

Abstract
Despite its specific clinical relevance, the field of hematopoietic stem cell mobilization has received broad attention, owing mainly to the belief that pharmacologic stem cell mobilization might provide clues as to how stem cells are retained in their natural environment, the bone marrow ‘niche’. Inherent to this knowledge is also the desire to optimally engineer stem cells to interact with their target niche (such as after transplantation), or to lure malignant stem cells out of their protective niches (in order to kill them), and in general to decipher the niche’s structural components and its organization. Whereas, with the exception of the recent addition of CXCR4 antagonists to the armamentarium for mobilization of patients refractory to granulocyte colony-stimulating factor alone, clinical stem cell mobilization has not changed significantly over the last decade or so, much effort has been made trying to explain the complex mechanism(s) by which hematopoietic stem and progenitor cells leave the marrow. This brief review will report some of the more recent advances about mobilization, with an attempt to reconcile some of the seemingly inconsistent data in mobilization and to interject some commonalities among different mobilization regimes.
Keywords: mobilization, CXCR4, G-CSF, hematopoietic stem cell, bone marrow, niche
HEMATOPOIETIC STEM CELL MOBILIZATION: IMPACT BEYOND CLINICAL APPLICATION
Despite its specific clinical relevance, the field of hematopoietic stem cell (HSC) mobilization has received broad attention, owing mainly to the belief that pharmacologic stem cell mobilization might provide clues as to how stem cells are retained in their natural environment, the bone marrow (BM) ‘niche’. Inherent to this knowledge is also the desire to optimally engineer stem cells to interact with their target niche (such as after transplantation), or to lure malignant stem cells out of their protective niches (in order to kill them), and in general to decipher the niche’s structural components and its organization. Whereas, with the exception of the recent addition of CXCR4 antagonists to the armamentarium for mobilization of patients refractory to granulocyte colony-stimulating factor (G-CSF) alone,1 clinical stem cell mobilization has not changed significantly over the last decade or so, much progress has been made trying to explain the complex mechanism(s) by which hematopoietic stem and progenitor cells (HSPCs) leave the marrow. This brief review will report some of the more recent advances about mobilization, with an attempt to reconcile some of the seemingly inconsistent data in mobilization and to interject some commonalities among different mobilization regimes. In addition, we will discuss putative mechanisms opposing mobilization, as the vast majority of HSPCs are retained inside the marrow and is not mobilized. Therefore, because of its restrictive nature, this is not an exhaustive review of mobilization, as this need is supplanted by several recent reviews, including in the excellent Spotlight Series ‘Perspective on Stem cell Homing and Mobilization’ in this journal, dedicated to Dr McCulloch (http://www.nature.com/leu/spotlights/stem_cell_mobilization.html).2–13 (See also Cancelas et al.76 and Winkler and Levesque94 for some thorough reviews focussing on specific pathways in mobilization).
WHERE THE MOBILIZED HSCS COME FROM: BONE AND VASCULAR NICHES FOR HSCS
With respect to the microanatomic location of HSC niches—close to the bone (endosteal niche)14 or close to vessels (vascular niche)15,16—evidence has been provided by protagonists from either fraction. Alternatively, the presence of HSCs in both compartments has been described,17,18 although HSCs close to vessels could represent HSCs in transition, preparing either to leave the bone or to engage in niches favoring proliferation/differentiation. In a recent perspective article by Bianco,19 we were reminded of the significant body of evidence that in normal cancellous bone, vessels and inner bone surfaces are never more than a few micrometers apart. The conclusion of that manuscript was that it should be evident that the endosteal vs vascular niche concept applies neither to the bulk of murine primitive hematopoiesis (which is predominantly located in cancellous bone) nor to adult human hematopoiesis, which is exclusively located in the axial skeleton and the proximal and distal ends of long bones, entirely made up of spongious bone. ‘The apparent multiplicity of the landmarks of the scene can be profitably reinterpreted by assuming that the HSC niche does not have a fixed anatomy’.19 We fully subscribe to these views and therefore suggest that the anatomical situation dictates that HSCs are at any given time simultaneously perivascularly and endosteally located and simultaneously receive cues both from the bone and blood. We propose that it is precisely this flexibility that allows the stem cell niche to respond and adapt to systemic cues.
IS THE STEM CELL NICHE REALLY HYPOXIC?
The high abundance of capillaries in cancellous bone is in our opinion incompatible with the sometimes proposed hypoxia in BM HSC niches. Also, conceptually it is fair to surmise that a highly proliferative organ like hematopoietic BM requires considerable quantities of oxygen and nutrients. Blood flow to marrow was estimated as 0.2 ml/min*g,20 which is more similar to cerebral blood flow (0.5 ml/min*g), that is considered an oxygen-rich environment, than to adipose tissue blood flow (0.04 ml/min*g), which is unarguably relatively hypoxic.21 Lower oxygen tension in BM than in arterial blood (or in a standard incubator, where it is 160 mm Hg) is an inevitable consequence of physics, but an inflationary use of the term hypoxia to describe this fact should be avoided. The estimated oxygen partial pressure in BM is 55 mm Hg.22 In a recent review, Suda et al.23 discuss the matter of hypoxic stem cell niches in some detail and refer to the oxygen pressure of 55 mm Hg as ‘severely hypoxic’. Given the average normal pO2 in the cerebrum of 15–30 mm Hg, this interpretation has only a relative meaning.24 As is further argued in that manuscript, their strong antioxidative machinery as well as the evidence provided of their largely anaerobic metabolism protect HSCs from oxidative stress. Were their natural habitat truly hypoxic, why would such machinery be required? Furthermore, even though localizing HSCs in situ remains challenging, a look at a histological section of hematopoietic BM and all images of HSCs in situ thus far presented very much suggest that HSCs do not dwell in anatomically separate compartments but are distributed throughout the BM17 likely in immediate proximity to their highly proliferative immature and mature progeny. Published data demonstrating relative hypoxia in a G-CSF-stimulated marrow25 are possibly more indicative of increased oxygen consumption owing to the induced proliferation than of G-CSF-induced reduction of oxygenation. Moreover, compared with hematopoietic marrow, fatty marrow such as is found in adult long bones of larger mammals is likely very hypoxic, considering its low capillary density, yet in this hypoxic environment HSCs are no longer found. On the contrary, after long-bone fracture, an abundance of vessels sprout into the fracture zone, so that bone repair can take place, and transiently hematopoietic marrow repopulates the fracture cleft until its consolidation, as was formally documented in the rabbit and monkey models several decades ago.26 Thus active hematopoietic marrow seems to seek out, or possibly induce comparatively high-oxygen BM environments. Furthermore, no evidence has been provided as yet that the self-renewal of HSCs in purportedly hypoxic areas is very different from that in higher-oxygen environments, that is, BM HSCs vs circulating HSCs. Also, as stem cells and progenitor cells are mobilized at the same time by many different approaches and quantitatively fluctuate in tandem, this can be interpreted to suggest anatomic proximity in BM. Therefore, if we consider the evidence of high perfusion and high-oxygen pressure in BM, the putative proximity between HSCs and the enormously efficient and energy-consuming proliferation/differentiation/egress machinery for mature blood cells, as well as the ample armamentarium against oxygen stress with which HSCs are equipped, it is hard to defend the severely hypoxic niche concept.
At least in long bones, BM regeneration after ‘myeloablative’ conditioning, such as after lethal irradiation, occurs from the inner bone surface toward the central region, apparently because vessels transcending the bone are relatively preserved and sprout into the necrotic marrow cavity (Figure 1). In the postirradiation situation thus indeed the region to which transplanted cells must home and engraft is close to the bone, while the niche repairs itself and expands to regenerated marrow regions.27 Thus, in discussing issues such as stem cell localization, care should be taken to consider inherent differences between normal and radiation-damaged BM hematopoiesis (issues discussed in Jiang et al.27).
Figure 1.
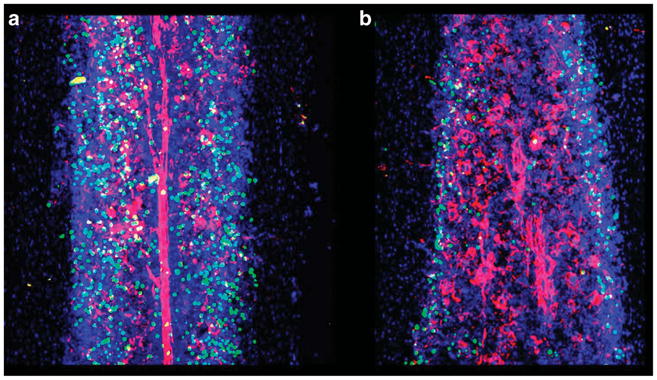
Irradiation destroys marrow integrity and shifts the stem cell niche toward the endosteal region: mice were lethally irradiated (b) or not (a). Identical numbers of carboxyfluoroscein succinimidyl ester (green)-labeled lineage-negative/kit + cells were injected intravenously within 3 h of irradiation. Note the severe vascular distortion, particularly in the central regions of bones, and the preferentially endosteal localiztion of transplanted cells in tibiae of irradiated mice. Moreover, the total number of cells homing to non-irradiated BM was considerably larger than in irradiated hosts, in keeping with previous reports. Z-stack sections (a total of consecutive 25 sections from tibiae) were done 20 h after cell transfer. Blood vessels were labeled with CD31 (red) and nuclei were counter-stained with 4′,6-diamidino-2-phenylindole.
CIRCULATING CELLS DURING STEADY STATE
Unlike most other organs, the hematopoietic system is distributed throughout the body. The function of the system must be orchestrated by soluble factors, which will stochastically induce on average similar amounts of activation, proliferation and differentiation in all the different sites. If each spongious bone compartment was a separate vessel for a small number of stem cells without the ability to replenish them from the outside, then in that bone failed self-renewal divisions could extinguish hematopoiesis. At other bone sites, excessive self-renewal could lead to accumulation of HSCs until their number exceeds the hematopoietic capacity of their retention. Therefore, in order to maintain approximate equal numbers at all sites at all times, a certain ‘leakiness’ of stem cell retention, that is, low-level release of HSCs, must occur from marrow to circulation to replenish distal sites of hematopoiesis. This way, a continuous exchange of HSCs from distant marrow sites is enabled, which is expected to stabilize the system. The mechanisms responsible for allowing egress of the few circulating stem/progenitor cells at baseline, as well as the modestly elevated numbers of circulating stem/progenitor cells during minor hematopoietic stresses, such as infection or exercise, are mechanistically not well understood. Enforced mobilization has been studied in much greater detail. Whether neuronal (sympathetic) or humoral (cytokines, chemokines, prostaglandins, sphingolipids, endotoxin from gut biota and many others) signals implicated in enforced mobilization are integrated in very similar ways, as during baseline mobilization, implying thereby an exaggeration of (certain facets of) physiological stem cell egress, is not entirely clear.
It is likely an adaptive process to have higher levels of circulating HSPCs during minor hematopoietic stress, as they can contribute to recruitment of stem cells into proliferation/differentiation pathways in many sites. During hematopoietic stress, a transient local skewing of the ratio between self-renewal and differentiating cell divisions could occur, resulting in exhaustion of the stem cell pool in this bone. Thus, circulating stem cells originating from distant sites can immigrate into and occupy vacant stem cell niches, to maintain immature hematopoiesis at all bone sites life long. Partial body irradiation experiments performed many years ago clearly demonstrate the ability of circulating HSPCs (rather mobilized by systemic stimuli) to home to radiation-damaged bone from distant marrow sites and to regenerate completely the destroyed hematopoiesis.28,29 Engraftment of circulating HSPCs to distant bone sites was also demonstrated decades ago in parabiotic mice.30–33 In contrast, recruitment of stroma-regenerating cells to the damaged bone has been largely undocumented.34–36 As the example of allogeneic stem cell transplantation indicates, BM stroma ‘stem’ cells are fairly resilient to chemotherapy and total body irradiation. With the exception of BM transplantation in osteogenesis imperfecta mice and possibly even patients, which are characterized by severe osteoblast deficiency,37–40 after HSC transplantation the BM stroma remains exclusively of recipient type, even when cell products relatively abundant in stroma cells are transplanted (that is, a BM cell transplant, as opposed to one by mobilized peripheral blood (PB) stem cells), or when transplants are performed in patients with putative hematopoietic stroma insufficiency, such as in aplastic anemia.41–44 Stromal cell repair and their competence to provide supportive function for HSPCs is achieved likely by mutual interactions (cross talk) between hematopoietic cells and host stroma cells.
Compared with most other organs in the body, the two components participating in hematopoiesis, blood and bone, are characterized by highly dynamic behavior and considerable plasticity. Both are known to integrate hematopoietic stimuli, as evidenced by effects of G-CSF on the bone, marrow stroma and hematopoiesis during mobilization (see below).
ENFORCED STEM CELL EGRESS OR MOBILIZATION: THE QUICK MOBILIZERS AND THEIR TARGETS
When suitable agents are used, mobilization can occur briskly with a very short time lag. Thus, in the case of the CXCR4 antagonist AMD3100, cells appear in circulation within 30 min of administration;1,45 mobilization with Groβ or IL-8 is even faster.46,47 Importantly, this observation would suggest that HSCs do not have to migrate very far before reaching the blood stream. Thus, some HSCs would be located close to BM vasculature, thereby representing vascular niches for ‘mobilizable’ HSCs.
The molecular mechanism by which AMD3100 works, as advocated in all published work, is by blocking CXCR4 signaling.48 However, as to which cells (hematopoietic or mesenchymal) are targeted by AMD3100 to bring about the egress of HSPCs, alternative hypotheses have been proposed.1,48,49
If we believe that AMD3100 has no way of selecting CXCR4 on one cell over another, then blockade of CXCR4 on hematopoietic stem/progenitor cells will be achieved concurrently with CXCR4 blockade on many other cell types expressing the receptor, including the bone/BM stroma cells. Once CXCR4 is blocked, HSPCs lose their sensitivity to CXCL12. In this situation, the pericellular concentration of CXCL12 should be quite irrelevant for their behavior or fate, as AMD3100-treated HSPCs cannot sense CXCL12. Incubation experiments of HSPCs with AMD3100 followed by studies of CXCL12-dependent functions in vitro indeed support this notion.45 The overwhelmingly preferred mode of action of CXCR4 antagonists therefore is by blocking CXCR4 on HSPCs and thus directly depriving HSPCs of a retention signal.1 This proposed mode of action is also entirely compatible with data generated in mice with CXCR4-deficient hematopoietic cells but CXCR4-competent stroma (see below).15,50,51 Subsequently, the CXCR4-unresponsive cells are either attracted into the blood by a positive stimulus (none has been identified, but Sphingosine 1 phosphate (S1P) might be a candidate),11,52–55 repulsed by a repellent (although no candidates have been proposed) or they passively migrate into blood once deprived of a retaining signal. As relevant stem cell mobilization was achieved with the pan-Gi-protein blocker Pertussis toxin,56 which blocks CXCR4 and S1P (edg) receptors alike, as well as most other chemokine receptors, the latter option appears the most likely.
An alternative mechanism that was put forth implies that after AMD3100 treatment, HSPCs retain their responsiveness to CXCL12, as AMD3100 targets only stromal cells producing CXCL12, thereby altering CXCL12 concentrations in BM and indirectly inducing HSPC egress.49 How selective binding of AMD3100 to stromal cells could be achieved is hard to fathom. Loss or inversion of CXCL12 gradient between BM and PB has been shown by competitive displacement of stroma-bound CXCL12 by dextran sulfate.57–59
For the purpose of discussion, we propose a third theoretically conceivable but never seriously contemplated mechanism. AMD3100 might not actually cause mobilization, but that by binding to ‘spontaneously’ circulating HSPCs it might trap cells in the blood stream or prolong their transit time until clearance from the circulation. If this were the case, then the ‘spontaneous’ turnover of HSPCs between blood and tissues would have to be extremely rapid, and a number of other points also argue against this hypothesis. This hypothesis would also imply that in the mouse at least 2000 or so (2000 if homing under AMD3100 was exactly zero, more if some residual clearance from blood remained possible) hematopoietic progenitor cells would spontaneously leave the marrow every hour, so that at least once every 20 days the entire pool of hematopoietic progenitor cells would have recirculated once. However, the kinetics of the natural turnover of HSPCs between marrow and blood have been a controversial point, and accumulation of circulating HSPCs with repeated injections of AMD3100 would have been expected and is in contrast to experimental data.1,45 Furthermore, given the accumulated evidence indicating that homing of HSPCs is largely independent of CXCR4/CXCL12 (see below), how CXCR4 blockade should trap cells in blood is unclear.
Direct imaging of qualitative and quantitative occupation of CXCR4 by AMD3100 on HSPCs in blood or marrow or on BM stroma cells after in vivo application has not been achieved, because thus far attempts at coupling AMD3100 to fluorescent markers have failed. Such studies would provide significant direct insight of the working mechanism. To this end, however, recent studies with human donors treated with AMD3100 showed differential binding of CXCR4 antibodies 1D9 and 12G5.60 Both bind to CXCR4, one to the N-terminal tail and the other to the transmembrane loops. Both are displaced by CXCL12, but only the latter by the much smaller AMD3100. The antibody staining pattern on cells from AMD3100-mobilized individuals—1D9 upregulated and 12G5 blocked—very much suggests that indeed AMD3100 is bound to the cells and blocks subsequent CXCL12 binding. Upregulation of 1D9 binding is interpreted as compensatory in nature, as the cells no longer sensing CXCL12 (as the receptors are blocked by the antagonist) mobilize additional CXCR4 from intracellular stores. In aggregate, the accumulated evidence suggests that AMD3100 binding to HSPCs (and possibly other cells) renders them insensitive to CXCL12-mediated retention and hence directly triggers their egress from marrow.
The precise mechanisms of mobilization by Groβ and IL-8 are not fully understood; neutrophil protease-mediated effects have been proposed,46,61 with CXCL12 as a potential downstream target.62 IL-8-mediated mobilization failed in G-CSF receptor-deficient mice.63 These agents, as well as AMD3100,48 do clearly indicate that proliferation is not required for mobilization. The kinetics of mobilization would simply not support preceding proliferation. On the same vein alternative evidence, that proliferation by itself is not sufficient for mobilization, has been provided by G-CSF receptor-deficient mice, which after treatment with cyclophosphamide show dramatic proliferation of HSPCs in marrow but fail to mobilize efficiently.63 Thus, neither proliferation per se is a prerequisite for mobilization nor is its absence an impediment to mobilization. It does, however, serve to increase the size of mobilizable HSPC pools and thus in this context is probably quite relevant for its efficacy.
Recently, certain sphingolipids have garnered attention with respect to their role in HSPC mobilization.11,54,55 Given that these are among the few chemoattractants for HSPCs53 and that their blood levels are the highest of any tissue (because of significant S1P production by erythrocytes), S1P could theoretically be the opposing force to CXCL12 in BM, with HSPCs tethered in BM between these two forces in a labile equilibrium (see above).56 Recent data by the Ratajczak laboratory would be compatible with that hypothesis.55 However, like other mobilizing agents, an alternative mode of action of S1P has been proposed, that is, the targeting by S1P of CXCL12-producing marrow stroma cells, with reduction of CXCL12 and consequent HSPC egress.52 We are looking forward to future definitive experiments stringently testing these hypotheses.
ROLES OF CXCR4/CXCL12 IN STEM CELL MAINTENANCE
The effects of CXCL12 on primitive cells are complex and despite significant progress remain only partly understood because of some shortcomings of experimental models. On the one hand, CXCL12, elaborated by stromal cells, such as osteo-lineage cells64 or other mesenchymal cells like the chemokine-abundant reticular cells15,65 or nestin-positive stroma cells,66 serves as ‘glue’ that attaches HSCs to niche cells (it is not an adhesion molecule per se, but attracts HSCs to stromal surfaces while activating several integrins). CXCL12 downregulation, or blockade of its cognate receptor, is sufficient to allow egress of HSCs. How can this be achieved? Link and colleagues67 recently proposed that immature granulocytes are tethered in a skewed, yet labile, equilibrium between CXCL12 in BM (retaining the cells) and CXCL2 in PB. According to this view, as granulocytes mature, their sensitivity to CXCL12 decreases, resulting in relative weakening of the forces keeping granulocytes in BM and increasing net granulocyte egress into blood. Thus, the authors currently favor the model of HSCs as a naturally itinerant specimen that will spontaneously move into the PB unless specifically restrained by niche cues, including but probably not limited to CXCL12. Considering the considerable risks that cells with self-renewal capacity, telomerase activity and vulnerability to oncogenic mutations are subjected to under peripheral-blood high-oxygen conditions, it would seem the safest to keep these cells away from PB environment. Modest downregulation of CXCL12 in BM and a concomitant increase in PB have been observed after several days of treatment with G-CSF.68 Although marrow concentrations of CXCL12 remained several orders of magnitude higher than in blood (that is, no gradient inversion was observed), a 10-fold smaller excess of BM-CXCL12 was achieved compared with baseline and it is conceivable that this weakening of restraining forces alone might be sufficient to allow net HSC egress from marrow. That after G-CSF treatment the CXCR4/CXCL12 axis remains partly intact is evidenced by the fact that addition of AMD3100 to G-CSF results in additive-to-synergistic augmentation of mobilization.1
The molecular pathways of homing have sometimes been proposed to represent the opposite (or mirror image) of mobilization. While its role in mobilization is well documented, the role of CXCR4/CXCL12 in homing of HSCs after intravenous application is more complex and therefore merits some discussion. As an unspecific damage signal,69,70 after systemic chemotherapy and/or total body irradiation, CXCL12 is upregulated in all tissues, not only in BM. If chemoattraction only affected the trafficking of HSCs, then in conditioned hosts HSCs would be rather less likely to preferentially home to marrow than any other tissue. From studies with CXCR4-deficient BM hematopoietic cells, as well as with BM HSPCs treated with CXCR4 antagonists or Pertussis toxin, it is clear that the CXCR4/CXCL12 pathway does not critically mediate homing to BM, as homing was not impaired under these experimental conditions,50,51,71,72 although controversial data have been published.73 Thus, with respect to CXCL12 it may be hard to defend the mirror image theory of homing and mobilization. However, homing of mobilized PB HSPCs or cytokine-incubated HSPCs was AMD3100 or Pertussis toxin sensitive71,74 or sensitive to anti-functional CXCR4 antibodies,73 and cord blood HSPC homing was completely blocked by anti-functional antibodies.73 Potential differences between sources of HSPCs used in these experiments might be attributed to reduced integrin expression and/or avidity on circulating vs BM-residing cells. Lack of Vav1(ref. 75) or Rac1/2 expression,76 both displaying normal homing, but defective retention, also do not seem to support the mirror image metaphor of homing vs mobilization.
In addition to acting as a retention factor, CXCL12 has been proposed to mediate, directly or indirectly, quiescence of hematopoietic cells or to bias HSCs against differentiation. After genetic ablation of CXCR4, HSCs were elevated in PB,15,50,51 through lack of retention, lack of quiescence or both. This phenotype is reminiscent to one in mice treated with CXCR4 inhibitors.1,45,77,78 As germline CXCR4 ablation is perinatally lethal, several different models of conditional CXCR4 ablation were presented, by generation of radiation chimerae with CXCR4−/− cells or induced ablation in adult mice. The reported phenotypes of animals with CXCR4-deficient hematopoiesis are surprisingly divergent (reviewed in Rettig et al.13). CXCR4−/− fetal liver cells provided reduced radioprotection, and mice reconstituted with these cells developed marrow failure over time.50 A similar phenotype was observed when ablation was induced in CXCR4f/f.mx-cre + mice by injection of poly I:C.15 By contrast, CXCR4Δ/Δ.tam-cre + mice not only maintained marrow hematopoiesis, but actually had increased HSC numbers in BM despite high numbers of circulating HSPCs.51 The latter phenotype—high HSPCs in marrow despite very high circulating HSPCs—is reminiscent to that of mice deficient for alpha4 integrin in their hematopoietic system: circulating HSPCs are significantly increased life-long; spleens are large and harbor large numbers of transplantable cells.79 Moreover, we have provided evidence that, like deletion of CXCR4, deletion of VLA4 is associated with slightly increased proliferation in the stem/progenitor cell population, which apparently suffices to compensate for the retention deficit of VLA4-deficient HSCs and to maintain normal hematopoiesis at homeostasis. However, under stress conditions, restoration of hematopoiesis impaired. The reasons why early hematopoiesis is impaired in CXCR4−/− HSCs are not entirely clear, although several hypotheses have been put forth. Absence of CXCR4 could lead to HSC cell death, to their emigration from marrow, or to their exhaustion through increased downsteam differentiative divisions. To clarify some of these issues, effects of reversible CXCR4 inhibition, for example, by long-term pharmacological suppression of CXCR4, or by studies in genetic on–off models may be insightful.
HSPCs themselves as well as their mature progeny are also a source of CXCL12. Autocrine stimulation of blood cells with CXCL12 has been demonstrated,80 but the role of cell-intrinsic CXCL12 for HSPCs in vivo has not been fully defined. Studies in mice lacking the ligand (CXCL12) were hampered by the lethal phenotype of total CXCL12 deficiency. Stroma-specific ablation in different types of ‘niche’ cells would be informative to test whether hematopoietic phenotypes of stroma-deleted CXCL12-deficient and hematopoiesis-deleted CXCR4 mice are similar. Thorough analyses of hematopoietic-specific CXCL12 knockouts in adults are not yet available. However, transplantation of CXCL12-deficient fetal liver cells was not associated with a dramatic phenotype.81 CXCL12-deficient HSCs engrafted with equivalent efficiency as wild-type HSCs and were retained and maintained stemness post transplant in wild-type recipients with a CXCL12-competent stroma. However, one generally assumes that nature is not wasteful, so that some role for HSPC-derived CXCL12 may exist.
G-CSF-MEDIATED MOBILIZATION
After several days of stimulation with G-CSF, BM cellularity is markedly increased, and egress of mature and immature cells including true HSCs is observed. Left behind is a marrow environment partially destroyed by proteases, with attenuated bone (endosteal surfaces), depleted osteo-macs (mature hematopoietic cells that serve as nursing cells for osteoblasts), suppressed osteoblasts, leaky blood vessels, as well as a host of dead hematopoietic cells.3,5,62,82–86 Some reports suggest that the hematopoietic self-renewing stem cells also take a transient hit, so that the repopulating capacity of post-G-CSF BM is attenuated, but this remains controversial.1,87,88
Levesque et al.88 recently suggested that egress of the most immature cells actually precedes peak progenitor cell mobilization by at least 1 day. Interpretation of this observation is not straightforward. Either HSCs are truly mobilized first (that is, reside closest to vessels or are retained the least firmly) or they are relatively diluted in BM by the ongoing stimulation with G-CSF, which favors myeloid differentiation. However, even in the best of circumstances (continuous infusion of G-CSF for 5 days), in C57Bl/6 mice only about 20 000 CFU-C/ml or 40 000 CFU-C/mouse, representing 4% of the CFU-Cs contained in the entire animal (ca. 10E6), are found in circulation, while the rest stay behind. For the proposed mechanisms involved in G-CSF-mediated mobilization, which include downregulation and cleavage of CXCL12 and disruption of VCAM-1/VLA4 by cleavage of the former and downregulation of the latter, it is clear that in G-CSF-mediated mobilization these pathways are only partially disrupted, as additional direct interference with either pathway (G-CSF +AMD3100 or G-CSF +anti-VLA4) resulted in additive-to-synergistic mobilization.1,45,77,78 The basis for the additive-to-synergistic mobilization likely is the enlargement of mobilizable pools of immature hematopoietic cells under the influence of G-CSF. We would therefore propose that the cumulative data indicate that G-CSF is a modestly potent mobilizing agent and that its quite satisfactory clinical efficacy hinges largely on combined effects of proliferation48 (expansion of mobilizable pools) and mobilization.
G-CSF-MEDIATED MOBILIZATION AND THE VEGETATIVE NERVOUS SYSTEM
Several recent studies have suggested roles of sympathetic output on circulating HSPCs at steady state and under G-CSF in mice.89 Experiments were presented showing that sympathectomy or pharmacologic or genetic reduction of β-adrenergic output or β-adrenergic receptor blockade reduced spontaneous and enforced egress, while stimulation of these pathways augmented mobilization.49,89,90 According to these studies, sympathetic nerve endings abound in BM, with a direct impact on circulating HSPC numbers. An alternative hypothesis that β-adrenergic signals maintain niche (cell) integrity and that their perturbation thus indirectly leads to stem cell egress is also possible. Most recently, the hypothesis was put forth that G-CSF affects mobilization by acting as a potent noradrenalin reuptake inhibitor and data were presented showing modestly augmented G-CSF-mediated mobilization in mice under extended treatment with a classic non-selective tricyclic antidepressant that acts as a noradrenalin reuptake inhibitor.91 Use of such compounds in human donors was proposed to augment clinical mobilization responses to G-CSF. The proposition that G-CSF had reuptake inhibitor effects was surprising, as reuptake inhibitors are associated with characteristic, frequent and rather dramatic idiosyncratic adverse effects, particularly during treatment initiation. Despite careful observation, no such effects were ever reported in donors.92,93 Within our database of mobilized volunteer stem cell donors, we therefore took the opportunity to identify donors taking noradrenalin reuptake inhibitors or β-receptor blockers, in order to investigate potential effects thereof on G-CSF-induced stem cell mobilization in humans. The mobilization response (concentration of circulating CD34 + cells) of volunteer donors taking (because of some pre-existing condition) a drug from either group was compared to that in concurrently treated volunteer donors receiving neither class of agents. Careful analyses confirmed that all donors had received the same dosing schedule of G-CSF and that the three groups were indistinguishable with respect to donor demographics. According to the hypotheses put forth for mice, the former should have mobilized better and the latter should have mobilized less well. However, not even a trend towards differential mobilization was observed (Figure 2), suggesting at least that different mechanisms are involved in mobilization between mice and men.
Figure 2.

Modification of sympathetic input does not affect mobilization efficiency with G-CSF: volunteer stem cell donors receiving noradrenalin reuptake inhibitors for depression (NRI, n =14) or β-blockers for hypertension (β-Bl., n =24), or 559 concurrent donors not receiving any drugs interfering with sympathetic tone (ctrl.) received a 5-day course of standard-dose G-CSF (9 doses q12 h, 7.5–10 μg/kgBW*day) in preparation for matched-unrelated stem cell apheresis at our facility. Circulating CD34 + cells were enumerated using single-platform flow cytometry 2–4 h after the ninth dose, as described.102 Donors receiving NRI or β-blockers were typical for MURD donors,92 that is, in terms of donor epidemiology (age, sex and body mass index), there were no differences between the groups. Mobilization was neither enhanced by NRI nor suppressed by β-blockers (mean + s.d.).
CELLULAR AND MOLECULAR TARGETS FOR STEM CELL MOBILIZATION
Many careful reviews, including recent ones, have summarized the knowledge surrounding stem cell mobilization by G-CSF and alternative mobilizing agents.2–7 Whereas, many cellular targets have been identified for G-CSF (osteoblasts,64 osteocytes, CAR cells15 and nestin-positive Mesenchymal Stem Cells66) and mature hematopoietic cells (osteo-macs94 and granulocytes63), our knowledge about molecular targets and mediators remains only partially defined. As discussed above, almost all proposed mechanisms converge on interference with the CXCL12/CXCR4 pathway, predominantly by reducing stromal CXCL12, which is then proposed to unleash HSPCs and result in their egress. It is apparently not necessary to reduce CXCL12 levels to levels below or at blood/plasma levels; a modest reduction suffices to allow egress of HSPCs. This observation seems to be compatible with the ‘labile equilibrium’ hypothesis we proposed above.
Similar to the mechanism of G-CSF-induced downregulation of CXCL12 in BM, implemented through a complex chain of events (discussed above), is the mechanism of cyclophosphamide-mediated mobilization. Reduction of CXCL12 in BM stroma was also proposed as the mechanism of mobilization by CXCR4 antagonists,49 although this is not universally accepted and a specific cellular target has not been proposed. Additional mediators purportedly targeting CXCL12, which have, however, not achieved clinical relevance, include components of the complement system (through elaboration of proteolytic enzymes from mature cells), and signals from the sympathetic nervous system (by directly targeting BM stroma cells—see above).
The only other molecular mechanism, partly independent of CXCL12/CXCR4, consistently implicated in HSPC mobilization with G-CSF is the modification of adhesive interactions of HSPCs with marrow stroma, through downregulation of integrins, either by cleavage of integrin receptors as a consequence of mature blood cell-derived proteases induced by G-CSF, or other undefined mechanisms. Integrins can also be targeted directly with anti-functional antibodies or small-molecule inhibitors, where it appeared that competition of the antagonists for integrin-binding sites displaced HSPCs from marrow.77,78,95 Thus roles for several beta1 and beta2 integrins have been reported. Although probably too weak to gain relevance for clinical mobilization, these observations are insightful from the point of view of the molecular tapestry of the HSC niche.
Of importance, with all the described manipulations, CXCL12 concentrations always remain significantly higher in BM than in PB, and CXCR4, on all mobilized cells ever studied, is expressed and retains or quickly regains its responsiveness to CXCL12. Thus the fact that these comparatively subtle changes in BM result in HSPC egress may indicate that the equilibrium between HSPC retention and egress is very labile, and minor environmental changes suffice to induce HSPC egress.
As a paradigm for such a mechanism, Link and colleagues67 recently proposed a model by which granulocytes are induced to leave the marrow: the cells are suspended in an equilibrium between retaining (CXCL12-mediated) and mobilizing (CXCL2-mediated) forces. Changes in either—reduced retaining or enhanced mobilizing forces—can induce granulocytes to leave the marrow. Conceivably, similar principles might apply to HSPCs. Unlike the granulocytes, the ‘pulling’ force for HSPCs has not been identified. HSPCs are notoriously finicky in their responsiveness to chemoattractants.96 Ratajczak et al. have provided updated information about chemoattractant properties of components of the complement system,13,97–101 but the cognate receptors are not expressed on HSPCs. Recently, a role for S1P in HSPC mobilization was suggested, although whether S1P targets HSPCs directly, or whether S1P exerts its effect on HSPC egress indirectly, through mature hematopoietic or BM stroma cells currently remains elusive (see above).
That CXCL12/CXCR4 will be confirmed as the one central molecular mechanism upon which all mobilizing agents converge should not always be taken for granted, but current evidence implies at least a strong contributory role in almost all mobilizing schemes we know today.
SUMMARY
The considerable interest in the molecular and cellular pathways involved in stem cell mobilization continues to fuel active research by a number of excellent groups, which continue to provide novel and surprising insights. At a close look, however, we should realize that of the dozens of potential targets that have been proposed, based mostly on genetic but also pharmacological data in mice, for liberating HSCs from marrow, very few have been formally tested and even fewer have shown the predicted success in humans. As far as the evidence goes in humans, a number of hematopoietic cytokines have been associated with proliferation of immature cells and differentiation to mature cells that can facilitate egress by enzymatically destroying marrow integrity, including some stromal retention molecules (primarily G-CSF, but also GM-CSF and SCF), direct interference with CXCR4 has been successfully translated into the clinic and interference with the alpha4 integrin has at least provided proof-of-principle data. Targeting of the same three pathways (activation of G-CSF receptor, inhibition of CXCR4 or VLA4) is currently being tested as a means of improving the efficiency of chemotherapy for various hemoblastoses. As scientists will hone in on the HSC in its niche in years to come, we predict that many of the proposed retention (as well as stemness promoting) mechanisms do nothing more than maintaining the architectural integrity of the niche, and their disruption induces HSCs to ‘perish’ or to ‘abandon ship’.
Acknowledgments
Susanne Bräuninger and Darja Karpova are acknowledged for assembly of the mobilization data shown in Figure 1. HB acknowledges research funding by the German Federal Ministry for Education and Research (BMBF, Ci3), the Federal State of Hesse (LOEWE CGT and LOEWE OSF), Deutsche Krebshilfe 108031 and DFG BO 3553/1-1. TP’s research is funded through NIH Grants HL58734 and DK094702.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208:421–428. doi: 10.1084/jem.20110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenbaum AM, Link DC. Mechanisms of G-CSF-mediated hematopoietic stem and progenitor mobilization. Leukemia. 2011;25:211–217. doi: 10.1038/leu.2010.248. [DOI] [PubMed] [Google Scholar]
- 4.Hoggatt J, Pelus LM. Many mechanisms mediating mobilization: an alliterative review. Curr Opin Hematol. 2011;18:231–238. doi: 10.1097/MOH.0b013e3283477962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levesque JP, Winkler IG. Mobilization of hematopoietic stem cells: state of the art. Curr Opin Organ Transplant. 2008;13:53–58. doi: 10.1097/MOT.0b013e3282f42473. [DOI] [PubMed] [Google Scholar]
- 6.Mohty M, Ho AD. In and out of the niche: perspectives in mobilization of hematopoietic stem cells. Exp Hematol. 2011;39:723–729. doi: 10.1016/j.exphem.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Ratajczak MZ, Kim C. The use of chemokine receptor agonists in stem cell mobilization. Expert Opin Biol Ther. 2012;12:287–297. doi: 10.1517/14712598.2012.657174. [DOI] [PubMed] [Google Scholar]
- 8.Ratajczak MZ, Kim CH, Wojakowski W, Janowska-Wieczorek A, Kucia M, Ratajczak J. Innate immunity as orchestrator of stem cell mobilization. Leukemia. 2010;24:1667–1675. doi: 10.1038/leu.2010.162. [DOI] [PubMed] [Google Scholar]
- 9.Levesque JP, Helwani FM, Winkler IG. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia. 2010;24:1979–1992. doi: 10.1038/leu.2010.214. [DOI] [PubMed] [Google Scholar]
- 10.Ratajczak MZ. Spotlight series on stem cell mobilization: many hands on the ball, but who is the quarterback? Leukemia. 2010;24:1665–1666. doi: 10.1038/leu.2010.181. [DOI] [PubMed] [Google Scholar]
- 11.Ratajczak MZ, Kim CH, bdel-Latif A, Schneider G, Kucia M, Morris AJ, et al. A novel perspective on stem cell homing and mobilization: review on bioactive lipids as potent chemoattractants and cationic peptides as underappreciated modulators of responsiveness to SDF-1 gradients. Leukemia. 2012;26:63–72. doi: 10.1038/leu.2011.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winkler IG, Pettit AR, Raggatt LJ, Jacobsen RN, Forristal CE, Barbier V, et al. Hematopoietic stem cell mobilizing agents G-CSF, cyclophosphamide or AMD3100 have distinct mechanisms of action on bone marrow HSC niches and bone formation. Leukemia. 2012;26:1594–1601. doi: 10.1038/leu.2012.17. [DOI] [PubMed] [Google Scholar]
- 13.Rettig MP, Ansstas G, DiPersio JF. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia. 2012;26:34–53. doi: 10.1038/leu.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis SL, Grassinger J, Jones A, Borg J, Camenisch T, Haylock D, et al. The relationship between bone, hemopoietic stem cells, and vasculature. Blood. 2011;118:1516–1524. doi: 10.1182/blood-2010-08-303800. [DOI] [PubMed] [Google Scholar]
- 15.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 16.Kiel MJ, Morrison SJ. Maintaining hematopoietic stem cells in the vascular niche. Immunity. 2006;25:862–864. doi: 10.1016/j.immuni.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- 18.Lo CC, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianco P. Bone and the hematopoietic niche: a tale of two stem cells. Blood. 2011;117:5281–5288. doi: 10.1182/blood-2011-01-315069. [DOI] [PubMed] [Google Scholar]
- 20.Szabo Z, Szabo G. The effect of haemorrhage and bone fracture on bone marrow circulation. Res Exp Med (Berl) 1978;172:7–17. doi: 10.1007/BF01851061. [DOI] [PubMed] [Google Scholar]
- 21.Karpe F, Fielding BA, Ardilouze JL, Ilic V, Macdonald IA, Frayn KN. Effects of insulin on adipose tissue blood flow in man. J Physiol. 2002;540(Pt 3):1087–1093. doi: 10.1113/jphysiol.2001.013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99:394. doi: 10.1182/blood.v99.1.394. [DOI] [PubMed] [Google Scholar]
- 23.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Pennings FA, Schuurman PR, van den MP, Bouma GJ. Brain tissue oxygen pressure monitoring in awake patients during functional neurosurgery: the assessment of normal values. J Neurotrauma. 2008;25:1173–1177. doi: 10.1089/neu.2007.0402. [DOI] [PubMed] [Google Scholar]
- 25.Levesque JP, Winkler IG, Hendy J, Williams B, Helwani F, Barbier V, et al. Hematopoietic progenitor cell mobilization results in hypoxia with increased hypoxia-inducible transcription factor-1 alpha and vascular endothelial growth factor A in bone marrow. Stem Cells. 2007;25:1954–1965. doi: 10.1634/stemcells.2006-0688. [DOI] [PubMed] [Google Scholar]
- 26.Van DD, Harris N. Bone marrow reactions to trauma. Stimulation of erythropoietic marrow by mechanical disruption, fracture or endosteal curettage. Blood. 1969;34:257–275. [PubMed] [Google Scholar]
- 27.Jiang Y, Bonig H, Ulyanova T, Chang K, Papayannopoulou T. On the adaptation of endosteal stem cell niche function in response to stress. Blood. 2009;114:3773–3782. doi: 10.1182/blood-2009-05-219840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Croizat H, Frindel E, Tubiana M. The effect of partial body irradiation on haemopoietic stem cell migration. Cell Tissue Kinet. 1980;13:319–325. doi: 10.1111/j.1365-2184.1980.tb00470.x. [DOI] [PubMed] [Google Scholar]
- 29.Murate T, Utsumi M, Hotta T, Yamada H. Hematopoietic stem cell migration and proliferation after partial body irradiation: significant role of the spleen in hematopoietic recovery. Nihon Ketsueki Gakkai Zasshi. 1983;46:867–875. [PubMed] [Google Scholar]
- 30.Abkowitz JL, Robinson AE, Kale S, Long MW, Chen J. Mobilization of hematopoietic stem cells during homeostasis and after cytokine exposure. Blood. 2003;102:1249–1253. doi: 10.1182/blood-2003-01-0318. [DOI] [PubMed] [Google Scholar]
- 31.McBride RA, Simonsen M. Cellular and humoral phenomena during the inductive phase of parabisos tolerance. Transplantation. 1965;3:140–154. doi: 10.1097/00007890-196503000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Walker DG. Osteopetrosis cured by temporary parabiosis. Science. 1973;180:875. doi: 10.1126/science.180.4088.875. [DOI] [PubMed] [Google Scholar]
- 33.Harris JE, Ford CE, Barnes DW, Evans EP. Evidence from parabiosis for an afferent stream of cells. Nature. 1964;201:886–887. doi: 10.1038/201886a0. [DOI] [PubMed] [Google Scholar]
- 34.Barisic-Dujmovic T, Boban I, Clark SH. Fibroblasts/myofibroblasts that participate in cutaneous wound healing are not derived from circulating progenitor cells. J Cell Physiol. 2010;222:703–712. doi: 10.1002/jcp.21997. [DOI] [PubMed] [Google Scholar]
- 35.Boban I, Barisic-Dujmovic T, Clark SH. Parabiosis model does not show presence of circulating osteoprogenitor cells. Genesis. 2010;48:171–182. doi: 10.1002/dvg.20602. [DOI] [PubMed] [Google Scholar]
- 36.Stute N, Fehse B, Schroder J, Arps S, Adamietz P, Held KR, et al. Human mesenchymal stem cells are not of donor origin in patients with severe aplastic anemia who underwent sex-mismatched allogeneic bone marrow transplant. J Hematother Stem Cell Res. 2002;11:977–984. doi: 10.1089/152581602321080646. [DOI] [PubMed] [Google Scholar]
- 37.Otsuru S, Hofmann TJ, Rasini V, Veronesi E, Dominici M, Horwitz EM. Osteopoietic engraftment after bone marrow transplantation: effect of inbred strain of mice. Exp Hematol. 2010;38:836–844. doi: 10.1016/j.exphem.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dominici M, Marino R, Rasini V, Spano C, Paolucci P, Conte P, et al. Donor cell-derived osteopoiesis originates from a self-renewing stem cell with a limited regenerative contribution after transplantation. Blood. 2008;111:4386–4391. doi: 10.1182/blood-2007-10-115725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marino R, Martinez C, Boyd K, Dominici M, Hofmann TJ, Horwitz EM. Transplantable marrow osteoprogenitors engraft in discrete saturable sites in the marrow microenvironment. Exp Hematol. 2008;36:360–368. doi: 10.1016/j.exphem.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le BK, Gotherstrom C, Ringden O, Hassan M, McMahon R, Horwitz E, et al. Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation. 2005;79:1607–1614. doi: 10.1097/01.tp.0000159029.48678.93. [DOI] [PubMed] [Google Scholar]
- 41.Scopes J, Ismail M, Marks KJ, Rutherford TR, Draycott GS, Pocock C, et al. Correction of stromal cell defect after bone marrow transplantation in aplastic anaemia. Br J Haematol. 2001;115:642–652. doi: 10.1046/j.1365-2141.2001.03134.x. [DOI] [PubMed] [Google Scholar]
- 42.Laver J, Jhanwar SC, O’Reilly RJ, Castro-Malaspina H. Host origin of the human hematopoietic microenvironment following allogeneic bone marrow transplantation. Blood. 1987;70:1966–1968. [PubMed] [Google Scholar]
- 43.Hollings PE, Fitzgerald PH, Heaton DC, Beard ME. Host origin of in vitro bone marrow fibroblasts after marrow transplantation in man. Int J Cell Cloning. 1984;2:348–355. doi: 10.1002/stem.5530020603. [DOI] [PubMed] [Google Scholar]
- 44.Wilson FD, Konrad PN, Greenberg BR, Klein AK, Walling PA. Cytogenetic studies on bone marrow fibroblasts from a male-female hematopoietic chimera. Evidence that stromal elements in human transplantation recipients are of host type. Transplantation. 1978;25:87–88. doi: 10.1097/00007890-197802000-00010. [DOI] [PubMed] [Google Scholar]
- 45.Bonig H, Chudziak D, Priestley G, Papayannopoulou T. Insights into the biology of mobilized hematopoietic stem/progenitor cells through innovative treatment schedules of the CXCR4 antagonist AMD3100. Exp Hematol. 2009;37:402–415. doi: 10.1016/j.exphem.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King AG, Horowitz D, Dillon SB, Levin R, Farese AM, MacVittie TJ, et al. Rapid mobilization of murine hematopoietic stem cells with enhanced engraftment properties and evaluation of hematopoietic progenitor cell mobilization in rhesus monkeys by a single injection of SB-251353, a specific truncated form of the human CXC chemokine GRObeta. Blood. 2001;97:1534–1542. doi: 10.1182/blood.v97.6.1534. [DOI] [PubMed] [Google Scholar]
- 47.Laterveer L, Lindley IJ, Hamilton MS, Willemze R, Fibbe WE. Interleukin-8 induces rapid mobilization of hematopoietic stem cells with radioprotective capacity and long-term myelolymphoid repopulating ability. Blood. 1995;85:2269–2275. [PubMed] [Google Scholar]
- 48.Zhao M, Ross JT, Itkin T, Perry JM, Venkatraman A, Haug JS, et al. FGF signaling facilitates post-injury recovery of mouse hematopoietic system. Blood. 2012;120:1831–1842. doi: 10.1182/blood-2011-11-393991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dar A, Schajnovitz A, Lapid K, Kalinkovich A, Itkin T, Ludin A, et al. Rapid mobilization of hematopoietic progenitors by AMD3100 and catecholamines is mediated by CXCR4-dependent SDF-1 release from bone marrow stromal cells. Leukemia. 2011;25:1286–1296. doi: 10.1038/leu.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foudi A, Jarrier P, Zhang Y, Wittner M, Geay JF, Lecluse Y, et al. Reduced retention of radioprotective hematopoietic cells within the bone marrow microenvironment in CXCR4−/− chimeric mice. Blood. 2006;107:2243–2251. doi: 10.1182/blood-2005-02-0581. [DOI] [PubMed] [Google Scholar]
- 51.Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205:777–783. doi: 10.1084/jem.20072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golan K, Vagima Y, Ludin A, Itkin T, Cohen-Gur S, Kalinkovich A, et al. S1P promotes murine progenitor cell egress and mobilization via S1P1-mediated ROS signaling and SDF-1 release. Blood. 2012;119:2478–2488. doi: 10.1182/blood-2011-06-358614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juarez JG, Harun N, Thien M, Welschinger R, Baraz R, Pena AD, et al. Sphingosine-1-phosphate facilitates trafficking of hematopoietic stem cells and their mobilization by CXCR4 antagonists in mice. Blood. 2012;119:707–716. doi: 10.1182/blood-2011-04-348904. [DOI] [PubMed] [Google Scholar]
- 54.Ratajczak MZ, Lee H, Wysoczynski M, Wan W, Marlicz W, Laughlin MJ, et al. Novel insight into stem cell mobilization-plasma sphingosine-1-phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia. 2010;24:976–985. doi: 10.1038/leu.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ratajczak J, Kucia M, Mierzejewska K, Liu R, Kim CH, Natarajan N, et al. A novel view of paroxysmal nocturnal hemoglobinuria pathogenesis: more motile PNH hematopoietic stem/progenitor cells displace normal HSPCs from their niches in bone marrow due to defective adhesion, enhanced migration and mobilization in response to erythrocyte-released sphingosine-1 phosphate gradient. Leukemia. 2012;26:1722–1725. doi: 10.1038/leu.2012.46. [DOI] [PubMed] [Google Scholar]
- 56.Papayannopoulou T, Priestley GV, Bonig H, Nakamoto B. The role of G-protein signaling in hematopoietic stem/progenitor cell mobilization. Blood. 2003;101:4739–4747. doi: 10.1182/blood-2002-09-2741. [DOI] [PubMed] [Google Scholar]
- 57.Sweeney EA, Papayannopoulou T. Increase in circulating SDF-1 after treatment with sulfated glycans. The role of SDF-1 in mobilization. Ann N Y Acad Sci. 2001;938:48–52. doi: 10.1111/j.1749-6632.2001.tb03573.x. [DOI] [PubMed] [Google Scholar]
- 58.Sweeney EA, Lortat-Jacob H, Priestley GV, Nakamoto B, Papayannopoulou T. Sulfated polysaccharides increase plasma levels of SDF-1 in monkeys and mice: involvement in mobilization of stem/progenitor cells. Blood. 2002;99:44–51. doi: 10.1182/blood.v99.1.44. [DOI] [PubMed] [Google Scholar]
- 59.Sweeney EA, Priestley GV, Nakamoto B, Collins RG, Beaudet AL, Papayannopoulou T. Mobilization of stem/progenitor cells by sulfated polysaccharides does not require selectin presence. Proc Natl Acad Sci USA. 2000;97:6544–6549. doi: 10.1073/pnas.97.12.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–3924. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van PM, van OR, Velders GA, Hagoort H, Heegaard PM, Lindley IJ, et al. Serpina1 is a potent inhibitor of IL-8-induced hematopoietic stem cell mobilization. Proc Natl Acad Sci USA. 2006;103:1469–1474. doi: 10.1073/pnas.0510192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111:187–196. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu F, Poursine-Laurent J, Link DC. The granulocyte colony-stimulating factor receptor is required for the mobilization of murine hematopoietic progenitors into peripheral blood by cyclophosphamide or interleukin-8 but not flt-3 ligand. Blood. 1997;90:2522–2528. [PubMed] [Google Scholar]
- 64.Christopher MJ, Liu F, Hilton MJ, Long F, Link DC. Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood. 2009;114:1331–1339. doi: 10.1182/blood-2008-10-184754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagasawa T, Omatsu Y, Sugiyama T. Control of hematopoietic stem cells by the bone marrow stromal niche: the role of reticular cells. Trends Immunol. 2011;32:315–320. doi: 10.1016/j.it.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 66.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106:3020–3027. doi: 10.1182/blood-2004-01-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN, Dabeva MD, et al. HGF SDF-1, and MMP-9 are involved in stress-induced human CD34 + stem cell recruitment to the liver. J Clin Invest. 2003;112:160–169. doi: 10.1172/JCI17902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110:3300–3305. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 71.Bonig H, Priestley GV, Papayannopoulou T. Hierarchy of molecular-pathway usage in bone marrow homing and its shift by cytokines. Blood. 2006;107:79–86. doi: 10.1182/blood-2005-05-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wiesmann A, Spangrude GJ. Marrow engraftment of hematopoietic stem and progenitor cells is independent of Galphai-coupled chemokine receptors. Exp Hematol. 1999;27:946–955. doi: 10.1016/s0301-472x(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 73.Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 74.Bonig H, Priestley GV, Nilsson LM, Jiang Y, Papayannopoulou T. PTX-sensitive signals in bone marrow homing of fetal and adult hematopoietic progenitor cells. Blood. 2004;104:2299–2306. doi: 10.1182/blood-2004-04-1605. [DOI] [PubMed] [Google Scholar]
- 75.Sanchez-Aguilera A, Lee YJ, Lo CC, Ferraro F, Brumme K, Mondal S, et al. Guanine nucleotide exchange factor Vav1 regulates perivascular homing and bone marrow retention of hematopoietic stem and progenitor cells. Proc Natl Acad Sci USA. 2011;108:9607–9612. doi: 10.1073/pnas.1102018108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cancelas JA, Jansen M, Williams DA. The role of chemokine activation of Rac GTPases in hematopoietic stem cell marrow homing, retention, and peripheral mobilization. Exp Hematol. 2006;34:976–985. doi: 10.1016/j.exphem.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 77.Bonig H, Watts KL, Chang KH, Kiem HP, Papayannopoulou T. Concurrent blockade of alpha4-integrin and CXCR4 in hematopoietic stem/progenitor cell mobilization. Stem Cells. 2009;27:836–837. doi: 10.1002/stem.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramirez P, Rettig MP, Uy GL, Deych E, Holt MS, Ritchey JK, et al. BIO5192, a small molecule inhibitor of VLA-4, mobilizes hematopoietic stem and progenitor cells. Blood. 2009;114:1340–1343. doi: 10.1182/blood-2008-10-184721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scott LM, Priestley GV, Papayannopoulou T. Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol. 2003;23:9349–9360. doi: 10.1128/MCB.23.24.9349-9360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKalpha-dependent NF-kappaB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol. 2012;188:2380–2386. doi: 10.4049/jimmunol.1102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawabata K, Ujikawa M, Egawa T, Kawamoto H, Tachibana K, Iizasa H, et al. A cell-autonomous requirement for CXCR4 in long-term lymphoid and myeloid reconstitution. Proc Natl Acad Sci USA. 1999;96:5663–5667. doi: 10.1073/pnas.96.10.5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lapidot T, Petit I. Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp Hematol. 2002;30:973–981. doi: 10.1016/s0301-472x(02)00883-4. [DOI] [PubMed] [Google Scholar]
- 83.Fruehauf S, Seggewiss R. It’s moving day: factors affecting peripheral blood stem cell mobilization and strategies for improvement [corrected] Br J Haematol. 2003;122:360–375. doi: 10.1046/j.1365-2141.2003.04483.x. [DOI] [PubMed] [Google Scholar]
- 84.Cashen AF, Link D, Devine S, DiPersio J. Cytokines and stem cell mobilization for autologous and allogeneic transplantation. Curr Hematol Rep. 2004;3:406–412. [PubMed] [Google Scholar]
- 85.Levesque JP, Hendy J, Takamatsu Y, Williams B, Winkler IG, Simmons PJ. Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol. 2002;30:440–449. doi: 10.1016/s0301-472x(02)00788-9. [DOI] [PubMed] [Google Scholar]
- 86.Thomas J, Liu F, Link DC. Mechanisms of mobilization of hematopoietic progenitors with granulocyte colony-stimulating factor. Curr Opin Hematol. 2002;9:183–189. doi: 10.1097/00062752-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 87.Elfenbein GJ, Sackstein R. Primed marrow for autologous and allogeneic transplantation: a review comparing primed marrow to mobilized blood and steady-state marrow. Exp Hematol. 2004;32:327–339. doi: 10.1016/j.exphem.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 88.Levesque JP, Barbier V, Nowlan B, McCarhty D, Winkler I. Impairment of hematopoietic stem cell (HSC) niche by G-CSF is associated with rapid mobilization of serially reconstituting HSC and reduced competitive repopulation of mobilized bone marrow. Blood. 2011;118a:1889. [Google Scholar]
- 89.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 90.Lucas D, Battista M, Shi PA, Isola L, Frenette PS. Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell. 2008;3:364–366. doi: 10.1016/j.stem.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lucas D, Bruns I, Battista M, Mendez-Ferrer S, Magnon C, Kunisaki Y, et al. Nor-epinephrine reuptake inhibition promotes mobilization in mice: potential impact to rescue low stem cell yields. Blood. 2012;119:3962–3965. doi: 10.1182/blood-2011-07-367102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mueller MM, Bialleck H, Bomke B, Brauninger S, Varga C, Seidl C, et al. Safety and efficacy of healthy volunteer stem cell mobilization with filgrastim G-CSF and mobilized stem cell apheresis: esults of a prospective longitudinal 5-year follow-up study. Vox Sang. 2012 doi: 10.1111/j.1423-0410.2012.01632.x. e-pub ahead of print 24 July 2012. [DOI] [PubMed] [Google Scholar]
- 93.Holig K, Kramer M, Kroschinsky F, Bornhauser M, Mengling T, Schmidt AH, et al. Safety and efficacy of hematopoietic stem cell collection from mobilized peripheral blood in unrelated volunteers: 12 years of single-center experience in 3928 donors. Blood. 2009;114:3757–3763. doi: 10.1182/blood-2009-04-218651. [DOI] [PubMed] [Google Scholar]
- 94.Winkler IG, Levesque JP. Mechanisms of hematopoietic stem cell mobilization: when innate immunity assails the cells that make blood and bone. Exp Hematol. 2006;34:996–1009. doi: 10.1016/j.exphem.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 95.Bonig H, Wundes A, Chang KH, Lucas S, Papayannopoulou T. Increased numbers of circulating hematopoietic stem/progenitor cells are chronically maintained in patients treated with the CD49d blocking antibody natalizumab. Blood. 2008;111:3439–3441. doi: 10.1182/blood-2007-09-112052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Broxmeyer HE, Kim CH. Regulation of hematopoiesis in a sea of chemokine family members with a plethora of redundant activities. Exp Hematol. 1999;27:1113–1123. doi: 10.1016/s0301-472x(99)00045-4. [DOI] [PubMed] [Google Scholar]
- 97.Jalili A, Shirvaikar N, Marquez-Curtis L, Qiu Y, Korol C, Lee H, et al. Fifth complement cascade protein (C5) cleavage fragments disrupt the SDF-1/CXCR4 axis: further evidence that innate immunity orchestrates the mobilization of hematopoietic stem/progenitor cells. Exp Hematol. 2010;38:321–332. doi: 10.1016/j.exphem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee HM, Wysoczynski M, Liu R, Shin DM, Kucia M, Botto M, et al. Mobilization studies in complement-deficient mice reveal that optimal AMD3100 mobilization of hematopoietic stem cells depends on complement cascade activation by AMD3100-stimulated granulocytes. Leukemia. 2010;24:573–582. doi: 10.1038/leu.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reca R, Cramer D, Yan J, Laughlin MJ, Janowska-Wieczorek A, Ratajczak J, et al. A novel role of complement in mobilization: immunodeficient mice are poor granulocyte-colony stimulating factor mobilizers because they lack complement-activating immunoglobulins. Stem Cells. 2007;25:3093–3100. doi: 10.1634/stemcells.2007-0525. [DOI] [PubMed] [Google Scholar]
- 100.Ratajczak MZ, Kim C, Wu W, Shin DM, Bryndza E, Kucia M, et al. The role of innate immunity in trafficking of hematopoietic stem cells-an emerging link between activation of complement cascade and chemotactic gradients of bioactive sphingolipids. Adv Exp Med Biol. 2012;946:37–54. doi: 10.1007/978-1-4614-0106-3_3. [DOI] [PubMed] [Google Scholar]
- 101.Janowska-Wieczorek A, Marquez-Curtis LA, Shirvaikar N, Ratajczak MZ. The role of complement in the trafficking of hematopoietic stem/progenitor cells. Transfusion. 2012 doi: 10.1111/j.1537-2995.2012.03636.x. e-pub ahead of print 9th April 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dauber K, Becker D, Odendahl M, Seifried E, Bonig H, Tonn T. Enumeration of viable CD34( +) cells by flow cytometry in blood, bone marrow and cord blood: results of a study of the novel BD stem cell enumeration kit. Cytotherapy. 2011;13:449–458. doi: 10.3109/14653249.2010.529894. [DOI] [PubMed] [Google Scholar]