

Abstract
The calpain family of calcium-dependent proteases has been implicated in a variety of diseases and neurodegenerative pathologies. Prolonged activation of calpains results in proteolysis of numerous cellular substrates including cytoskeletal components and membrane receptors, contributing to cell demise despite coincident expression of calpastatin, the specific inhibitor of calpains. Pharmacological and gene knockout strategies have targeted calpains to determine their contribution to neurodegenerative pathology; however, limitations associated with treatment paradigms, drug specificity, and genetic disruptions have produced inconsistent results and complicated interpretation. Specific, targeted calpain inhibition achieved by enhancing endogenous calpastatin levels offers unique advantages in studying pathological calpain activation. We have characterized a novel calpastatin overexpressing transgenic mouse model, demonstrating a substantial increase in calpastatin expression within nervous system and peripheral tissues and associated reduction in protease activity. Experimental activation of calpains via traumatic brain injury resulted in cleavage of α-spectrin, collapsin response mediator protein-2, and voltage-gated sodium channel, critical proteins for the maintenance of neuronal structure and function. Calpastatin overexpression significantly attenuated calpain-mediated proteolysis of these selected substrates acutely following severe controlled cortical impact injury, but with no effect on acute hippocampal neurodegeneration. Augmenting calpastatin levels may be an effective method for calpain inhibition in TBI and neurodegenerative disorders.
Keywords: calcium, calpain, CRMP-2, protease, sodium channel, spectrin
INTRODUCTION
The calcium-dependent cysteine proteases, calpains, are multi-faceted regulators of normal cell function. Although 15 isoforms of calpains are known to exist within cells (Sorimachi et al., 2011), the most commonly studied are the ubiquitously expressed isoforms, μ-calpain and m-calpain. While these two isoforms share a similar heterodimeric structure, they differ in their calcium requirements. Micromolar range concentrations of ionic calcium are necessary to activate μ-calpains in vitro while millimolar calcium concentrations activate m-calpains. Regulation of calpains’ proteolytic activity occurs both by intracellular free calcium concentrations and by a common endogenous inhibitor, calpastatin. Calpastatin is an intracellular 110 kDa protein consisting of an N-terminal leader domain followed by four identical inhibitory domains, each able to specifically inhibit one molecule of calpain (Maki et al., 1987). When free calcium levels rise and activate calpains, a conformational change in the protease allows for inhibitor binding across the active site of calpain, blocking its access to substrates (Moldoveanu et al., 2008). Under physiologic conditions, calpains participate in cytoskeletal alterations, cell cycle and differentiation processes, apoptosis, and long-term potentiation (Goll et al., 2003), indicative of their importance to normal cell function.
Calpain activation contributes to the evolution of neurodegeneration in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis as well as damage associated with stroke, traumatic brain injury (TBI), and spinal cord injury (Camins et al., 2006). Under pathological conditions, altered intracellular calcium homeostasis leads to calpain activation, resulting in the cleavage of cellular substrates including cytoskeletal elements, membrane receptors, cytosolic proteins, and cell death mediators (Saatman et al., 2010). As the most well characterized calpain substrate following TBI, the cytoskeletal component α-spectrin is a valuable surrogate marker of calpain activation and its early proteolysis may indicate the severity of cellular damage and subsequent neuronal death (Saatman et al., 1996). Through the use of calpain inhibitors and identification of calpain-specific breakdown products (BDPs), the number of calpain substrates verified in models of TBI is expanding. Collapsin response mediator protein-2 (CRMP-2) proteolysis was detected in response to excitotoxic insult and attenuated with in vitro calpain inhibitor application. Identical calpain-mediated CRMP-2 cleavage patterns were identified in brain homogenates after experimental TBI (Zhang et al., 2007). Similarly, voltage-gated sodium channel cleavage, triggered by exogenous calpain activation or using an in vitro model of TBI, was reversed with viral-mediated calpastatin overexpression or treatment with the calpain inhibitor MDL28170 (von Reyn et al., 2009). Limited cleavage characteristic of calpains may modulate ion flux and receptor function, contributing to exacerbated calcium dysfunction, further calpain activation, and neuronal damage associated with brain injury.
Genetic manipulation of calpastatin to enhance endogenous inhibitory mechanisms enables suppression of both μ- and m-calpain, providing a powerful research tool for understanding the role of pathological calpain proteolysis. Transgenic mice with calcium/calmodulin-dependent protein kinase II α (CaMKIIα)-driven calpastatin expression exhibited a 3-fold reduction in in vitro m-calpain activity and significantly less hippocampal cell death in response to excitotoxic insult (Higuchi et al., 2005). Using these same mice, we recently showed that following severe contusion TBI, calpastatin overexpression reduced acute spectrin proteolysis and select behavioral deficits but did not affect cortical tissue damage (Schoch et al., 2012).
Subsequently, we developed a novel transgenic mouse with human calpastatin (hCAST) under constitutive control of the ubiquitous prion promoter (Prp) in order to produce a more widespread cellular distribution of calpastatin overexpression. Here we demonstrate that this hCAST transgenic mouse has cortical and hippocampal calpastatin levels approximately 80-fold greater than wildtype (WT) mice and use this new transgenic tool to verify the effectiveness of calpastatin in reducing calpain-mediated damage after TBI. To this end, we subjected WT and calpastatin overexpressing (Prp-hCAST) transgenic mice to severe controlled cortical impact (CCI) injury and evaluated acute posttraumatic proteolysis of three proteins critical for neuronal structure and function: α-spectrin, CRMP-2, and voltage-gated sodium channel 1.2 (Nav1.2). In addition, we assessed acute regional hippocampal neurodegeneration in brain-injured WT and Prp-hCAST transgenic mice.
METHODS
Human calpastatin overexpressing transgenic mice
Human calpastatin (hCAST) cDNA (Genbank accession number D16217) in the pTicCS plasmid was obtained from Dr. Masatoshi Maki (Higuchi et al., 2005). The hCAST sequence was cloned into the unique XhoI site of the MoPrP.Xho expression vector (Borchelt et al., 1996), containing ~12 kb of the mouse prion protein (Prp) gene including the promoter, the smaller of the two 5′ introns, and the 3′ untranslated sequences. The transgene expression cassette was released from the Prp-hCAST plasmid with NotI, and the resulting purified fragment was microinjected into the pronuclei of fertilized FVB/N oocytes. Transgenic founder animals were identified by PCR screening of tail genomic DNA using primers phgPrP5′ (5′GAACTGAACCATTTCAACCGAG3′) and phgPrP3′ (5′AGAGCTACAGGTGGATAACC3′). The MoPrP.Xho vector has previously been used to produce transgenic mice expressing coding sequences under the control of mouse Prp gene transcriptional elements (Li et al., 2007, Angers et al., 2009); thus, widespread hCAST expression in the CNS of transgenic mice was expected. Calpastatin expression in the CNS of founder mice was assessed by immunoblotting and compared with previously generated transgenic mice in which the hCAST was expressed under the CaMKIIα promoter (Higuchi et al., 2005).
Mice were maintained as heterozygotes by breeding FVB/N females (Harlan Laboratories, Indianapolis, IN) with hCAST transgenic males, exhibiting normal litter sizes and survival rates with no overt phenotype. Mice were housed in controlled conditions under a 14:10 light:dark photoperiod and provided with chow diet and water ad libitum. For experimental procedures, young adult transgenic and WT littermates were used. For qualitative measures, both males and females were analyzed. Although no notable sex-specific expression patterns were observed, efforts were made to utilize a consistent gender (male or female) within each quantitative experiment when possible in order to minimize any potential variation. All husbandry care and surgical procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee and were consistent with federal guidelines (Institute of Laboratory Animal Resources (U.S.). Committee on Care and Use of Laboratory Animals.) with all possible effort made to minimize pain and discomfort of the animal.
Controlled cortical impact injury
Contusion brain injury was modeled using a CCI device (TBI-0310 Impactor, Precision Systems and Instrumentation, Fairfax Station, VA) as previously described (Schoch et al., 2012). In brief, mice were placed in a stereotaxic head frame, receiving inhalant isoflurane anesthesia. A midline scalp incision exposed the skull and a 5 mm craniotomy was drilled on the left hemisphere, lateral to the central suture between bregma and lambda. A rounded, 3.0 mm diameter steel impactor, controlled by a pneumatically driven cylinder, was programmed to contact the exposed dura at a 1.0 mm depth, 3.5 m/s velocity, and 500 msec dwell time. These parameters result in a severe injury, defined by progressive cortical and hippocampal cell losses over the first 24 h after insult (Pleasant et al., 2011). Following the impact, a small cranioplast made of dental acrylic was placed over the craniotomy site and secured to the skull. The incision was sutured and the animal was placed on a 37°C heating pad until ambulating. Sham-injured mice received all surgical procedures except the impact injury.
Tissue preparation
Tissue designated for calpastatin inhibitory assays or immunoblots was obtained following carbon dioxide asphyxiation and immediate decapitation. Contralateral and ipsilateral cortical and hippocampal tissues were separately dissected and rapidly frozen in cold (−80°C) methanol. Samples were stored at −80°C until use.
For immunochemistry and Fluoro-jade C experiments, mice were perfused transcardially with 0.9% heparinized sterile saline and 10% neutral buffered formalin under anesthesia (65 mg/kg sodium pentobarbital, intraperitoneally). Heads were placed in formalin for 24 h after which the brain was removed for additional overnight fixation. Following cryoprotection in 30% sucrose solution, the brain was frozen in cold isopentanes (−25°C to −35°C). Brain tissue was cut on a sliding microtome (Dolbey-Jamison, Pottstown, PA) in the coronal plane at a thickness of 40 μm. Unused sections were stored in a cryoprotectant solution (30% ethylene glycol, 30% glycerol) at −20°C.
Calpastatin inhibitory assay in naïve mice
Frozen cortical tissue was sonicated in a calcium-free buffer solution (20 mM Tris, 1 mM EDTA, 100 mM KCl, 0.1% 2-mercaptoethanol) without protease inhibitors and centrifuged at 21,500 g for 20 min at 4°C. Supernatants were assayed for calpastatin inhibitory activity in the presence of porcine kidney m-calpain (14.06 μg/ml, Calbiochem, Gibbstown, NJ) and a fluorescently-conjugated BODIPY-FL casein calpain substrate (EnzChek® Protease Assay Kit, Invitrogen, Carlsbad, CA). In the presence of proteases, the casein substrate is cleaved to emit a fluorescent signal, which is read on a spectrafluorometer (Synergy HT Multi-Mode Microplate Reader, Biotek Instruments, Winooski, VT) at 485 nm excitation and 528 nm emission wavelengths. Fluorescence values were normalized to the amount of protein in the sample as determined by BCA assay (Pierce BCA protein assay kit, Thermo Scientific, Rockford, IL) and reported as units/mg of total protein. An average fluorescent reading was obtained from cortical samples run in duplicate.
Calpastatin immunohistochemistry in naïve mice
Tissue sections were rinsed in Tris-buffered saline (TBS) and treated with 3% hydrogen peroxide solution in methanol and water for 30 min to quench endogenous peroxidases associated with blood-brain barrier breakdown and normal vasculature. Following additional TBS washes, the tissue was blocked in 5% normal horse serum in TBS/0.1% Triton X-100 for 30 min. Tissue was incubated overnight in 4°C with primary antibody recognizing calpastatin of both mouse and human origin (Table 1). Biotinylated donkey anti-rabbit IgG secondary antibody (1:5000, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was applied for 1 h at room temperature, subsequently amplified by avidin and biotin complex (1:50, Vectastain Elite ABC kit, Vector Laboratories, Inc., Burlingame, CA), and developed in diaminobenzidine (DAB). Negative control tissue was treated identically but without addition of primary antibody. Tissue was viewed under a light microscope (Eclipse 50i, Nikon Corporation, Japan).
Table 1.
Primary antibodies used in immunohistochemistry and immunoblot experiments.
| Primary antibody | Species | Concentration | Source |
|---|---|---|---|
| α-spectrin | Mouse | 1:5000 | Millipore Co. |
| β-actin | Mouse | 1:1000 | Sigma-Aldrich |
| β-tubulin | Rabbit | 1:1000 | Abcam |
| Calpain-1 | Rabbit | 1:1000 | Calbiochem |
| Calpain-2 | Rabbit | 1:1000 | Abcam |
| Calpastatin | Rabbit | 1:500 | Santa Cruz Biotechnology |
| Caspase-3 | Rabbit | 1:500 | Abcam |
| CRMP-2 | Mouse | 1:1000 | IBL-America |
| ERK1/2 | Rabbit | 1:2000 | Cell Signaling Technology |
| GAPDH | Mouse | 1:10000 | Millipore Co. |
| MAP-2 | Mouse | 1:1000 | Sigma-Aldrich |
| Nav1.2 I-II loop | Rabbit | 1:1000 | Alomone Labs |
| p35/25 | Rabbit | 1:600 | Cell Signaling Technology |
| Pan NaCh | Mouse | 1:1000 | Sigma-Aldrich |
| Phospho-ERK1+2 | Rabbit | 1:1000 | Invitrogen |
Immunoblot of proteins from naïve and injured mice
Tissue was homogenized by sonication in a lysis buffer (20 mM Tris, 150 mM NaCl, 5 mM EGTA, 10 mM EDTA, 10 mM HEPES, 1% Triton-X, 10% glycerol) containing protease inhibitors (Complete MiniTM Protease Inhibitor Cocktail tablet, Roche Applied Science, Indianapolis, IN). Samples were centrifuged at 19,000 g for 20 minutes at 4°C and the soluble fraction collected for protein concentration determination. For analysis, equal amounts of protein were run on polyacrylamide gels (3-8% Tris-Acetate, 4-20% Tris-HCl, or 10% Tris-HCl Criterion™ Precast Gels, Bio-Rad Laboratories, Hercules, CA), transferred to PVDF or nitrocellulose membrane, and blocked in a 5% dry milk/TBS/0.05% Tween 20 solution for 1 h. Membranes were incubated in primary antibody (Table 1) diluted in 5% dry milk/TBS/0.05% Tween 20 overnight at 4°C. Secondary antibodies conjugated to either horseradish peroxidase (goat anti-mouse IgG 1:3000; Bio-Rad Laboratories) or infrared dye (goat anti-mouse IgG, goat anti-rabbit IgG 1:5,000-10,000, Rockland Immunochemicals, Gilbertsville, PA) were similarly prepared in 5% dry milk/TBS/Tween 20 solution and applied for a 1 h incubation at room temperature. Membranes probed for α-spectrin were visualized on an Odyssey Li-Cor imaging station (Li-Cor Biosciences, Lincoln, NE) and quantified by Odyssey imaging software. All other membranes were visualized by enhanced chemiluminescence on a Kodak imager and quantified by ImageJ software. Membranes were subsequently probed for β-actin, β-tubulin, or GAPDH (Table 1) to verify equal protein loading across lanes.
Fluoro-jade C staining and analysis of injured tissue
The fluorochrome Fluoro-jade C (FJC) was used to detect neuronal degeneration using methodology previously described (Schoch et al., 2012). In brief, three 40 μm tissue sections were selected at 400 μm intervals between approximate bregma level − 1.4 to −2.5 mm corresponding to the injury epicenter (Paxinos and Franklin, 2001). Tissue sections were initially exposed to DAB to quench endogenous peroxidases, thereby eliminating nonspecific fluorescence associated with hemorrhage, and were subsequently mounted on gelatin-coated slides. Following overnight drying, tissues were rehydrated in 1% NaOH and alcohol gradient, treated with 0.06% potassium permanganate, and stained with 0.01% Fluoro-jade C (Millipore Co., Billerica, MA) in 0.1% acetic acid. Tissue sections were dried on a slide warmer before immersion in xylenes and covered with Cytoseal XYL (Richard-Allen Scientific, Kalamazoo, MI).
Image analysis was performed by an observer blinded to genotype using a microscope equipped with epifluorescence (AX80, Olympus America Inc., Melville, NY). Fluoro-jade C-positive cells (FJC+) were counted separately within the dentate gyrus, CA3/CA3c, and CA1 regions of the hippocampus and averaged across the three selected tissue sections. Although degenerating neurons were also evident in the cortex of injured tissue, these FJC+ cells were not quantified.
Statistical analysis
All data are presented as mean + SEM and analyzed using Statistica software (StatSoft, Tulsa, OK). Calpastatin inhibitory activity was evaluated by one-way ANOVA. α-spectrin BDPs, 150 kDa and 145 kDa, were analyzed by nested (triplicate samples) two-way ANOVA (genotype × injury condition). Quantification of CRMP-2 BDPs was evaluated with a two-way ANOVA (genotype × injury condition). Full-length sodium channel and its proteolytic fragments were compared by nested (duplicate samples) one-way ANOVA. Numbers of Fluoro-jade C-positive cells within each hippocampal region were analyzed by unpaired t-test. A value of p<0.05 was considered significant and Newman-Keuls post-hoc testing performed when appropriate.
RESULTS
Calpastatin expression in wildtype and Prp-hCAST transgenic mice
Three Prp-hCAST transgenic founders were mated with FVB/N mice to produce lines hemizygous for the transgene array, referred to as 7472+/−, 7473+/−, and 7474+/−. Using an antibody that detects both mouse and human calpastatin, expression levels of calpastatin in whole brains of 7474+/− mice were estimated to be ~1.8-fold higher than levels in the brains of previously characterized calpastatin overexpressing transgenic mice (CaMKIIα promoter) which express hCAST exclusively in forebrain regions (Higuchi et al., 2005) (Figure 1A). Calpastatin was not detectable in the CNS of mice derived from the two remaining transgenic founders.
Figure 1.

Calpastatin expression in tissue homogenates obtained from naive wildtype (WT) and Prp-hCAST transgenic (Tg) mice. A) Calpastatin expression in whole brain of three founder Tg mice (7472+/−, 7474+/−, and 7473+/−) compared to an alternative calpastatin Tg model (CaMKIIα-Tg). Calpastatin expression in cortical and hippocampal tissue of Tg and WT mice with B) equal amounts (5 μg) of protein loaded or C) a 40-fold difference in protein load to enhance detection of mouse calpastatin (mCAST). D) Calpastatin expression identified in cerebellar and spinal cord tissue and peripheral tissues, including gastrocnemius and heart tissue from Tg and WT mice (5 μg protein/region).
Regional calpastatin expression levels were assessed in cortical and hippocampal homogenates from Prp-hCAST transgenic and WT littermates (n=4/genotype). Immunoblots performed with equal protein loading (5 μg) and developed to visualize the strong calpastatin signal in hCAST transgenic mice produced no discernible calpastatin signal in WT homogenates (Figure 1B). To compare endogenous levels of calpastatin in WT mice to the much higher levels in hCAST transgenic mice, differential amounts of protein from WT and Prp-hCAST cortical homogenates (n=4/genotype) were loaded for immunoblot analysis. With a 40-fold increase in protein loading of WT homogenates compared to transgenic homogenates, the signal in Prp-hCAST mice was approximately double the signal from WT samples, indicating a nearly 80-fold human calpastatin expression above endogenous, mouse calpastatin levels (Figure 1C). Blots also revealed a slight difference in the molecular weight of mouse calpastatin (mCAST) and hCAST, allowing differentiation of the two proteins. In addition to cortical and hippocampal brain regions, various central and peripheral tissues were also examined for calpastatin expression (n=4/genotype, 5 μg protein). Reactivity for hCAST was noted in the cerebellum and spinal cord of Prp-hCAST homogenates (Figure 1D). Consistent with previous literature identifying high levels of calpastatin within the cerebellum compared to other brain regions (Sato et al., 2011), endogenous mCAST was detectable in cerebellar homogenates from WT mice. In peripheral tissue such as the gastrocnemius and heart muscle, calpastatin reactivity was observed exclusively in samples from Prp-hCAST mice (Figure 1D), again indicative of a many-fold overexpression of calpastatin in the transgenic mice compared to WT. Calpastatin was not evident in the liver, lung, kidney, and spleen (data not shown).
Immunohistochemical labeling (n=3/genotype) using an antibody that recognizes both mouse and human calpastatin demonstrated a low level of reactivity in gray matter regions of WT mouse brain (Figure 2A), with faint neuronal labeling in the cortex (Figure 2C) and neuropil staining within the hippocampus (Figure 2E). Brain sections from Prp-hCAST transgenic mice exhibited a robust increase in calpastatin immunoreactivity compared to WT mice in cell bodies, neuropil, and white matter tracts (Figure 2B). All cortical layers, especially layers III and V, showed specific labeling within the neuronal cytoplasm (Figure 2D, inset). Reactivity within neuronal cell bodies was also clearly evident in CA1, CA3, and dentate gyrus hilar regions of the hippocampus (Figure 2F).
Figure 2.

Calpastatin localization in naïve wildtype and Prp-hCAST transgenic mice. Immunohistochemical labeling using an antibody recognizing mouse and human calpastatin in A) wildtype and B) Prp-hCAST transgenic mice demonstrates a robust increase in calpastatin in transgenic mice. Magnified areas of cortical layers II-V (C, D) and the hippocampus (E, F) are shown below each respective genotype, illustrating the neuronal localization of human calpastatin in Prp-hCAST mice. Scale bars represent 1 mm for images A-B, 100 μm for images C-F and 20 μm for insets.
Immunoblot and immunohistochemistry analyses included both male and female mice and no notable differences between genders were observed.
In vitro calpastatin inhibitory activity
The functionality of the hCAST construct within Prp-hCAST transgenic mice was assessed using an in vitro fluorogenic assay. Under baseline physiological conditions, calpain activity is low, consistent with its normally inactive conformation in the neuron. With the addition of exogenous calpain to mimic the elevated protease activity seen with pathological insults such as TBI, contralateral (or uninjured) cortical homogenates from WT mice (n=5, male) exhibited high protease activity measured in fluorescence units per total amount of protein (Figure 3A). In contrast, cortical homogenates from Prp-hCAST transgenic mice (n=6, male) exhibited an approximately 7-fold decrease in protease activity compared to WT littermates (p<0.001), indicative of effective inhibition by the overexpressed calpastatin protein.
Figure 3.

Characterization of the inhibitory activity of calpastatin and basal levels of putative calpain substrates in wildtype (WT) and Prp-hCAST transgenic (Tg) mice. A) Calpastatin inhibitory activity in contralateral (uninjured) cortical homogenates following the addition of exogenous calpain to mimic a state of elevated calpain activity. Data are represented as mean + SEM; ##p<0.001. B) Expression of select proteases and calpain substrates in cortical and hippocampal homogenates obtained from naïve WT and Tg mice.
Expression of calpains and calpain substrates in naïve mice
Inhibition of calpain proteases by constitutive calpastatin overexpression may produce compensatory changes in protease or calpain substrate expression. To test whether Prp-hCAST transgenic mice exhibited alterations in protease expression, levels of calpain-1, calpain-2, and caspase-3 proteins were assessed in homogenates from Prp-hCAST and WT mice (n=4/genotype). Protease expression did not differ between WT and Prp-hCAST mice in either the cortex or hippocampus (Figure 3B). Similarly, selected calpain substrates including p35, β-tubulin, MAP-2, ERK, and phospho-ERK were unaltered by calpastatin overexpression in naïve Prp-hCAST mice (Figure 3B).
Posttraumatic calpain-mediated proteolysis of α-spectrin
To understand the action of calpastatin during in vivo activation of calpains, male WT and Prp-hCAST transgenic mice were subjected to severe CCI brain injury. Calpains are activated acutely after experimental brain injury in the mouse, resulting in early proteolysis of α-spectrin and subsequent increase in spectrin BDPs. To correspond to the suggested peak activation of μ-calpain after CCI injury (Kampfl et al., 1996) and maximal breakdown of spectrin after brain injury (Deng et al., 2007), calpain-mediated spectrin proteolysis was analyzed at both 6 and 24 h following severe CCI. Calpains cleave intact α-spectrin (280 kDa) into 150 and 145 kDa BDPs; the 145 kDa product is specific to calpain proteolysis while the 150 kDa fragment can result from calpain and caspase activity (Pike et al., 1998). As expected, severe CCI injury resulted in calpain-mediated spectrin proteolysis evident by an increase in cortical 145 kDa BDP levels in WT mice at 6 h (n=5) and 24 h (n=7) following injury (p<0.001 compared to sham controls). In contrast, in brain-injured Prp-hCAST mice, levels of the calpain-specific proteolytic fragment (145 kDa) were maintained at sham control levels at both 6 h (n=6) and 24 h (n=6) (Figure 4A). The attenuation of calpain-specific spectrin proteolysis in Prp-hCAST mice relative to WT mice was statistically significant at 6 h and 24 h after injury (p<0.001).
Figure 4.

Cortical and hippocampal α-spectrin proteolysis following severe controlled cortical impact (CCI) injury in wildtype (WT) and Prp-hCAST transgenic (Tg) mice. Calpain-mediated spectrin breakdown (150 kDa and 145 kDa) in A) cortical and B) hippocampal homogenates at 6 and 24h after CCI injury. Breakdown product (BDP) optical densities are represented as mean + SEM; *p<0.05, **p<0.001 vs. sham and ##p<0.001 vs. WT. Representative blots with tubulin loading control are shown to the right.
Spectrin proteolysis in WT mice was also evident within the hippocampus following CCI, with increases in the 145 kDa fragment at 6 h and 24 h (p<0.001 compared to sham). Similar to the effect observed in the cortex, hCAST overexpression suppressed the proteolysis of spectrin to the 145 kDa fragment to near sham levels (Figure 4B). Although elevated breakdown was noted at 6 h (p<0.001) and 24 h post-injury (p<0.05) compared to sham, 145 kDa spectrin BDP levels were significantly reduced in injured Prp-hCAST mice compared to injured WT mice in the hippocampus at 24 h post-injury (p<0.001) (Figure 4B).
Severe brain injury resulted in increased levels of the 150 kDa fragment in both cortical and hippocampal homogenates above those in sham (p<0.001; n=4-5/genotype); however, calpastatin overexpression did not significantly alter this BDP (Figure 4). Caspase-mediated spectrin fragments (120 kDa) were notably absent in all time points and regions analyzed.
Posttraumatic calpain-mediated proteolysis of collapsin response mediator protein-2
Due to its potential role in trauma-induced neurodegeneration (Taghian et al., 2012), posttraumatic axon sprouting (Wilson et al., 2012) and NMDA trafficking (Brittain et al., 2011), CRMP-2 was investigated in WT and calpastatin overexpressing transgenic mice following TBI. From its full-length form, CRMP-2 is cleaved by calpains to a fragment of approximately 55 kDa in response to apoptotic stimuli, excitotoxic challenge, and TBI (Zhang et al., 2007). Immunoblot analysis of cortical and hippocampal homogenates from WT mice (n=3-5/condition, female) demonstrated the progressive accumulation from 6 h to 24 h following CCI brain injury of a 55 kDa CRMP-2 fragment (Figure 5), consistent with calpain-mediated proteolysis. Calpastatin overexpression (n=3-4/condition, female) prevented the accumulation of this BDP for up to 24 h in the cortex, resulting in a significant decrease in levels of CRMP-2 fragments in brain-injured Prp-hCAST mice relative to brain-injured WT mice (p<0.05 and p<0.0005 at 6 h and 24 h, respectively; Figure 5A). Within the hippocampus, CRMP-2 breakdown in Prp-hCAST mice was reduced at 6h, and completely inhibited at 24h post-injury (p<0.005, Figure 5B). These results both confirm the calpain-specific cleavage of CRMP-2 and further support the protective effect of calpastatin overexpression following in vivo calpain activation.
Figure 5.

Collapsin response mediator protein-2 (CRMP-2) breakdown following severe controlled cortical impact injury in wildtype (WT) and Prp-hCAST transgenic (Tg) mice. Quantification of a 55 kDa CRMP-2 fragment in A) cortical and B) hippocampal homogenates of WT and Prp-hCAST mice at 6 and 24 h after injury. Breakdown product (BDP) optical densities are represented as mean + SEM; **p<0.005 vs. sham and #p<0.05, ##p<0.005 vs. WT. Representative blots with GAPDH loading control are shown to the right. Full-length CRMP-2 bands (bracketed) appear overexposed in representative immunoblot images in order to identify the CRMP-2 BDP.
Posttraumatic calpain-mediated proteolysis of voltage-gated sodium channel
To investigate Nav1.2 α subunit proteolysis in vivo, a separate cohort of female WT and Prp-hCAST transgenic mice was subjected to severe CCI injury. Based on spectrin and CRMP-2 proteolysis data in which the most robust effect was identified in cortical homogenates, our analysis of sodium channel cleavage was restricted to the cortex. Immunoblots probed with a pan sodium channel antibody (Table 1) identified the full-length Nav1.2 α subunit protein (260 kDa) as well as a fragment at approximately 100 kDa (Figure 6A). Brain injury resulted in a progressive loss in full-length protein which was statistically significant at 24 h post-injury in WT (p<0.001) and Prp-hCAST (p<0.05) cortical homogenates. WT mice (n=4-5) exhibited a pronounced (p<0.01 compared to sham controls), but transient, increase in the 100 kDa BDP that was suppressed in Prp-hCAST mice (n=4). Use of an antibody that detects an epitope within the intracellular loop between domains I and II of the Nav1.2 α subunit yielded fragments of 170, 111, 100, and 85 kDa (Figure 6B). Brain injury resulted in a statistically significant increase in the 85 kDa fragment in WT homogenates (p<0.01), which was greatly attenuated in Prp-hCAST mice at both 6 and 24 h post-CCI (p<0.05 and p<0.01 compared to WT, respectively).
Figure 6.

Cleavage of the voltage-gated sodium channel (Nav1.2) α subunit in cortical homogenates following severe controlled cortical impact (CCI) injury. A) A pan sodium channel (NaCh) antibody detected full-length and a 100 kDa molecular weight fragment induced by CCI injury at 6 and 24 h post-CCI injury. B) Detection of 170, 111, 100, and 85 kDa fragment accumulation with an antibody that recognizes an epitope within the I-II loop of the Nav1.2 α subunit. Breakdown product (BDP) and intact optical densities are represented as mean + SEM; *p<0.05, **p<0.01 vs. sham and #p<0.05, ##p<0.01 vs. WT. Representative blots with GAPDH loading control are shown to the right. Black outlines delineate samples that were run on the same blot (i.e. sham and 6 h samples) from samples run on a separate blot (i.e. 24 h samples). Optical density analysis was performed on injured (ipsilateral, I) cortical samples. Contralateral (C) cortical samples are shown in adjacent lanes for reference only.
Acute hippocampal neurodegeneration in brain-injured mice
Reductions in calpain-mediated proteolysis of selected substrates with calpastatin overexpression may result in enhanced neuronal survival after CCI brain injury. To address this possibility, tissue sections obtained 24 h following severe CCI from male and female WT (n=9) and Prp-hCAST transgenic (n=10) mice were stained for Fluoro-jade C, identifying neuronal degeneration. Positive cellular staining was evident within the cortex and hippocampus of both WT and Prp-hCAST transgenic mice. Qualitative comparison of cortical Fluoro-jade C staining did not reveal an overt difference in acute neurodegeneration between genotypes. In the hippocampus, Fluoro-jade C-positive (FJC+) cells were found predominantly in the dentate gyrus and CA3/CA3c, with far fewer within the CA1 region. In all hippocampal regions analyzed, no statistically significant differences in numbers of degenerating neurons were identified between WT and Prp-hCAST transgenic mice (Figure 7).
Figure 7.
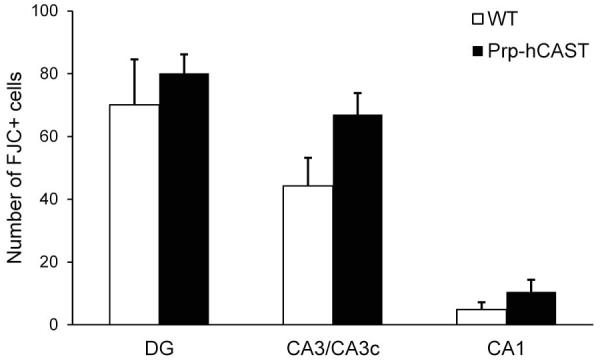
Hippocampal neurodegeneration assessed using Fluoro-jade C staining of tissue obtained from wildtype (WT) and Prp-hCAST transgenic mice 24 h following severe controlled cortical impact injury. Numbers of Fluoro-jade C-positive (FJC+) cells were analyzed in the dentate gyrus (DG), CA3/CA3c, and CA1 regions of the hippocampus and are shown as average counts per section (mean + SEM).
DISCUSSION
Calpains are important mediators of neuronal damage and death under conditions of neurodegenerative disease and traumatic insults. Due to the risk calpain activation poses toward neuronal cell viability, we have investigated an avenue of calpain inhibition using overexpression of calpastatin in a new transgenic mouse line. Our results demonstrate that Prp-hCAST mice robustly express calpastatin throughout the brain, yielding potent inhibition of exogenous calpain. Following in vivo activation of calpains by experimental TBI, calpastatin overexpression reduced calpain-mediated proteolysis of the substrates α-spectrin, CRMP-2, and voltage-gated sodium channel. These findings confirm calpains as a pathological target and validate calpastatin as an agent for modulating calpain activity following TBI.
Prp-hCAST transgenic mice were found to express calpastatin at levels on the order of 80-fold higher than endogenous levels in WT mice in the cortex and hippocampus, with near complete inhibition of in vitro protease activity. Other transgenic models of constitutive calpastatin overexpression have also demonstrated significant elevations in calpastatin levels or inhibitory function. Neuronal expression of calpastatin via the CaMKIIα promoter produced a 3-fold greater calpastatin inhibitory activity, although hCAST and mCAST protein levels were not quantitatively compared (Higuchi et al., 2005). Use of the Prp promoter to initiate gene expression may yield a more widespread distribution throughout the brain, particularly concentrated in axons and terminals (Barmada et al., 2004). In addition to its central distribution, calpastatin expression was also noted in peripheral tissues, making this mouse an attractive tool for many models of both central and peripheral nerve injury and other pathologies involving non-neuronal tissues. An alternative calpastatin transgenic mouse, developed using the Thy1.1 promoter, achieved an approximately 15-fold greater calpastatin expression over WT resulting in effective calpain-2 inhibition but also in changes in basal levels of calpain-1 and calpain-2 and certain calpain substrates (Rao et al., 2008). Altered basal levels of proteases or calpain substrates was not evident in the Prp- or CaMKIIα-driven models, suggesting constitutive overexpression of calpastatin did not produce compensatory upregulation of proteases or widespread changes in substrate regulation. Both transgenic models (using CaMKIIα and Thy1.1 promoters) reduced pathological activation of calpain and proteolysis of multiple calpain substrates after excitotoxic stimulus. Prp-driven hCAST overexpression was similarly successful in abating posttraumatic calpain-mediated proteolysis of structurally and functionally relevant proteins.
α-spectrin is an essential protein component of the neuronal cytoarchitecture that functions in both structural support and membrane protein anchoring. Its cleavage may contribute to neuronal pathology due to alterations in membrane stability or membrane-associated protein function. Severe TBI led to increased α-spectrin proteolysis into its characteristic 150 and 145 kDa fragments in WT mice, consistent with previous literature documenting potent and early spectrin cleavage in neurons of affected brain regions following experimental contusion injury (Saatman et al., 1996, Pike et al., 1998). Calpastatin overexpression in Prp-hCAST transgenic mice prevented appearance of the calpain-specific 145 kDa BDP in the cortex and hippocampus up to 24 h after severe CCI injury. Targeting calpain activity via administration of pharmacological calpain inhibitors has produced inconsistent results in the ability to decrease spectrin breakdown following TBI (Posmantur et al., 1997, Saatman et al., 2000, Kupina et al., 2001, Thompson et al., 2010), which may be a reflection of the differences in treatment parameters or limitations associated with the drugs themselves. Notably, some calpain inhibitors are not solely selective for calpains, have poor blood-brain barrier permeability, and are metabolically unstable (Carragher, 2006). Newer inhibitors designed to mimic calpastatin show promise in preventing calpain-specific spectrin breakdown in response to elevated intracellular calcium (McCollum et al., 2006) or ischemic insult (Anagli et al., 2009).
Calpastatin overexpression in our transgenic model was unable to inhibit spectrin proteolysis into the 150 kDa fragment. Although a 150 kDa fragment can be generated through caspase activity, which is not inhibited by calpastatin, it is unlikely that caspase-3 contributed significantly to the accumulation of a 150 kDa BDP given the absence of the signature 120 kDa fragment (Pike et al., 1998). Rather, the 150 kDa product may represent the initial cleavage product of spectrin, later cleaved to a 145 kDa fragment (Zhang et al., 2009). Why calpain inhibition would be effective in inhibiting the second, but not the initial cleavage of α-spectrin is not clear. The 145 kDa BDP has been suggested to be a more sensitive marker for calpain activation (Zhang et al., 2009) and a biomarker for injury severity in experimental TBI (Pike et al., 2001, Ringger et al., 2004) and human TBI patients (Mondello et al., 2010). Thus, inhibition of spectrin breakdown in Prp-hCAST mice may be an indication of reduced injury severity.
The collapsin response mediator proteins are a family of intracellular proteins expressed within the CNS during development and periods of axonal growth (Quinn et al., 1999). CRMP-2, in particular, is important in protein trafficking (Rahajeng et al., 2010), microtubule assembly (Fukata et al., 2002), and neurite outgrowth (Quinn et al., 2003). Calpain digestion of CRMP-2 results in generation of a 55 kDa BDP which is inhibited with SJA6017 application in cortical lysates digested with calpain-2 (Zhang et al., 2007). Here we demonstrate that CCI injury results in the appearance and subsequent accumulation of a 55 kDa CRMP-2 fragment as previously described (Zhang et al., 2007), which is attenuated by calpastatin overexpression. CRMP-2 cleavage by calpains following TBI may disrupt the protein’s interactions with axonal transport proteins (Touma et al., 2007) and act to down-regulate surface expression of NMDA receptors (Bretin et al., 2006) and voltage-gated calcium channels (Brittain et al., 2009). Thus, CRMP-2 processing may not only affect axonal function, but also modulate receptors and channels, potentiating the characteristic ionic imbalance of TBI and neurodegenerative conditions. Inhibition of CRMP-2 cleavage in the presence of the calcium channel-binding domain prevents hippocampal cell death following TBI (Brittain et al., 2011), supporting a link between CRMP-2 cleavage and neuronal death.
Multiple voltage-gated sodium channel isoforms are present within the CNS, participating in action potential generation and propagation. The Nav1.2 protein is localized to axons and terminals of neurons (Westenbroek et al., 1989) and, with injury, is cleaved by calpains (Iwata et al., 2004, von Reyn et al., 2009). Due to the sodium channel’s pivotal role in action potential generation and ion flux, injury-induced Nav1.2 damage or dysfunction may result in increased sodium influx and prolonged membrane depolarization, further exacerbating calcium dysregulation and calpain activation (Yuen et al., 2009). Severe CCI resulted in α-subunit breakdown evidenced by the appearance of fragments of similar size to in vitro findings. Calpastatin overexpression inhibited the accumulation of two distinct channel fragments (85 kDa, 100 kDa) up to 24 h post-CCI, providing strong support that these products are calpain specific. We are the first to demonstrate attenuated sodium channel proteolysis into select fragments with calpain inhibition by calpastatin in vivo, confirming results with application of the calpain inhibitor MDL28170 after in vitro stretch injury (von Reyn et al., 2009, von Reyn et al., 2012). Slight differences in the molecular weights of fragments between in vivo and neuronal stretch injury were evident, likely reflecting differences in injury pathology or gel migration patterns.
In cases where multiple breakdown products were evident, calpastatin overexpression in Prp-hCAST transgenic mice inhibited the appearance of some but not all cleavage products. In particular, Nav1.2 proteolysis into various fragments was coincident with loss of full length protein expression suggesting that calpastatin overexpression is most effective in inhibiting the progressive breakdown of the channel into smaller-sized fragments. Alternatively, cleavages may indicate the activity of alternative proteolytic pathways that function independent of calpain activation and are therefore unaffected by calpastatin overexpression. It is unknown whether partial breakdown of spectrin, CRMP-2, or sodium channel imparts irreversible damage to the protein and cell or whether the fragments may exhibit independent functions in mediating cell death. Our results demonstrate an absence of acute hippocampal neuroprotection, consistent with previous data using CaMKIIα-driven calpastatin overexpression (Schoch et al., 2012). Nevertheless, reduced calpain-mediated proteolysis may delay neuronal damage, allowing the cell to repair itself or expanding the therapeutic window for additional survival interventions. While the accumulation of calpain-mediated fragments assessed in this study was definitively reduced in Prp-hCAST mice, subacute cell survival and functional improvements will need to be investigated.
Calpastatin overexpression within Prp-hCAST mice ensures inhibition of multiple calpain isoforms thereby allowing for broad inhibition of calpains’ proteolytic activity following TBI. However, we are unable to draw conclusions about the differential roles of calpain-1 versus calpain-2 in mediating posttraumatic damage. This study introduces a novel calpastatin overexpressing transgenic mouse, and demonstrates reduced posttraumatic proteolysis of key cellular substrates, spectrin, CRMP-2, and sodium channel. Boosting endogenous calpastatin through genetic manipulation promotes inhibitory mechanisms at the initiation of damage and maintains this inhibition long past the primary insult, a strategy that may be critical for acute, overwhelming calpain activity. Continued studies investigating the ability of calpastatin overexpression to reduce TBI and neurodegenerative pathology may lead to the development of superior agents for calpain inhibition in vivo.
Acknowledgements
This work was funded by NIH grants F31 NS071804 (KMS), T32 DA0222738 (KMS), P30 NS051220, P01 NS058484 (KES), and P01 NS056202 (DFM) and Kentucky Spinal Cord and Head Injury Research Trust (KSCHIRT) 6-12 (KES).
Abbreviations used
- BDP
Breakdown product
- CaMKIIα
calcium/calmodulin-dependent protein kinase II α
- CRMP-2
collapsin response mediator protein-2
- CCI
controlled cortical impact
- DAB
diaminobenzidine
- FJC
Fluoro-jade C
- hCAST
human calpastatin
- mCAST
mouse calpastatin
- Prp
prion protein
- TBI
traumatic brain injury
- TBS
Tris-buffered saline
- Nav1.2
voltage-gated sodium channel 1.2
- WT
wildtype
Footnotes
The authors have no conflicts of interest.
Contributor Information
Kathleen M. Schoch, Spinal Cord and Brain Injury Research Center (SCoBIRC) University of Kentucky B416 Biomedical and Biological Sciences Research Building (BBSRB) 741 South Limestone Street Lexington, KY 40536-0509 (859) 323-5187 (859) 257-5737 (fax) kmscho6@uky.edu.
Catherine R. von Reyn, Janelia Farm Research Campus HHMI 19700 Helix Drive Ashburn, VA 20147 (571) 209-4000 vonreync@janelia.hhmi.org.
Jifeng Bian, Department of Microbiology, Immunology, and Pathology Colorado State University 1619 Campus Delivery Fort Collins, CO 80523 (970) 491-2968 Jifeng.Bian@colostate.edu.
Glenn C. Telling, Department of Microbiology, Immunology, and Pathology Colorado State University 1619 Campus Delivery Fort Collins, CO 80523 (970) 491-2968 glenn.telling@colostate.edu.
David F. Meaney, Department of Bioengineering University of Pennsylvania 240 Skirkanich Hall 210 South 33rd Street Philadelphia, PA 19104-6321 (215) 573-2726 (215) 573-2071 (fax) dmeaney@seas.upenn.edu.
Kathryn E. Saatman, Spinal Cord and Brain Injury Research Center (SCoBIRC) University of Kentucky B473 Biomedical and Biological Sciences Research Building (BBSRB) 741 South Limestone Street Lexington, KY 40536-0509 (859) 323-5145 (859) 257-5737 (fax) k.saatman@uky.edu.
REFERENCES
- Anagli J, Han Y, Stewart L, Yang D, Movsisyan A, Abounit K, Seyfried D. A novel calpastatin-based inhibitor improves postischemic neurological recovery. Biochem Biophys Res Commun. 2009;385:94–99. doi: 10.1016/j.bbrc.2009.04.141. [DOI] [PubMed] [Google Scholar]
- Angers RC, Seward TS, Napier D, Green M, Hoover E, Spraker T, O’Rourke K, Balachandran A, Telling GC. Chronic wasting disease prions in elk antler velvet. Emerg Infect Dis. 2009;15:696–703. doi: 10.3201/eid1505.081458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada S, Piccardo P, Yamaguchi K, Ghetti B, Harris DA. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis. 2004;16:527–537. doi: 10.1016/j.nbd.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Bretin S, Rogemond V, Marin P, Maus M, Torrens Y, Honnorat J, Glowinski J, Premont J, Gauchy C. Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. Journal of neurochemistry. 2006;98:1252–1265. doi: 10.1111/j.1471-4159.2006.03969.x. [DOI] [PubMed] [Google Scholar]
- Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, Molosh AI, You H, Hudmon A, Shekhar A, White FA, Zamponi GW, Brustovetsky N, Chen J, Khanna R. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2) The Journal of biological chemistry. 2011;286:37778–37792. doi: 10.1074/jbc.M111.255455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. The Journal of biological chemistry. 2009;284:31375–31390. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camins A, Verdaguer E, Folch J, Pallas M. Involvement of calpain activation in neurodegenerative processes. CNS Drug Rev. 2006;12:135–148. doi: 10.1111/j.1527-3458.2006.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher NO. Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr Pharm Des. 2006;12:615–638. doi: 10.2174/138161206775474314. [DOI] [PubMed] [Google Scholar]
- Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–165. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (U.S.) NIH publication, p v . Committee on Care and Use of Laboratory Animals. Guide for the care and use of laboratory animals. U.S. Dept. of Health and Human Services, Public Health Service; Bethesda, Md.: [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampfl A, Posmantur R, Nixon R, Grynspan F, Zhao X, Liu SJ, Newcomb JK, Clifton GL, Hayes RL. mu-calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem. 1996;67:1575–1583. doi: 10.1046/j.1471-4159.1996.67041575.x. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW, Wang KK, Hall ED. The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma. 2001;18:1229–1240. doi: 10.1089/089771501317095269. [DOI] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. Embo J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki M, Takano E, Mori H, Sato A, Murachi T, Hatanaka M. All four internally repetitive domains of pig calpastatin possess inhibitory activities against calpains I and II. FEBS Lett. 1987;223:174–180. doi: 10.1016/0014-5793(87)80531-8. [DOI] [PubMed] [Google Scholar]
- McCollum AT, Jafarifar F, Lynn BC, Agu RU, Stinchcomb AL, Wang S, Chen Q, Guttmann RP. Inhibition of calpain-mediated cell death by a novel peptide inhibitor. Exp Neurol. 2006;202:506–513. doi: 10.1016/j.expneurol.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Gehring K, Green DR. Concerted multi-pronged attack by calpastatin to occlude the catalytic cleft of heterodimeric calpains. Nature. 2008;456:404–408. doi: 10.1038/nature07353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondello S, Robicsek SA, Gabrielli A, Brophy GM, Papa L, Tepas J, Robertson C, Buki A, Scharf D, Jixiang M, Akinyi L, Muller U, Wang KK, Hayes RL. alphaII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. Journal of Neurotrauma. 2010;27:1203–1213. doi: 10.1089/neu.2010.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Pike BR, Flint J, Dutta S, Johnson E, Wang KK, Hayes RL. Accumulation of non-erythroid alpha II-spectrin and calpain-cleaved alpha II-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. Journal of neurochemistry. 2001;78:1297–1306. doi: 10.1046/j.1471-4159.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL. Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. Neuroreport. 1998;9:2437–2442. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- Pleasant JM, Carlson SW, Mao H, Scheff SW, Yang KH, Saatman KE. Rate of neurodegeneration in the mouse controlled cortical impact model is influenced by impactor tip shape: implications for mechanistic and therapeutic studies. Journal of Neurotrauma. 2011;28:2245–2262. doi: 10.1089/neu.2010.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posmantur R, Kampfl A, Siman R, Liu J, Zhao X, Clifton GL, Hayes RL. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience. 1997;77:875–888. doi: 10.1016/s0306-4522(96)00483-6. [DOI] [PubMed] [Google Scholar]
- Quinn CC, Chen E, Kinjo TG, Kelly G, Bell AW, Elliott RC, McPherson PS, Hockfield S. TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:2815–2823. doi: 10.1523/JNEUROSCI.23-07-02815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn CC, Gray GE, Hockfield S. A family of proteins implicated in axon guidance and outgrowth. J Neurobiol. 1999;41:158–164. [PubMed] [Google Scholar]
- Rahajeng J, Giridharan SS, Naslavsky N, Caplan S. Collapsin response mediator protein-2 (Crmp2) regulates trafficking by linking endocytic regulatory proteins to dynein motors. The Journal of biological chemistry. 2010;285:31918–31922. doi: 10.1074/jbc.C110.166066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, Cataldo AM, Haroutunian V, Nixon RA. Marked calpastatin (CAST) depletion in Alzheimer’s disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J Neurosci. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringger NC, O’Steen BE, Brabham JG, Silver X, Pineda J, Wang KK, Hayes RL, Papa L. A novel marker for traumatic brain injury: CSF alphaII-spectrin breakdown product levels. Journal of Neurotrauma. 2004;21:1443–1456. doi: 10.1089/neu.2004.21.1443. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7:31–42. doi: 10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Zhang C, Bartus RT, McIntosh TK. Behavioral efficacy of posttraumatic calpain inhibition is not accompanied by reduced spectrin proteolysis, cortical lesion, or apoptosis. J Cereb Blood Flow Metab. 2000;20:66–73. doi: 10.1097/00004647-200001000-00010. [DOI] [PubMed] [Google Scholar]
- Sato K, Minegishi S, Takano J, Plattner F, Saito T, Asada A, Kawahara H, Iwata N, Saido TC, Hisanaga S. Calpastatin, an endogenous calpain-inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. Journal of neurochemistry. 2011;117:504–515. doi: 10.1111/j.1471-4159.2011.07222.x. [DOI] [PubMed] [Google Scholar]
- Schoch KM, Evans HN, Brelsfoard JM, Madathil SK, Takano J, Saido TC, Saatman KE. Calpastatin overexpression limits calpain-mediated proteolysis and behavioral deficits following traumatic brain injury. Experimental neurology. 2012;236:371–382. doi: 10.1016/j.expneurol.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi H, Hata S, Ono Y. Calpain chronicle-an enzyme family under multidisciplinary characterization. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87:287–327. doi: 10.2183/pjab.87.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghian K, Lee JY, Petratos S. Phosphorylation and cleavage of the family of collapsin response mediator proteins may play a central role in neurodegeneration after CNS trauma. Journal of Neurotrauma. 2012;29:1728–1735. doi: 10.1089/neu.2011.2063. [DOI] [PubMed] [Google Scholar]
- Thompson SN, Carrico KM, Mustafa AG, Bains M, Hall ED. A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J Neurotrauma. 2010;27:2233–2243. doi: 10.1089/neu.2010.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma E, Kato S, Fukui K, Koike T. Calpain-mediated cleavage of collapsin response mediator protein(CRMP)-2 during neurite degeneration in mice. The European journal of neuroscience. 2007;26:3368–3381. doi: 10.1111/j.1460-9568.2007.05943.x. [DOI] [PubMed] [Google Scholar]
- von Reyn CR, Mott RE, Siman R, Smith DH, Meaney DF. Mechanisms of calpain mediated proteolysis of voltage gated sodium channel alpha-subunits following in vitro dynamic stretch injury. Journal of neurochemistry. 2012;121:793–805. doi: 10.1111/j.1471-4159.2012.07735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH, Siman R, Meaney DF. Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:10350–10356. doi: 10.1523/JNEUROSCI.2339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Xiong W, Wang Y, Ping X, Head JD, Brittain JM, Gagare PD, Ramachandran PV, Jin X, Khanna R. Prevention of posttraumatic axon sprouting by blocking collapsin response mediator protein 2-mediated neurite outgrowth and tubulin polymerization. Neuroscience. 2012;210:451–466. doi: 10.1016/j.neuroscience.2012.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Browne KD, Iwata A, Smith DH. Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. Journal of neuroscience research. 2009;87:3620–3625. doi: 10.1002/jnr.22161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Larner SF, Liu MC, Zheng W, Hayes RL, Wang KK. Multiple alphaII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis. 2009;14:1289–1298. doi: 10.1007/s10495-009-0405-z. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Ottens AK, Sadasivan S, Kobeissy FH, Fang T, Hayes RL, Wang KK. Calpain-mediated collapsin response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. Journal of Neurotrauma. 2007;24:460–472. doi: 10.1089/neu.2006.0078. [DOI] [PubMed] [Google Scholar]