

Abstract
Synaptic vesicle exocytosis is mediated by the vesicular Ca2+-sensor synaptotagmin-1. Synaptotagmin-1 interacts with the SNARE protein syntaxin-1A and with acidic phospholipids such as phosphatidylinositol 4,5-bisphosphate (PIP2). However, it is unclear how these interactions contribute to triggering membrane fusion. Using both PC12 cells from Rattus norvegicus and artificial supported bilayers we now show that synaptotagmin-1 interacts with the polybasic linker region of syntaxin-1A independent of Ca2+ via PIP2. This interaction allows both Ca2+-binding sites of synaptotagmin-1 to bind to phosphatidylserine (PS) in the vesicle membrane upon Ca2+-triggering. We determined the crystal structure of the C2B-domain of synaptotagmin-1 bound to phosphoserine, allowing for developing a high-resolution model of synaptotagmin bridging two different membranes. Our results suggest that PIP2 clusters organized by syntaxin-1 act as molecular beacons for vesicle docking, with the subsequent Ca2+-influx bringing the vesicle membrane close enough for membrane fusion.
Neuronal exocytosis is mediated by SNARE proteins. The vesicular SNARE synaptobrevin-2 binds to an acceptor SNARE complex consisting of SNAP-25 and syntaxin-1A in the plasma membrane (review1–4). During binding, a tight α-helical bundle is formed that pulls the membranes together and overcomes the energy barrier for membrane fusion. Syntaxin-1A and SNAP-25 are not uniformly distributed in the plasma membrane, but cluster in ~70 nm-sized membrane domains5–7. We recently showed that polyanionic PIP2 is the dominant inner-leaflet lipid in these membrane domains enriched in syntaxin-1A7. Furthermore, we showed that syntaxin-1A clustering is driven by electrostatic interactions between PIP2 and a juxtamembrane polybasic linker region of syntaxin-1A that connects the SNARE domain and the transmembrane helix (260KARRKK265)7. The significance of these syntaxin–PIP2 clusters for fast exocytosis, however, has remained unclear.
Synaptotagmin-1 acts as a main Ca2+-sensor of neuronal exocytosis (review2–4). It consists of an N-terminal unstructured luminal domain, a single transmembrane helix and a cytoplasmic domain that contains a 61-residue cytoplasmic linker connecting the transmembrane domain to two calcium-binding C2-domains, called C2A and C2B. C2A and C2B bind 3 and 2 Ca2+-ions, respectively, with affinities ranging from 0.1 to 2 mM2,3. Despite intense efforts, it is still controversially discussed how synaptotagmin-1 triggers Ca2+-evoked exocytosis, with the mechanism of action likely involving interactions of synaptotagmin-1 with both SNARE proteins and membranes. Synaptotagmin-1 binds to SNAP-25, syntaxin-1A, and the binary and ternary SNARE complexes (review2–4,8). Synaptotagmin-1 interactions with SNAREs are observable in absence of Ca2+ (i.e. with an excess of the Ca2+-chelator EGTA) and are enhanced by Ca2+. It has remained elusive for a long time how synaptotagmin-1 precisely binds to SNARE proteins (review2), especially since regions of synaptotagmin-1 that were implied in SNARE binding are also involved and compete with membrane binding9. These discrepancies were recently overcome in a single-molecule study, where synaptotagmin-1 was proposed to interact only loosely with the SNARE complex via weak electrostatic interactions in a configuration that left the lipid-binding domains free for interactions with membranes8. A recent electron paramagnetic resonance (EPR) study lend further support to a not very structured but rather dynamic binding of synaptotagmin-1 to the SNAREs10. Nevertheless, there are still some unresolved questions concerning binding of synaptotagmin to SNAREs. For instance an unstructured and dynamic binding mode is difficult to reconcile with the reported ability of synaptotagmin-1 to replace complexin-1 from the SNARE complex11,12 and it does not explain the Ca2+-dependency of SNARE-binding (review2–4,8). Binding of synaptotagmin-1 to membranes containing anionic phospholipids is better understood. In the absence of Ca2+, a conserved polybasic lysine stretch located on the C2B-domain and other surface exposed basic residues interact with anionic phospholipids such as monovalent PS and polyvalent PIP2. In the presence of Ca2+, both Ca2+-binding sites of the C2-domains interact with anionic phospholipids2–4,10,13–27. In its Ca2+-bound state, the C2-domains of synaptotagmin-1 are able to tether two membranes17–20,22,24, but how exactly the C2-domains are able to simultaneously bind to two membranes and whether such tethering is also relevant in the activation mechanism of synaptotagmin is still unclear. We performed a series of controlled binding experiments to resolve the interactions of synaptotagmin-1 with syntaxin-1 as well as with the vesicular and plasma membranes.
Results
Synaptotagmin-1 binds to syntaxin-1A via PIP2
Since synaptotagmin-1 binds to both PIP2 and syntaxin-1A, we tested whether synaptotagmin-1 can also bind to membrane clusters that are enriched in both syntaxin-1A and PIP26,7. Indeed, a bacterially expressed soluble domain of synaptotagmin-1 (called C2AB-domain; residues 97–421) bound to the syntaxin-1A clusters on inverted plasma membrane sheets of PC12 cells (a neuroendocrine cell line) irrespective of Ca2+ (Fig. 1a). In these experiments, freshly prepared (non fixed) membrane sheets were briefly (< 5 min) incubated with a monoclonal antibody HPC-1 against syntaxin-1 labeled with Alexa fluor 488 (anti-Sx1-AF488) and C2AB labeled at position S342C with the far-red fluorophore KK114. Super-resolution STED microscopy with HPC-1 and a fluorescently labeled secondary antibody on fixed membrane sheets showed that the C2AB clusters and syntaxin-1A membrane domains overlapped and were of similar size (~75 nm; Fig. 1b–d, Supp. Fig 1a–b). Addition of Ca2+ to a final concentration of 200 µM did increase binding of C2AB to the syntaxin-1A clusters and strongly increased C2AB binding to the areas between the clusters (Fig. 1e–g, Supp. Fig. 1c). To probe the mobility of membrane bound C2AB under these conditions we applied fluorescence correlation spectroscopy in combination with STED (STED-FCS)28. STED-FCS allows for determining the apparent diffusion coefficient D for different observation areas ranging from 50 to 250 nm (full-width at half-maximum intensity; FWHM) revealing hindered diffusion in case of any deviation from a constant dependence D(FWHM)28,29. As expected29, a fluorescent lipid phosphatidylcholine analog (Atto647N-PC) showed free Brownian diffusion with constant values D = 0.5 µm2 s−1 (Fig. 2a). In contrast, the dynamics of C2AB bound to the syntaxin-1A clusters were too slow to be evaluated by point FCS measurements, because the cluster bound population strongly photobleached during a typical excitation period of 10 s. C2AB bound to the areas between the clusters was mobile but diffusion was strongly anomalous (Fig. 2a). The decrease of D for small FWHM reveals transient binding (trapping) of C2AB to slow or immobilized membrane constituents and was observed previously for lipids with large polar head groups29, indicating that we probed the diffusion of a C2AB-PIP2 complex. Indeed, a fluorescent PIP2-analog (Top-Fluor PIP2) partitioned to the same membrane clusters where C2AB bound (Fig. 2b–c). Moreover, overexpression of the phosphatase domain of synaptojanin-1, fused to a CAAX-box to target it to the plasma membrane30, strongly reduced both Ca2+-independent and Ca2+-dependent C2AB binding to PC12 membrane sheets (Supp. Fig. 2). Synaptojanin-1 is a polyphosphoinositide 5-phosphatase, and the expression of the construct completely removes PIP2 from the plasma membrane30 and results in dispersion of the syntaxin-1 membrane domains7. Thus, synaptotagmin-1 binds selectively to the PIP2–syntaxin-1 clusters at the plasma membrane and this raises the question which parts of synaptotagmin-1 (C2AB) are involved in binding to these clusters.
Figure 1.

Synaptotagmin-1 binds to syntaxin-1 clusters in inverted sheets of PC12-cell derived plasma membranes. (a) Representative confocal images of unfixed membrane sheets showing immunolabeled syntaxin-1 (anti-Sx1-AF488; green) and the bound C2AB fragment of synaptotagmin-1, labeled with the fluorescent dye KK114 (C2AB-KK114; magenta). Right: fluorescence intensity profiles through the lines marked in the images (counts per pixel; yellow bars: clustered regions). In the absence of Ca2+, 100 nM C2AB bound to >80% of the syntaxin-1 enriched membrane clusters. (b) Representative STED image of C2AB-KK114 on an unfixed PC12 membrane sheet. (c) Fluorescence intensity profiles through the lines marked in b showing size and density of C2AB clusters. (d) Size distribution of the C2AB and syntaxin-1A clusters as determined from the STED microscopy (green: syntaxin-1A, diameter 80 ± 23 nm, similar to that reported previously5; magenta: C2AB, diameter 75 ± 26 nm; FWHM; ± s.d., 150 domains, 6 sheets, 2 independent preparations). (e) Distribution of C2AB-KK114 without Ca2+ (left, purple) and after addition of 200 µM Ca2+ (middle, blue), and after chelating previously added Ca2+ by adding EGTA (orange, right). (f,g) Fluorescence signal in the areas between (f) and at (g) the puncta determined from the images of C2AB-KK114 (peak counts ± range from 6 sheets of 3 independent preparations; *: P = 6 × 10−4 – 8 × 10−4, two-sided t-test; see Supp. Fig. 1c for distributions of C2AB binding to the puncta). (scale bars, 2 µm)
Figure 2.
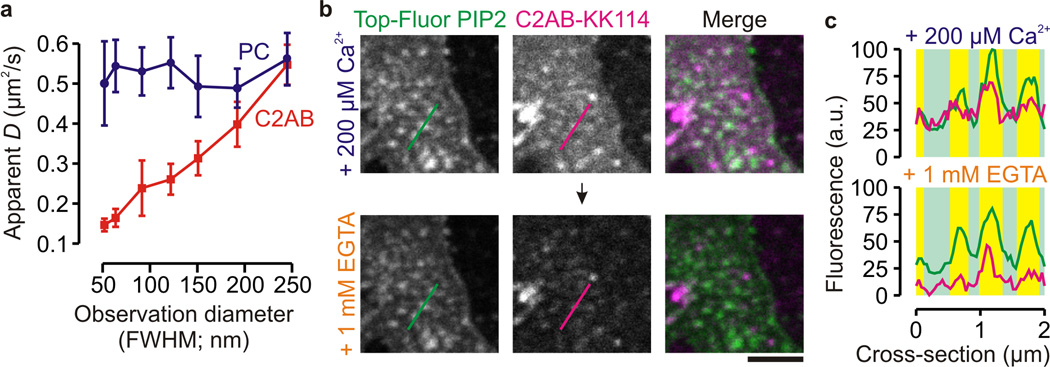
Synaptotagmin-1 binding to syntaxin-1 clusters is mediated by PIP2. (a) Dependence of the apparent diffusion coefficient D on the FWHM of the observation area obtained by STED-FCS of C2AB-KK114 (C2AB; red) and PC-Atto647N (PC; blue) on PC12 membrane sheets in the presence of 200 µM Ca2+, indicating free diffusion for the PC and transient molecular interactions for the C2AB fluorescent analogs. (± s.e.m. from 4 sheets) (b) Confocal images of unfixed PC12 membrane sheets preincubated with Top-Fluor PIP2 to localize PIP2-clusters (green), followed by incubation with labeled synaptotagmin-1 (100 nM C2AB-KK114; magenta) with 200 µM Ca2+ (upper panels) and after chelating Ca2+ by an excess of EGTA (lower panels) (scale bar, 2 µm) (c) Fluorescence intensity profiles through the lines marked in b (see Fig. 1). Binding of C2AB to the PIP2 clusters was observed in absence of Ca2+.
C2AB mutants disrupted in Ca2+-binding to either the C2A- (D178A D230A D232A) or the C2B-domain (D309A D363A D365A) were still able to bind to the syntaxin-1A clusters on PC12 membrane sheets (Fig. 3a). However, Ca2+-dependent binding of C2AB to the areas between the domains was substantially reduced compared to wild-type. These results are likely explained by reduced Ca2+-dependent membrane binding where the Ca2+-binding sites of the C2A- and C2B-domains insert in the membrane in a cooperative manner13,21. In sharp contrast to the Ca2+-binding mutants, C2AB with 2 lysines of the polybasic lysine stretch converted to alanines (K326A K327A; the KAKA mutant15) no longer bound to the syntaxin-clusters in absence of Ca2+ (Fig. 3a). However, again in contrast to the Ca2+-binding mutants, this mutant was still capable of Ca2+-dependent binding to the syntaxin-1A domains and the areas between the syntaxin-1A domains. These findings clearly indicate that the polybasic lysine stretch of synaptotagmin-1 binds to the syntaxin-1A clusters via PIP2, and syntaxin-1 and synaptotagmin-1 may interact with one or more PIP2 molecule simultaneously (Fig. 3b) or alternatively PIP2 may unbind from syntaxin-1 before binding to the C2B-domain. Synaptotagmin-1 binding to syntaxin-1A viaPIP2 is in line with competitive inhibition experiments of SNARE binding by inositol hexabisphosphate (IP6)16,31. Of course, we cannot exclude other modes of (Ca2+-mediated) binding of synaptotagmin-1 to SNAREs, such as to SNAP-25 or to the anionic surface of binary or ternary SNARE complexes2,3,8,10, but apparently these binding sites are not easily accessible when the SNAREs are in a native environment.
Figure 3.

Synaptotagmin-1 binds to the syntaxin-1 clusters via the conserved polybasic patch. (a) Representative confocal images of unfixed PC12 cell membrane sheets incubated with 100 nM of different AF488-labeled C2AB mutants, either in the presence of 200 µM Ca2+ (blue) or after chelating the Ca2+ by an excess of EGTA (orange). Right: fluorescence intensity profiles through the lines marked in the images (see Fig. 1). The mutants with disrupted Ca2+-binding sites in the C2A- (A-mutant) or C2B- (B-mutant) domain showed reduced binding to the areas between but not at the membrane puncta. In contrast, the KAKA-mutant no longer bound to the membrane puncta in absence of Ca2+, confirming that binding to the PIP2–syntaxin clusters is mediated by the polybasic lysine stretch. (b) Model of Ca2+-independent binding of the basic stretch of synaptotagmin-1 (purple) to membrane domains enriched in PIP2 (blue) and syntaxin-1A (Syx1; red). Green: SNAP25; cyan: synaptobrevin-2 (Syb2). (scale bar, 5 µm)
We recently demonstrated the reconstitution of PIP2–syntaxin-1A clusters in artificial membranes7. Here we studied binding of synaptotagmin-1 to these clusters using supported lipid bilayers. Main advantages of supported lipid bilayers are that they allow precise control of the lipid composition and are easy to image by fluorescence microscopy. We prepared stacked bilayers of 2 – 3 membranes by spin-coating lipid mixtures in chloroform on microscope cover glass. Artifacts due to electrostatic and hydrophobic interactions with the glass support can be circumvented by studying lipid bilayers that are on top of the basal bilayer32. PIP2–syntaxin-1A membrane clusters were reconstituted in membranes composed of 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC), with or without 20% PS, 30% cholesterol, 1 – 2% PIP2 and syntaxin-1A257–288 that consisted of a conserved polybasic juxtamembrane linker region and the C-terminal transmembrane helix of syntaxin-1A (Fig. 4a–d, Supp. Fig. 3a). As shown previously7, syntaxin-1A257–288 clustering was dependent on the presence of PIP2 (Supp. Fig. 3b). Moreover, PIP2–syntaxin-1A257–288 clusters were also present when an excess of PS or cholesterol was used (Supp. Fig. 3c) and fluorescent Top-Fluor PIP2 was enriched at the syntaxin-1A257–288 clusters (Fig. 4e–f, Supp. Fig. 3d). Similar to PC12 membrane sheets (Fig. 1), C2AB bound to the PIP2–syntaxin-1A257–288 clusters in absence of Ca2+ and addition of Ca2+ to a final concentration of 100 – 200 µM increased C2AB binding to both the puncta and the areas between the puncta (Fig. 4a–d, Supp. Fig. 3a). Addition of the Ca2+-chelator EGTA to a final concentration of 1 mM did only slightly affect the clusters but substantially removed C2AB that was bound to the areas between the membrane clusters. In absence of syntaxin-1A257–288, C2AB bound homogeneously to membranes containing 2 mol% PIP2 but only when Ca2+ was present (Fig. 5a). Apparently, this concentration of PIP2 was not high enough for substantial C2AB-binding in absence of Ca2+ which required at least 5 mol% PIP2 (Supp. Fig. 3e). Control experiments without PIP2 and with Mg2+ showed that C2AB binding to supported lipid bilayers was specific for both PIP2 and Ca2+ (Supp. Fig. 3f–g).
Figure 4.

Synaptotagmin-1 binding to PIP2–syntaxin-1A clusters in artificial stacked supported membranes generated by spin-coating on glass surfaces. (a) Representative confocal images of membranes (69% DOPC, 30% cholesterol, 1% PIP2) containing syntaxin-1A257–288 labeled with Atto647N (SxTMH; magenta), which were incubated with 100 nM C2AB fragment (labeled with Alexa fluor 488; C2AB-AF488; green) in the absence (purple) or after addition of 200 µM Ca2+ (blue) and after chelating previously added Ca2+ with EGTA (orange). (b) Fluorescence intensity profiles through the lines marked in a (see Fig. 1). (c,d) Fluorescence (peak counts) between (c) and at (d) the puncta determined from the different images of C2AB-AF488 (± range; 6 positions, 3 independent bilayers; *: P = 0.006 – 0.01, two-sided t-test; see Supp. Fig. 3a for distributions of C2AB binding to the puncta). (e,f) Same as panels a and b in absence of Ca2+, but now with fluorescent Top-Fluor PIP2 (0.1 mol% TF-PIP2; green) instead of C2AB. PIP2 was clearly enriched in the SxTMH clusters. (scale bars, 2 µm)
Figure 5.

Synaptotagmin-1 binds to PIP2 clusters induced by the polybasic linker region of syntaxin-1 in artificial membranes. (a) Representative confocal images of supported lipid bilayers (DOPC, 2% PIP2) prelabeled with 0.1 mol% of the fluorescent lipid analog bodipy-FL PC (Lipid; green), followed by incubation with 100 nM C2AB (labeled with KK114; C2AB-KK114; magenta) in the presence of 200 µM Ca2+. (control; purple, left), after addition of 20 µM of the soluble syntaxin-1A260–265 linker peptide (KARRKK; blue, middle) and after addition of EGTA (with the peptide still present; orange, right). (b) Fluorescence intensity profiles through the lines marked in a (see Fig. 1). (c,d) Fluorescence peak counts between (c) and at (d) the puncta determined from the confocal images of C2AB-KK114 (± range; 6 positions, 3 independent bilayers; *: P = 2 × 10−3, two-sided t-test; see Supp. Fig. 4a for distributions of C2AB binding to the puncta). (e) Control experiments showing the same as in panel a, but using membranes containing PS instead of PIP2 (80% DOPC, 20% DOPS). Addition of the polybasic linker did not induce clustering of C2AB-KK114. (scale bars, 5 µm)
In order to test the nature of synaptotagmin-1 binding to membrane clusters of PIP2 and syntaxin-1A, we performed experiments with short cationic peptides that mimic the polybasic linker region of syntaxin-1A. A peptide corresponding to the polybasic region of syntaxin-1 (260KARRKK265) was able to cluster C2AB in artificial membranes in the presence of PIP2 (Fig. 5a–d, Supp. Fig. 4), but this clustering was not specific for the sequence of this region as it was also induced by pentalysine (Lys5). Membrane clustering by syntaxin-1A260–265 was not observed when PS instead of PIP2 was used (Fig. 5e) and was relatively insensitive to Ca2+ or Mg2+ (Fig. 5; Supp. Fig. 4). PIP2 clustering by polyvalent cationic peptides has also been observed previously33. Ca2+ is also known to cluster PIP234 and indeed C2AB was clustered at high Ca2+ concentrations above 0.5 mM in absence of the polybasic peptides (Supp. Fig. 5). However, the clusters induced by high Ca2+ concentrations were different from the clusters induced by the basic peptides since only in the latter PC was at least partially depleted from the clusters (compare Fig. 5a). More importantly, the Ca2+ but not the ligand-induced cluster formation was reversed when Ca2+ was subsequently chelated with EGTA. (Supp. Fig. 5). Together, we conclude that synaptotagmin-1 binds in a Ca2+-independent manner to membrane domains in which PIP2 is concentrated around the polybasic linker of syntaxin-1A7.
Vesicle recruitment to PIP2–syntaxin-1A membrane clusters
It is well established17–20,22,24 that the C2AB-fragment can crosslink two membranes in the presence of Ca2+. Therefore, we tested whether C2AB can recruit vesicles to the PIP2–syntaxin-1A clusters. Indeed, in the presence of C2AB we observed binding of 35 nm-sized17 artificial liposomes (composed of a 5:2:1:1 molar ratio of DOPC:DOPE:DOPS:cholesterol with 1% of the fluorescent lipid analog bodipy-FL PC) to PC12 membrane sheets (Fig. 6a). Binding was Ca2+-dependent but was only partially reversed by EGTA, which we attribute to short range (hydrophobic) interactions that occur after membrane binding. Liposome tethering was only observed when the liposomes contained > 5% of the anionic phospholipid 1,2-dioleoyl-sn-glycero-3-phosphatidylserine (DOPS; Fig. 6b). Tethering clearly overlapped with the sites where C2AB was bound (Fig. 6a). However, some tethering (albeit weaker) was also observed in the absence of C2AB (Fig. 6c). This was probably caused by endogenous synaptotagmin-1, because liposome tethering was substantially reduced by preincubation with antibodies specific for synaptotagmin-1 (Fig. 6d). In order to exclude interference by endogenous synaptotagmin-1, we performed experiments with supported lipid bilayers of precisely controlled composition (Fig. 7a–b, Supp. Fig. 6). Liposome recruitment to reconstituted PIP2–syntaxin-1 clusters in artificial membranes was clearly dependent on the presence of the C2AB-fragment or of full-length synaptotagmin-1 incorporated in the liposomes and was observed irrespective of the presence of PS in the supported bilayer.
Figure 6.

Ca2+-dependent recruitment of liposomes to the plasma membrane by synaptotagmin-1. (a) Representative confocal images of unfixed PC12 membrane sheets that were incubated with bodipy-labeled liposomes containing 11% phosphatidylserine (PS) (green; ~5 nM final liposome concentration17) and 100 nM C2AB labeled with KK114 (C2AB; magenta) in the absence (No Ca2+; upper panels) and after addition (+ Ca2+; lower panels) of 200 µM Ca2+. Right: fluorescence intensity profiles through the lines marked in the images (see Fig. 1). (b) Same as a in the presence of Ca2+, but here liposomes lacking PS (No PS) were used. (c) Same as a in the presence of Ca2+, but in the absence of C2AB. The membrane sheet was visualized with Atto647N-PE (Lipid; magenta). Liposomes still bound in absence of C2AB. (d) Same as a, but now endogenous synaptotagmin-1 was blocked with an Atto647N-labeled antibody (Anti-Syt1-A647N). Liposome tethering could be partially rescued by addition of 100 nM C2AB. (scale bars, 5 µm)
Figure 7.

Two membrane binding modes of synaptotagmin-1. (a) Representative confocal images of supported lipid bilayers (lipid composition given in Fig. 5) containing unlabeled syntaxin-1A257–288 and incubated with bodipy-labeled liposomes (Liposome; green; ~5 nM liposome concentration17) and 100 nM C2AB-KK114 (C2AB; magenta) in presence of 200 µM Ca2+. (b) Representative confocal images of bodipy-labeled liposomes bound to supported lipid bilayers with a more physiological composition (52% DOPC, 15% DOPS, 3% PIP2, 30% cholesterol) and for different unlabeled C2AB mutants: wild-type C2AB (C2AB), 1:1,000 protein-to-lipid ratio of full-length synaptotagmin-1 incorporated in the liposomes (FL Syt1), in absence of C2AB (No C2AB), the C2A-mutant (A-mut.), the C2B-mutant (B-mut.), the double mutant (AB-mut.), the KAKA mutant (KAKA), the RQRQ mutant (RQRQ), the C2A domain (C2A) and the C2B domain (C2B). The bar graphs indicate the liposome binding efficiency and show the membrane-bound liposome densities determined for 8 different regions from at least 2 independent experiments (error bars, range of values; see Supp. Fig. 6 for distributions of liposome binding to the puncta). We observed no binding in absence of C2AB, with the AB-mutant and with C2A-domain. (c) Model of vesicle recruitment by synaptotagmin-1 in the presence of Ca2+ (orange) where the C2B domain is oriented in a ‘parallel’ orientation with the Ca2+-binding site interacting with the plasma membrane. Color coding is as in Fig. 3b. (d) Same as c, but now with the C2B-domain in a ‘perpendicular’ orientation with the Ca2+-binding site of C2B interacting with the vesicular membrane. (scale bars, 5 µm)
We used artificial supported lipid membranes and different synaptotagmin-1 mutants to determine which of the binding sites of synaptotagmin-1 are involved in liposome recruitment (Fig. 7b, Supp. Fig. 6). Interestingly, liposome binding to the membrane was prevented when a mutant disrupted in Ca2+-binding to both C2A and C2B (AB-mutant: D178A D230A D232A D309A D363A D365A) was used (Fig. 7b). In contrast, mutating only one of the Ca2+-binding sites (A-mutant: D178A D230A D232A; or B-mutant: D309A D363A D365A) or only the polybasic patch (KAKA mutant: K326A K327A) did not suffice to markedly reduce liposome tethering. The C2B domain (residues 271 – 421), but not the C2A domain (residues 96 – 263), was able to tether liposomes, in agreement with earlier observations19 but contrasting references (20,22). Finally, liposome tethering was still observed with C2AB with two conserved arginines mutated that are located opposite the Ca2+-binding loop (RQRQ mutant: R398Q R399Q). This mutant displays severely disrupted Ca2+-stimulated synchronous release in neurons12. Arg398 and Arg399 potentially interact in trans with anionic membranes19,24, although this is controversial and no or minor interactions with membranes are also reported20,27. In fact, Arg398 and Arg399 are recently proposed to crosslink membranes via oligomerization of the C2AB fragment, although this oligomerization required higher concentrations of C2AB than the 100 nM used in our experiments20. Accordingly, we found no indications for such oligomerization of C2AB under our experimental conditions, because (i) liposomes could still be tethered when C2AB was pre-bound to the supported lipid bilayers and unbound C2AB was removed prior to addition of the liposomes (Supp. Fig. 7a), (ii) we did not find evidence for oligomerization by fluorescence cross-correlation spectroscopy using a mixture of C2AB labeled with different dyes regardless of the presence of Ca2+ or membranes (Supp. Fig. 7b) and (iii) the RQRQ mutant (with impaired oligomerization20) was still capable of liposome tethering (Fig. 7b).
Together, these data suggest that the C2AB fragment can tether liposomes in at least two possible conformations: In the first conformation, the C2B domain is oriented ‘parallel’ to the membrane, with both the polybasic lysine stretch and the Ca2+-binding site of the C2B-domain interacting with the plasma membrane, leaving Arg398, Arg39919,24 and the C2A domain free to interact in ‘trans’ with the vesicle membrane (Fig. 7c). This first configuration would explain the capability of the KAKA mutant to tether liposomes (Fig. 7b). In the second conformation, the C2B domain is oriented ‘perpendicular’ to the plasma membrane and the polybasic patch interacts with the plasma membrane whereas both Ca2+-binding sites interact with the vesicular membrane (Fig. 7d). This second configuration would explain the capability of the mutants with the Ca2+-binding sites of either (but not both) the C2A or C2B-domains disrupted to tether liposomes (Fig. 7b). The second conformation seems surprising given the close proximity of the polybasic patch and the Ca2+-binding site on the C2B-domain. Indeed, it was concluded from EPR measurements that the C2B domain bound in a ‘parallel’ orientation and PIP2 only resulted in tilting of the C2B domain towards the membrane24. On the other hand, the membrane-bound orientation of the structurally similar C2-domain of protein kinase C alpha (PKCα) is still controversial and both parallel35,36 and perpendicular37 binding are reported. Accordingly, while it is generally agreed that PIP2 changes the membrane orientation of C2-PKCα relative to the membrane, it is debated whether C2-PKCα tilts towards the membrane35 (i.e. similar to C2B24) or away from the membrane36. Our data now indicate that the C2B membrane can tether liposomes in both parallel and perpendicular conformations.
Membrane tethering of the C2AB-domain in a perpendicular conformation would imply that C2B interacts with both the plasma membrane and the vesicular membrane simultaneously. It seems logical that the polybasic patch of synaptotagmin-1 would interact with the plasma membrane, since PIP2 is the dominant inner-leaflet lipid in the plasma membrane at the sites of docked vesicles7 whereas it is believed to be absent from synaptic vesicles. The polybasic stretch is a conserved PIP2-binding motif, as shown previously for the C2-domains of rabphilin-3A and PKCα38,39 and in the recent NMR structure of synaptotagmin-1 with inositol 6-phosphate25. Our findings indicate that the Ca2+-binding site on the C2B-domain can interact with the vesicular membrane because a C2AB-mutant disrupted in Ca2+-binding still binds to the PIP2–syntaxin-1A membrane clusters, but no longer can tether liposomes. These interactions of the Ca2+-binding pocket may occur via PS or PIP2 which can complete the Ca2+-coordination site and increase the Ca2+-binding affinity of synaptotagmin-1, as is well described in the literature13,18,27,40.
Simultaneous interactions of C2B with PS and PIP2
To shed more light on how binding of the two adjacent sites to two opposing membranes can be accommodated, we crystallized the C2B fragment in presence of phosphoserine as an analogue for PS and determined the structure at 1.5 Å resolution (Fig. 8a and b, Supp. Fig. 8a, Table 1). Crystals belong to the tetragonal space group P4212 with two molecules in the asymmetric unit. Both molecules contain phosphoserine. The carboxyl group of phosphoserine is bound to a Ca2+-atom (calcium 1), which explains the Ca2+-dependency of PS binding. Binding of the phosphoserine carboxyl group completes the (pentagonal bipyramidal) coordination site of calcium 1 (Fig. 8a). In the apo structure (pdb: 1UOV) this site is occupied by a water molecule41. Phosphoserine is further stabilized by extensive interactions with the protein involving all of its functional groups. The amine group of phosphoserine forms a hydrogen bond with the carboxyl group of D309 and the phosphoserine carboxyl group forms a hydrogen bond with the amide nitrogen of Lys366 (Fig. 8a). Lys366 also participates in a salt bridge with the phosphoserine phosphate group, which is surface exposed. In the crystal structure, Lys332 is involved in crystal packing contacts by forming a salt bridge with Glu350 from a symmetry related molecule and is too far away to form a salt bridge with the phosphate group (6.5 Å), but might otherwise swing in and interact with the phosphate group. Both Lys332 and Lys366 are structurally conserved and correspond to Arg198 and Arg233 in the C2A-domain. Note that the interaction between phosphoserine and the C2B-fragment is completely different from the interaction described previously between PS and the C2-domain of PKCα (Supp. Fig. 8b)40. In the PKCα structure the phosphate group of PS interacts with calcium 1 and the PS carboxyl group forms a hydrogen bond to the amide nitrogen of Asn189 instead. In contrast to the only sparse interactions of the PS head group observed in the PKCα structure, phosphoserine extensively interacts with C2B in our structure. Note that we observed three and four calcium atoms bound in the two molecules of the asymmetric unit, whereas typically two bound calcium ions are reported for C2B26,41 (further discussed in Supp. Fig. 8c).
Figure 8.

Lipid binding modes of the C2B-domain. (a) Stereo view showing a close-up of the phosphoserine bound Ca2+-binding site of the C2B-domain from synaptotagmin-1. Coordination of calcium 1 is shown with red dashed lines and the interactions of phosphoserine with the protein are marked with black dashed lines. Calcium ions are drawn as yellow spheres. Distances are given in Å. (b) Overall structure of C2B-fragment with the four bound Ca2+-ions and phosphoserine (PS). We modeled the head group of PIP2 into the polybasic stretch based on reference (39) (pdb: 3GPE). (c) Snapshot from a coarse-grained molecular dynamics simulation run of the ‘parallel’ membrane binding conformation of the C2B-domain. Here, both the PS interacting Ca2+-binding site and the PIP2-interacting polybasic stretch interact with the same membrane, as shown by EPR24. Arg398 and Arg399 are shown in green. (Scale bar, 2 nm) (d) Snapshot of membrane cross-linking by the C2B-domain (i.e. in ‘perpendicular’ conformation). Note the perturbation of the C2B-fragment in the lower membrane (see Supp. Fig. 9). The membranes were composed of 80% DOPC and 20% DOPS. (scale bars; 5 nm)
Table 1.
Data collection and refinement statistics.
| C2B with phosphoserine | |
|---|---|
| Data collection1 | |
| Space group | P4 21 2 |
| Cell dimensions | |
| a, b, c (Å) | 97.0, 97.0, 89.0 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 50-1.50 (1.54-1.50) 2 |
| Rsym (%) | 3.5 (25.9) |
| I / σI | 43.9 (7.9) |
| Completeness (%) | 97.7 (81.7) |
| Redundancy | 12.2 (7.4) |
| Wilson B-factor (Å2) | 13.7 |
| Refinement | |
| Resolution (Å) | 50-1.50 |
| No. reflections | 66840 |
| Rwork / Rfree | 18.3/ 20.4 |
| No. atoms | |
| Protein | 2413 |
| Ligand (2 PS)/ions (7 Ca2+, 5 SCN−) | 22/ 22 |
| Water | 256 |
| B-factors (Å2) | |
| Overall | 13.5 |
| Protein | 12.6 |
| Ligand (2 PS)/ions (Ca2+, SCN−) | PS: 14.6 / Ca2+ 11.8 / SCN−: 14.4 |
| Water | 21.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.23 |
Data were collected from a single crystal.
Values in parentheses are for highest-resolution shell.
To better understand how the C2B domain can bind simultaneously to the PIP2-containing plasma membrane and the PS-containing vesicle membrane, we used the crystal structure of the C2-domain of PKCα with the headgroup of PIP239 to model PIP2 to the polybasic patch in our new Ca2+–PS-bound C2B-domain structure (Fig. 8b). We then used this structure as a starting point for carrying out molecular dynamics simulations. In these coarse-grained simulations, several atoms were represented by one simulation bead17,42, which allowed for simulations of relatively large systems of > 5,000 lipids. For the parallel binding mode (Fig. 8c), we incorporated both lipids into a membrane composed of 80% DOPC and 20% DOPS. For the perpendicular binding mode (Fig. 8d), we incorporated PIP2 in a planar membrane (i.e. mimicking the plasma membrane) and the PS in a vesicle of 40 nm size (i.e. mimicking a synaptic vesicle). Both systems were stable over simulation times of 500 ns – 1 µs, supporting our conclusion that both conformations of interaction of the C2B-domain with membranes are sterically feasible. Due to the proximity of the Ca2+–PS and PIP2 binding sites, perpendicular binding by C2B brings the membranes very close (below 2 nm from molecular dynamics simulations). PS-binding by the Ca2+-bound C2A-domain and electrostatic membrane interactions with other surface exposed cationic residues of synaptotagmin-1 such as Arg398 and Arg399 which face toward the vesicular membrane in the parallel orientation (Fig. 8c) may act synergistically in the cross-linking of the membranes19,24. Finally, in the perpendicular binding mode, several conserved loops perturb the plasma membrane (further discussed in Supp. Fig. 9) and it is conceivable that under these conditions some stress is exerted on the plasma membrane, which may contribute to a lowering of the energy barrier in the fusion process14.
Discussion
In this study, we found that the polybasic lysine stretch of synaptotagmin-1 interacts independent of Ca2+ with polyanionic PIP2 that is clustered in the membrane by electrostatic interactions with the polybasic juxtamembrane linker region of syntaxin-1A. This interaction can leave the Ca2+-binding sites of synaptotagmin-1 free to bind to PS in the vesicle membrane, thus bringing the vesicle membrane very close to the PIP2–syntaxin-1A membrane clusters upon Ca2+-influx. These findings shed new light on previous observations that so far were difficult to be integrated into a coherent picture. First, Ca2+-independent binding of synaptotagmin-1 to PIP2 positions synaptic vesicles in the vicinity of the acceptor SNAREs prior to membrane fusion, in agreement with previously reported roles of both synaptotagmin-143 and PIP2 (review44) in docking of synaptic vesicles. Second, since PIP2 binding increases the Ca2+-affinity of the C2B-domain more than 40-fold18, pre-binding of synaptotagmin-1 to the PIP2 clusters might explain why the Ca2+-sensitivity of exocytosis, for instance in the calyx of Held (10 – 25 µM)45, is well below the Kd of synaptotagmin-113. Indeed, the impaired binding of the KAKA mutant of synaptotagmin-1 to PIP2-enriched syntaxin-1 membrane domains may well explain the reduced vesicular release probability from hippocampal neurons of this mutant15. Third, our finding sheds new light on the interaction between syntaxin-1A and synaptotagmin-1 that is controversially discussed (see Introduction). Our data show that in an intact membrane this interaction is indirect as it is mediated by syntaxin-bound PIP2. Finally, our results lend further support to the view that synaptotagmin-1 exerts its function in the synapse by regulating the distance between the membranes: upon Ca2+-influx pre-docked vesicles are pulled very close to the plasma membrane (~4 – 9 nm by cryo-EM)19,22, thus promoting SNARE assembly and triggering fusion. Because cross-linking the membranes by the C2B-domain in the perpendicular orientation would result in a ~5 nm shorter membrane tethering distance compared to the parallel orientation (from structural considerations)17, such a model might explain why disruption of Ca2+-binding to the C2A-domain (which does not affect perpendicular binding) has a only minor effects on release whereas disruption of the C2B-domain is much more severe (review2,3). Although our model is still speculative, it agrees well with recent in vitro studies by us17 and others20 showing that membrane fusion is enhanced by orders of magnitude due to distance regulation by synaptotagmin-1. Furthermore, it provides a structural explanation for the ability of synaptotagmin to pull plasma and vesicle membranes closely together in a Ca2+- dependent manner.
Online Methods
Synaptotagmin purification
DNA constructs of full-length synaptotagmin-1 and mutant C2AB-fragments (rat synaptotagmin-197–421) are described13,17, except for the R398Q R399Q (RQRQ) mutant, for which we ordered a synthetic gene (Genscript) in the NdeI–EcoRI site of pET28a(+) (sequence in Supplementary Note). Proteins were expressed in Escherichia coli strain BL21-CodonPlus (DE3)-RIPL (Stratagene) and purified with Ni2+-affinity chomatography followed by ion-exchange chromatography as described13,17,18. The single cysteine mutant of C2AB (C278S S342C) was labeled by 20 min incubation with a 2–10-fold molar excess of Alexa fluor 488 C5-maleimide (Invitrogen) or KK114-maleimide46 in 50 mM HEPES, pH 7.4, 500 mM NaCl and 100 µM Tris(2-carboxyethyl)phosphine. The labeled protein was separated from the free dye by size exclusion chomatography (Sephadex G50, Sigma).
Antibody labeling
We labeled 250 µl of 0.2 mg/ml of the antibody HPC-1 (for syntaxin-1; Sigma-Aldrich, S0664)47 with ~250 nmol Alexa fluor 488 C5-maleimide (Invitrogen) and 100 µl of 1 mg/ml of the antibody 41.1 (for synaptotagmin-1; Synaptic Systems, 105011) with 5 µl of ~10 mM Atto647N NHS-ester (Attotec) by 20 min incubation in 10 mM Na-phosphate pH 7.6 with 250 mM NaCl followed by size exclusion chromatography (Sephadex G50) for removal of unreacted dye. The size exclusion chromatography step resulted in a ~5-fold dilution of the antibody concentration.
Peptide synthesis
Synthesis of Atto647N (Atto-tec) labeled syntaxin-1A257-288 is described7 and syntaxin-1A260-265 (KARRKK) was synthesized via solid phase peptide synthesis on Fmoc-Lys(Boc)-rink amide resin following standard Fmoc strategies. Purification was performed by HPLC on C18 material using water and acetonitrile as mobile phases. The peptides identity was confirmed by mass spectrometry.
Cell culture and immunofluorescence
PC12 cells (clone 251)48 were maintained and propagated as described5. Transfection was with lipofectamine-LTX as described7. Membrane sheets were generated as described5,7, except that the PC12-buffer contained no EDTA (20 mM K-Hepes pH 7.4, 120 mM K-gluconate, 20 mM K-acetate, 2 mM ATP and 0.5 mM DTT). All experiments with PC12 cells were carried out in this buffer. Immunostaining of unfixed membrane sheets was performed by blocking for 5 min with 3% (w/v) BSA in PC12-buffer and incubating with 2 µg/ml Alexa fluor 488-labeled antibody HPC-1 for < 5 min followed by brief washing. For blocking endogenous synaptotagmin-1, the PC12 membrane sheets were incubated with 4 µg/ml Atto647N-labeled antibody 41.1 for 30 min. PC12 membranes were labeled with Atto647N PE, Atto647N PC28,29, bodipy-FL PC, or Top-Fluor PIP2 (Avanti) by 5 min incubation with 1 µM of BSA-complexed lipid analog as described28,29. C2AB was used at a final concentration 100 nM for the incubation of membrane sheets or of artificial membranes. Liposomes were used at a final lipid concentration of ~50 µM (about 5 nM liposome concentration17,49).
Liposome preparation
Liposomes were prepared of a 5:2:1:1 molar ratio of DOPC:DOPE:DOPS:cholesterol with 1% bodipy-FL PC as described17. Briefly, the lipid mixture was dried in a rotary evaporator and resuspended to a total lipid concentration of 30 mM in 20 mM Hepes, pH 7.4, 150 mM KCl with 1.5% Na+-cholate. Liposomes were then formed by size exclusion chomatography (Sephadex G50) in the same buffer without Na+-cholate. Full-length synaptotagmin-1 was reconstituted in the liposomes at a protein-to-lipid molar ratio of 1:1,000 as described17.
Supported lipid bilayers
Supported lipid bilayers were generated by spin-coating (at 2,000 rpm) a 3 mg/ml solution in chloroform of the required cholesterol, DOPC, DOPS and brain PIP2 (all from Avanti) mixture with 0.1 mol% Atto647N phosphatidylethanolamine or bodipy-FL PC on glass50. In case syntaxin-1A257-288 was present, the lipids were dissolved to 1 mg/ml in methanol and mixed to a final protein-to-lipid ratio of approximately 1:100 of unlabeled peptide and 1:1,000 of Atto647N-labeled peptide in 2,2,2-trifluoroethanol (TFE) followed by spin-coating. This procedure gives 1–3 stacked supported lipid bilayers50. Microscopy experiments were performed in 20 mM Hepes, pH 7.4, with 150 mM KCl. C2AB and liposome binding assays were performed identical to the experiments with PC12 membrane sheets.
Confocal and STED microscopy
Confocal and STED imaging and STED-FCS were performed on a home-built confocal STED microscope that was in detail described previously29 except that the setup was expanded with a fast beam scanner (Yanus, Till-Photonics) and a blue excitation (≈ 80 ps pulse width, LDH-P-485B, PicoQuant) and STED laser (577 nm OPSL CW, Coherent) for fast STED imaging and STED-FCS of red-emitting (KK114 and Atto647N) and blue-emitting (Alexa fluor 488) dyes. The excitation intensity was in the range of 5–8 µW at a diffraction-limited focal diameter of approximately 250 nm (640 nm) and 190 nm (485 nm). The average STED power was 180 mW (770 nm) or 120 mW (577 nm) measured in the back aperture of the objective. A software gated detection scheme was used in case of the STED recordings using the blue continuous-wave STED laser51. For imaging, the resolution was approximately 50 nm (for both the 770 nm and 577 nm STED channels) as measured independently by imaging 20 nm-sized fluorescent beads.
Details of the STED-FCS analysis are described28,29. Briefly, we first recorded FCS data for different STED intensities, i.e. for observation areas of different diameters d. Fitting the FCS data as described28,29 resulted in the dependence of the average apparent diffusion coefficient D on the size of the observation area d. The diameter d of the observation area for each STED intensity was calibrated by STED-FCS measurements of a KK114-labeled phosphatidylethanolamine freely diffusing in a DOPC supported lipid bilayer.
Purification and crystallization of C2B fragment
For expression of the C2B-fragment, we ordered a synthetic gene (Genscript) in the NdeI – XhoI restriction sites of pET-28a(+). This gene codes for residues 271 – 421 of rat synaptotagmin-1 (Swiss-Prot P21707). The codon usage was optimized for expression in Escherichia coli (strain K12) (sequence in Supplementary Note). E. coli strain BL21(DE3) expressing C2B was grown in Luria Broth at 37°C to an optical density of 0.6 at 600 nm and induced with 1 mM IPTG and incubated at 30°C for four hours. The cells were harvested and resuspended in buffer A (25 mM HEPES, pH 7.4, 100 mM NaCl). Bacteria were lysed by sonication. Cell debris was removed by centrifugation (5,000 ×g for 20 min) and the supernatant was incubated with Ni-NTA beads for one hour at 4°C. The beads were washed with buffer A containing 30 mM imidazole and the protein was eluted with buffer A containing 300 mM imidazole. The protein was then diluted in a 1:1 ratio with buffer A supplemented with 5 mM CaCl2. Precipitant was removed by centrifugation. The protein was then concentrated and applied onto a Superdex-75 16/60 (GE Healthcare) column. Running buffer was buffer A with 5 mM CaCl2. The protein was concentrated to 30 mg/ml, aliquoted and then frozen in liquid nitrogen and stored at −80°C. A 100 mM phosphoserine (TCI Europe) stock solution was prepared with water and the pH was adjusted to pH 6 with NaOH. Crystals were grown at 20°C with the hanging drop vapor diffusion method. 2 µl of 16 mg/ml C2B in 0.1 M NaCl, 25 mM Hepes pH 7.4, 5 mM CaCl2 with 20 mM phosphoserine were mixed with 2 µl of the precipitant containing 0.2 M KSCN and 20% PEG 600. Crystals were transferred into a cryoprotectant consisting of 10% ethylene glycol, 26% PEG 600, 0.1 M KSCN, 5 mM CaCl2, 20 mM phosphoserine and then flash cooled in liquid nitrogen.
X-ray data collection and structure determination
Diffraction data were collected at a wavelength of 1.0 Å at 100 K on beamline X10SA at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland). Data were processed with XDS52. Crystals diffracted to 1.5 Å resolution and belong to the tetragonal space group P4212. There are two molecules in the asymmetric unit, which are related by pseudo translational non-crystallographic symmetry (pseudo translation vector: u = 0, v = 0, z = 0.5). The structure was determined by molecular replacement using BALBES53 with 1UOV41 as search model. The structure was refined with REFMAC554 and model building was done with COOT55. The final model comprises residues 271 – 418 for molecule A and residues 271 – 419 for molecule B. Structure was analyzed with PROCHECK56 and 90.9% residues are located in the core region of the Ramachandran plot and 9.1% in the allowed region. Figures were prepared with PYMOL (version 1.3. Schrödinger, LLC, 2002).
Molecular dynamics
The molecular dynamics simulations were performed with the GROMACS simulation package57 and are described in the Supplementary Notes.
Supplementary Material
Acknowledgements
We thank V. Belov (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany) for the KK114-maleimide dye and D. Cafiso (University of Virginia, Charlottesville, WV, USA) for comments. G.v.d.B is financed by the Human Frontier Science Program. This work was supported by the US National Institutes of Health (P01 GM072694, to R.J.) and the Deutsche Forschungsgemeinschaft (SFB803). We thank A. Schönle (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany) for the software ImSpector. X-ray diffraction data were collected at beamline X10SA at the Swiss Light Source and we thank the beamline staff for their help during data collection.
Footnotes
Accession codes
Coordinates and structure factors have been deposited in the PDB with accession code 2YOA.
Author Contributions
G.v.d.B, A.H., D.M. and R.J. designed and performed the experiments and wrote the paper. E.I., D.F. and K.K. performed the crystallographic structure determination. H.J.R. and H.G. performed the molecular dynamics simulations. S.M. and U.D. synthesized the peptide. S.W.H., V.M. and C.E. contributed to the microscopy and discussed data. All authors contributed to the manuscript.
Author Information
The authors declare no competing financial interests.
References
- 1.Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizo J, Chen X, Araç D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu. Rev. Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- 4.Jahn R, Fasshauer D. Exocytosis of synaptic vesicles – molecular machines, calcium, and beyond. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sieber JJ, et al. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- 6.Aoyagi K, et al. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 2005;280:17346–17352. doi: 10.1074/jbc.M413307200. [DOI] [PubMed] [Google Scholar]
- 7.van den Bogaart G, et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 2012;479:552–555. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi UB, et al. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araç D, et al. Facile detection of protein–protein interactions by one-dimensional NMR spectroscopy. Biochemistry. 2003;42:2774–2780. doi: 10.1021/bi0272050. [DOI] [PubMed] [Google Scholar]
- 10.Lai AL, Huang H, Herrick DZ, Epp N, Cafiso DS. Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J. Mol. Biol. 2011;504:696–706. doi: 10.1016/j.jmb.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang J, et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 12.Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C2B domain is fundamental for synaptotagmin-1 function. Nat.Struct.Mol. Biol. 2008;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radhakrishnan A, Stein A, Jahn R, Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2009;284:25749–25760. doi: 10.1074/jbc.M109.042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon HT, Kozlov MM, Martens S. Membrane curvature in synaptic vesicle fusion and beyond. Cell. 2010;140:601–605. doi: 10.1016/j.cell.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Li L, et al. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J. Biol. Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- 16.Schiavo G, Gu QM, Prestwich GD, Söllner TH, Rothman JE. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc. Natl. Acad. Sci. USA. 1996;93:13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Bogaart G, et al. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat. Struct. Mol. Biol. 2011;18:805–812. doi: 10.1038/nsmb.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases the Ca2+ affinity of synaptotagmin-1 40-fold. J. Biol. Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Araç D, et al. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 20.Hui E, et al. Mechanism and function of synaptotagmin-mediated membrane apposition. Nat. Struct. Mol. Biol. 2011;18:813–821. doi: 10.1038/nsmb.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrick DZ, Sterbling S, Rasch KA, Hinderliter A, Cafiso DS. Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry. 2006;45:9668–9674. doi: 10.1021/bi060874j. [DOI] [PubMed] [Google Scholar]
- 22.Connell E, et al. Cross-linking of phospholipid membranes is a conserved property of calcium-sensitive synaptotagmins. J. Mol. Biol. 2008;380:42–50. doi: 10.1016/j.jmb.2008.01.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JY, et al. Solution single-vesicle assay reveals PIP(2)-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J. 2012;31:2144–2155. doi: 10.1038/emboj.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo W, Herrick DZ, Cafiso DS. Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry. 2011;50:2633–2641. doi: 10.1021/bi200049c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joung MJ, Mohan SK, Yu C. Molecular level interaction of inositol hexaphosphate with the C2B domain of human synaptotagmin I. Biochemistry. 2012;51:3675–3683. doi: 10.1021/bi300005w. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez I, et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- 27.Vrljic M, et al. Post-translational modifications and lipid binding profile of insect cell-expressed full-length mammalian synaptotagmin 1. Biochemistry. 2011;50:9998–10012. doi: 10.1021/bi200998y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eggeling C, et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- 29.Mueller V, et al. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys. J. 2011;101:1651–1660. doi: 10.1016/j.bpj.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milosevic I, et al. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci. 2005;25:2557–2565. doi: 10.1523/JNEUROSCI.3761-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rickman C, et al. Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J. Biol. Chem. 2004;279:12574–12579. doi: 10.1074/jbc.M310710200. [DOI] [PubMed] [Google Scholar]
- 32.Jensen MH, Morris EJ, Simonsen AC. Domain shapes, coarsening, and random patterns in ternary membranes. Langmuir. 2007;23:8135–8141. doi: 10.1021/la700647v. [DOI] [PubMed] [Google Scholar]
- 33.Murray D, et al. Electrostatic properties of membranes containing acidic lipids and adsorbed basic peptides: theory and experiment. Biophys. J. 1999;77:3176–3188. doi: 10.1016/S0006-3495(99)77148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levental I, et al. Calcium-dependent lateral organization in phosphatidylinositol 4,5-bisphosphate (PIP2)- and cholesterol-containing monolayers. Biochemistry. 2009;48:8241–8248. doi: 10.1021/bi9007879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ausili A, Corbalán-García S, Gómez-Fernández JC, Marsh D. Membrane docking of the C2 domain from protein kinase Cα as seen by polarized ATR-IR. The role of PIP2. Biochim.Biophys.Acta. 2011;1808:684–695. doi: 10.1016/j.bbamem.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 36.Lai CL, Landgraf KE, Voth GA, Falke JJ. Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain: a combined molecular dynamics and experimental study. J. Mol. Biol. 2010;402:301–210. doi: 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen CH, et al. Configuration of PKCalpha-C2 domain bound to mixed SOPC/SOPS lipid monolayers. Biophys. J. 2009;97:2794–2802. doi: 10.1016/j.bpj.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montaville P, et al. The PIP2 binding mode of the C2 domains of rabphilin-3A. Protein Sci. 2008;17:1025–1034. doi: 10.1110/ps.073326608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerrero-Valero M, et al. Structural and mechanistic insights into the association of PKCα-C2 domain to PtdIns(4,5)P2. Proc. Natl. Acad. Sci. USA. 2009;106:6603–6607. doi: 10.1073/pnas.0813099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gómez-Fernández JC. Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 1999;18:6329–6238. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng Y, et al. Crystallographic identification of Ca2+ and Sr2+ coordination sites in synaptotagmin I C2B domain. Protein Sci. 2004;13:2665. doi: 10.1110/ps.04832604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI forcefield: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 43.de Wit H, et al. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 44.Martin TFJ. Role of PI(4,5)P2 in vesicle exocytosis and membrane fusion. Subcell. Biochem. 2012;59:111–130. doi: 10.1007/978-94-007-3015-1_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr.Opin.Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Kolmakov K, et al. Red-emitting rhodamine dyes for fluorescence microscopy and nanoscopy. Chemistry. 2010;16:158–166. doi: 10.1002/chem.200902309. [DOI] [PubMed] [Google Scholar]
- 47.Barnstable CJ, Hofstein R, Akagawa K. A marker of early amacrine cell development in rat retina. Brain Res. 1985;352:286–290. doi: 10.1016/0165-3806(85)90116-6. [DOI] [PubMed] [Google Scholar]
- 48.Heumann R, Kachel V, Thoenen H. Relationship between NGF-mediated volume increase and "priming effect" in fast and slow reacting clones of PC12 pheochromocytoma cells. Role of cAMP. Exp. Cell. Res. 1983;145:179–190. doi: 10.1016/s0014-4827(83)80019-6. [DOI] [PubMed] [Google Scholar]
- 49.Van den Bogaart G, et al. One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol. 2010;17:358–364. doi: 10.1038/nsmb.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mennicke U, Salditt T. Preparation of solid-supported lipid bilayers by spin-coating. Langmuir. 2002;18:8172–8177. [Google Scholar]
- 51.Vicidomini G, et al. Sharper low-power STED nanoscopy by time gating. Nat. Methods. 2011;8:571–573. doi: 10.1038/nmeth.1624. [DOI] [PubMed] [Google Scholar]
- 52.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 53.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr. D. Biol. Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 55.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 56.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 57.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.