2002.02001.x. [DOI] [PubMed] [Google Scholar]
- Shy ME, Arroyo E, Sladky J, Menichella D, Jiang H, Xu W, Kamholz J, Scherer SS. Heterozygous P0 knock-out mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP) J Neuropathol Exp Neurol. 1997;56:811–821. [PubMed] [Google Scholar]
- Shy ME, Balsamo J, Lilien J, Kamholz J. A molecular basis for hereditary motor and sensory neuropathy disorders. Curr Neurol Neurosci Rep. 2001;1:77–88. doi: 10.1007/s11910-001-0079-6. [DOI] [PubMed] [Google Scholar]
- Ulzheimer JC, Peles E, Levinson SR, Martini E. Altered expression of ion channel isoforms at the node of Ranvier in P0-deficient myelin mutants. Mol Cell Neurosci. 2004;25:83–94. doi: 10.1016/j.mcn.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Mizuno R, Nishimura T, Ogawa Y, Yoshikawa H, Fujimura H, Adachi E, Kishimoto T, Yanagihara T, Sakoda S. Cloning and expression of myelin-associated oligodendrocytic basic protein. A novel basic protein constituting the central nervous system myelin. J Biol Chem. 1994;269:31725–31730. [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa H, Dyck PJ. Uncompacted inner myelin lamellae in inherited tendency to pressure palsy. J Neuropathol Exp Neurol. 1991;50:649–657. doi: 10.1097/00005072-199109000-00009. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chow CY, Sahenk Z, Shy ME, Meisler MH, Li J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain. 2008;131:1990–2001. doi: 10.1093/brain/awn114. [DOI] [PMC free article] [PubMed] [Google Scholar]