

Abstract
The primary cilium is required during early embryo patterning, epithelial tubulogenesis, and growth factor-dependent signal transduction. The requirement for primary cilia during renal epithelial-mesenchymal tissue interactions that give rise to nephrons is undefined. Here, we used Cre-mediated recombination to generate mice with Kif3a deficiency targeted to the ureteric and/or metanephric mesenchyme cell lineages in the embryonic kidney. Gradual loss of primary cilia in either lineage leads to a phenotype of reduced nephron number. Remarkably, in addition to cyst formation, loss of primary cilia in the ureteric epithelial cell leads to decreased expression of Wnt11 and Ret and reduced ureteric branching. Constitutive expression of GLI3 repressor (Gli3Δ699/+) rescues these abnormalities. In embryonic metanephric mesenchyme cells, Kif3a deficiency limits survival of nephrogenic progenitor cells and expression of genes required for nephron formation. Together, our data demonstrate that Kif3a controls nephron number via distinct cell lineage-specific mechanisms.
Introduction
Primary cilia are microtubule-based organelles that function as signaling centers during development and cell differentiation [1]. The primary cilium arises in a quiescent cell from the basal body as a microtubule-based plasma membrane-invested cytoskeletal structure termed the axoneme. Cilia assembly and maintenance and growth of the axoneme is mediated by a kinesin motor protein-based transport process termed intraflagellar transport (IFT), by which particles are transported in a bidirectional manner along the axoneme [2]. Deficiency of KIF3A, a component of the kinesin II motor complex, disables anterograde IFT, and causes both failure of formation and maintenance of the primary cilium [3]. A critical role for the primary cilium during embryogenesis was initially demonstrated by the finding that mice with Kif3a deficiency lack nodal cilia and exhibit defects in left-right asymmetry [3]. Many human congenital malformation syndromes are caused by mutations in proteins that are localized to cilia and ciliary basal bodies [1]. Some of the mutated proteins are downstream effectors of the Hedgehog (Hh), WNT and FGF signaling pathways. Hh ligands signal by binding the cell surface protein Patched (PTC), which functions as a constitutive inhibitor of Smoothened (SMO). In the absence of Hh ligand, inactive SMO promotes the processing of full length GLI3 to a C-terminally truncated transcriptional repressor, GLI3 repressor (GLI3R). Hh activates SMO, leading to the blockage of GLI3 processing and the nuclear translocation of full-length GLI proteins to induce transcription. Several lines of evidence implicate the primary cilium in mammalian Hh signaling. First, disruption of Hh signaling generates a phenotype very similar to that described in embryos with deficiency of IFT proteins [4]. Second, PTC, SMO, and GLI are localized to the primary cilium [5]–[7]. Third, IFT proteins act downstream of PTC1 and SMO and upstream of GLI proteins [4], [8]. Cilia defects alter the ratio of GLI activator to GLI3R resulting in aberrant Hh signaling [1]. The primary cilium is also implicated in WNT signaling since NPHP2 (inversin), NPHP3, and GLIS2, each of which promotes noncanonical WNT signaling, are localized to the cilium. Inactivation of any of these noncanonical WNT effectors increases canonical WNT activity [9], [10]. In contrast to Hh and WNT signaling, the role of the primary cilium in regulating FGF signaling is largely unknown. FGFs have been shown to regulate cilia length [11] but a role for the cilium in regulating FGF signaling has not been demonstrated previously. However, the localization of FGF receptors to cilia in murine airway cells suggests a possible role for the cilium in regulating FGF signaling [12].
The discovery that proteins mutated in polycystic kidney diseases are localized to the primary cilium identified the primary cilium as critical to renal epithelial cell differentiation [13]. In direct support of this cilia-dependent function, kidney-specific inactivation of Kif3a in the ureteric epithelial cell lineage inhibits ciliogenesis and induces epithelial cysts [14]. The observation that nephron formation may be impaired in mice with deficiency of NPHP2 (inversin), a cilia-localized protein and polycystic kidney disease gene [9] suggests that primary cilia may function during stages of renal development that control nephron formation and which precede epithelial differentiation.
Nephron formation is dependent on inductive mesenchymal-epithelial tissue interactions between the ureteric bud (an epithelial tubule) and the metanephric blastema (a mesenchymal tissue). Metanephric mesenchyme cells adjacent to ureteric branch tips are induced to form nephrogenic precursors that constitute the mature nephron (glomerulus, proximal tubule, loop of Henle, and distal tubule), a process that has been termed nephrogenesis. In turn, the ureteric bud and its branches are stimulated to undergo successive branching events in response to signals by adjacent mesenchyme cells, resulting in formation of the collecting ducts, calyces and pelvis, a process termed renal branching morphogenesis [15]. Investigation of the molecular mechanisms that control nephrogenesis and branching morphogenesis has elucidated critical roles for signaling by Hh, WNT and FGF proteins. Sonic Hh (Shh) controls inductive tissue interactions during murine kidney development by inhibiting formation of GLI3 repressor [16]. During branching morphogenesis, GLI3R plays a critical role in distal ureteric branch tips by promoting the expression of Ret and Wnt11, both of which are required for ureteric branching [17]. Canonical WNT signaling is required for renal branching morphogenesis [18] and formation of nephrogenic precursors in response to WNT9b and WNT4 [19]–[21]. Nephron formation is also dependent on expression of FGF8 by metanephric mesenchyme cells. Deficiency of Fgf8 abrogates expression of Wnt4 and limits nephron formation to stages prior to the formation of the glomerulus [22].
Here, we tested our hypothesis that the primary cilium is required during growth factor-mediated renal mesenchymal-epithelial interactions. We investigated our hypothesis by generating mouse strains with deficiency in Kif3a in all kidney cells or in the ureteric or metanephric mesenchyme cell lineage. Our results demonstrate that Kif3a deficiency and subsequent loss of primary cilia is accompanied by a decrease in the number of nephrons. Analysis of mice with lineage-specific Kif3a deficiency showed that Kif3a performs distinct functions in ureteric and metanephric mesenchyme cells. In ureteric cells, Kif3a deficiency disrupts ureteric branching and expression of Ret and Wnt11, which act in concert to promote ureteric branching. Remarkably, constitutive expression of GLI3R in Kif3a-deficient ureteric cells rescues each of these abnormalities. Analysis of mice with Kif3a deficiency in metanephric mesenchyme cells revealed two further mechanisms by which Kif3a controls nephron number. First, Kif3a-deficient cells exhibit reduced survival, negatively impacting the mass of mesenchyme cells that can contribute to nephrons. Second, expression of FGF8 and its downstream effectors by Kif3a-deficient cells is markedly reduced. Yet, expression of Hh signaling effectors is unaffected. Together, these results demonstrate a fundamental role for Kif3a and the primary cilium in controlling nephron number during murine kidney development.
Results
Kif3a Deficiency Decreases Nephron Formation in the Murine Kidney
We initiated investigation of primary cilium function during renal morphogenesis by examining the cellular distribution of primary cilia in distinct lineages that give rise to the kidney. Acetylated alpha-tubulin (α-AcT) is expressed in the ciliary axoneme and the basal body from which the axoneme emerges; expression on the apical cell surface marks the primary cilium. Examination of α-AcT expression at E11.5, the stage at which the ureteric bud invades the metanephric blastema, demonstrated expression in virtually all ureteric and mesenchyme cells (Figure S1A’ and S1A’’). By E13.5 and E15.5, α-AcT could be clearly localized to the apical surface of ureteric cells (Figure S1B’, S1B’’, S1C’, and S1C”), as well as the mesenchyme-derived structures, condensing mesenchyme, vesicles and S-shaped bodies (Figure S1D’ and S1D”), that precede formation of the mature nephron. Together, these data indicate that primary cilia are formed during early stages of ureteric branching and nephron formation.
We investigated the functional contribution of the primary cilium to renal development by generating mice with loss of primary cilia in both ureteric and metanephric mesenchyme cells. KIF3A is a component of the microtubule heterotrimeric kinesin II motor complex, which mediates anterograde IFT. Kif3a deficiency disables anterograde IFT and leads to failure in formation and maintenance of cilia [3]. Since germline deficiency in Kif3a is embryonic lethal prior to the onset of kidney development [3], we used a conditional Kif3aloxP allele [3] and a Tamoxifen-inducible Cre mouse strain [23] to generate Cre-ER™;Kif3aloxp/− (termed Kif3a™ ) mice. Administration of Tamoxifen (3mg/40g body weight) prior to E10.5 induced embryonic demise prior to kidney formation. In contrast, embryos of pregnant dams treated with Tamoxifen at E10.5 survived until shortly after E13.5 (Figure 1A), thus providing a means to analyze primary cilium function.
Figure 1. Loss of primary cilia and decreased nephron number in mice with Tamoxifen-induced Kif3a deficiency.

(A) Chart showing embryonic stage at which Tamoxifen was injected (E10.5) and at which kidneys were retrieved (E13.5) for analysis (red line). (B, B’) KIF3A co-localizes with α-AcT in both ureteric bud and metanephric mesenchyme cells in WT kidney at E13.5. Insert box shows high-resolution image of KIF3A located in a primary cilium in a metanephric mesenchyme cell. (C, C’) Expression of Kif3a is largely undetectable 72 hours after Tamoxifen administration to pregnant Kif3aloxP/loxP mice which had been crossed to Cre-ER™;Kif3a+/− mice. (D, E) The number of primary cilia (green) is markedly decreased in both SIX2-positive cells (nephrogenic precursors) and in ureteric cells Kif3a™ kidney (E, asterisk) compared to WT kidney (D). (F, G) Imaging of NCAM-positive nephrogenic precursor structures (arrows). The number of precursors is decreased in Kif3a™ mice (G) compared to WT mice (F). (H) Quantitation of the number of cells with a primary cilium. Tamoxifen administration to Kif3a™ embryos decreases cilia number by approximately 50% in both ureteric and metanephric mesenchyme cells (***, P<0.001). (I) Quantitation of the number of NCAM-positive nephrogenic precursor structures in tissue sections reveals a significant decrease in Kif3a™ mice compared with WT mice (**, P<0.01). WT, wild type; Scale bar: 25 micrometer.
Loss of KIF3A in Tamoxifen-treated mice was confirmed using anti-KIF3A antibody (Figure 1B, 1C’). In control mice (WT mice injected with Tamoxifen at E10.5), KIF3A and α-AcT co-localized in ureteric and metanephric mesenchyme cells (Figure 1B, B’). In ureteric branches, characterized by a lumen, co-localization of KIF3A and α-AcT was restricted to the apical cell surface (Figure 1B, arrow located in UB apical domain). In contrast, expression of KIF3A was markedly diminished in Kif3a™ mice (Figure 1C’). Further, co-localization of KIF3A with α-AT in the apical domain of epithelial cells was not detected (Figure 1C). We assessed the impact of KIF3A deficiency on primary cilia by counting the fraction of ureteric cells and nephrogenic metanephric mesenchyme (SIX2-positive) cells with primary cilia by imaging these respective cells in three randomly selected optical fields in a 5 micrometer sagittal tissue section generated from the mid-point of each kidney (n = 6 mice/group) (Figure 1D and 1E). The number of cells with primary cilia was decreased by 54% in each of these cell populations in Kif3a-deficient mice compared with controls (Figure 1H). Next, we determined the effect of KIF3A and primary cilium loss on kidney development. Ureteric branches and nephrogenic precursors were identified in tissue sections with cytokeratin (CK) and NCAM, respectively (Figure 1F and 1G). Both ureteric branches and nephron precursors formed normally in mutant mice. The number of NCAM-positive nephrogenic precursors was determined for each kidney in five tissue sections - a mid-sagittal section and two sections generated 40 micrometer and 80 micrometer in both directions from the mid-sagittal section, resulting in a total of five sections. Quantitation of NCAM-positive nephrogenic precursors demonstrated a 34% decrease in mutant mice (no. NCAM-positive structures/section, WT versus Kif3a™: 15.2±2.96 versus 10±1.5, p = 0.001, n = 4 mice/group) (Figure 1I). These data demonstrate that Kif3a controls the number of nephrons formed during renal embryogenesis.
Kif3a Functions in a Cell-lineage Specific Manner to Control Nephron Number
The short-term viability of embryos in Tamoxifen-treated pregnant mice limited the availability to investigate mechanisms underlying the requirement for Kif3a during nephron formation. To address this limitation, we generated mice with loss of Kif3a targeted specifically to the ureteric (Kif3a−/−UB mice) or metanephric mesenchyme (Kif3a−/−MM mice) lineages, using Hoxb7-CreEGFP [24] and Rarb2-Cre [25] mouse strains, respectively. Kif3a−/−UB and Kif3a−/−MM mouse embryos were generated in the proportion predicted by Mendelian segregation and survived to birth. The efficiency and specificity of Kif3a deletion was demonstrated by analyzing Kif3a mRNA expression in ureteric bud and metanephric mesenchyme tissue fractions isolated at E11.5 using quantitative PCR. Kif3a mRNA was reduced by over 95% in ureteric and metanephric mesenchyme cells in Kif3a−/−UB and Kif3a−/−MM mice, respectively, but was not significantly decreased in cells that were not targeted by the respective Cre alleles (Figure S2). KIF3A protein expression was examined by immunofluorescence in embryonic kidney tissue. Comparison of anti-KIF3A antibody-generated signals on the apical surface of control and mutant cells revealed specific identification of KIF3A. In control embryos, KIF3A (green color) co-localized with α-AcT (red color) on the apical surface of ureteric cells (Figure 2A, UB: arrow) and in mesenchyme cells (Figure 2A, arrow in box). In Kif3a−/−UB mice, KIF3A was lost in ureteric cells (Figure 2A’, box inset) but was expressed in metanephric mesenchyme cells (Figure 2A’, upper arrow). In Kif3a−/−MM mice, KIF3A was lost in metanephric mesenchyme cells (box inset, Figure 2A”) but was expressed in ureteric cells (Figure 2A”, arrow in UB).
Figure 2. Gradual loss of primary cilia after induction of Kif3a deficiency in ureteric or metanephric mesenchyme cell lineages.

(A, A’, A’’) KIF3A expression in E13.5 kidney tissue. (A) KIF3A co-localizes with α-AcT (arrow) on the apical surface of ureteric cells and in metanephric mesenchyme cells (box) in WT mice. (A’) In Kif3a−/−UB mice, KIF3A expression (arrow) is markedly decreased in ureteric cells (box) but is comparable to WT in metanephric mesenchyme cells. (A”) In Kif3a−/−MM mice, KIF3A expression (arrow) is lost in metanephric mesenchyme cells (box) but is retained in ureteric cells. (B, B’, B’’) Primary cilia (arrow) in ureteric cells at E15.5. The number of cilia is decreased in Kif3a−/−UB mice (B’) but is comparable to WT (B) in Kif3a−/−MM mice (B”). (C, C’, C’’) Primary cilia (arrow) in proximal tubules at E15.5. The number of cilia is decreased in Kif3a−/−MM mice (C”) but is unaffected in Kif3a−/−UB mice (C’) compared to WT (C). (D, D’, D”) Primary cilia in collecting ducts at P0. Cilia (red) are absent from the collecting duct lumen and the cell body (asterisk) (D’) but are unaffected in Kif3a−/−MM mice. (E, E’, E’’) Primary cilia (arrow) in proximal tubules at P0. Cilia (arrows) are absent from the tubule lumen in Kif3a−/−MM mice (E’’). Expression of α-AcT (asterick) is visible within the body of some proximal tubule cells. Cilia are unaffected in the proximal tubule of Kif3a−/−UB mice (E’). (F–G”) SEM of collecting ducts in WT (F, G), Kif3a−/−UB (F’, G’), and Kif3a−/−MM (F”, G”) mice. In Kif3a−/−UB mice, cilia (arrow) are decreased in number at E15.5 (F’, box) and are absent at P0 (G’, box) compared to WT (F, G, boxes). Cilia are unaffected in Kif3a−/−MM mice (F”, G”). (H–I”) SEM of proximal tubules in WT (H, I), Kif3a−/−UB (H’, I’), and Kif3a−/−MM (H”, I”) mice. In Kif3a−/−MM mice, cilia (arrow) are shorter at E15.5 (H”, box) and are absent at P0 (I”, box) but are unaffected in Kif3a−/−UB mice (H’, I’) versus WT controls (H, I, boxes). MM, metanephric mesenchyme; UB, ureteric bud; WT, wild type. Scale bars: A–E’’, 25 micrometer, F–I’’, 5 micrometer.
Next, we determined the effect of KIF3A deficiency on cilia. Cilia number was quantitated in sagittal tissue sections generated from the mid-point of the kidney by counting the number of cells, identified by DAPI, associated with a primary cilium, identified with anti-α-AcT. Deletion of Kif3a in ureteric or metanephric mesenchyme cells resulted in a gradual loss of primary cilia during embryogenesis. Kif3a−/−UB mice demonstrated 64% fewer ureteric-derived cells with primary cilia at E13.5 (Figure 2A’ versus 2A; % cells with primary cilia, Kif3a−/−UB versus WT: 33±3.46 versus 97±1.23, n = 5 mice/group) and 59% fewer cells with primary cilia at E15.5 (Figure 2B’ versus 2B; % cells with primary cilia, Kif3a−/−UB versus WT: 38±4.37 versus 97±1.07, n = 5 mice/group). By P0, primary cilia could be detected in collecting ducts only rarely (Figure 2D’ versus 2D). Furthermore, scanning electron microscopy (SEM) of collecting duct cells revealed decreased cilia number at E15.5 and complete absence of cilia at P0 (Figure 2F’ versus 2F, and 2G’ versus 2G, boxes). In Kif3a−/−MM kidneys, the number of metanephric mesenchyme cells with primary cilia was decreased by 70% at E13.5 (Figure 2A’’ versus 2A, asterisk; % cells with primary cilia, Kif3a−/−MM versus WT: 29±5.67 versus 99±1.14, n = 5 mice/group) and by 67% in proximal tubule cells at E15.5 (Figures 2C’’ versus 2C; % cells with primary cilia, Kif3a−/−MM versus WT: 32±3.09 versus 99±1.12, n = 5 mice/group). By P0, cilia were virtually absent from proximal tubule cells (Figure 2E” versus 2E). These results were confirmed by SEM analysis of proximal tubules, identified by apical brush border villae, at E15.5 and P0 (Figure 2H’’ versus 2H and 2I’’ versus 2I, boxes). Taken together, these results indicate that deletion of Kif3a results in a cell-specific gradual loss of primary ciliaduring embryonic kidney development with complete absence of cilia by P0.
Kif3a+/−UB and Kif3a +/−MM heterozygote mice were viable and characterized by normal renal development (data not shown). In contrast, both Kif3a−/−UB and Kif3a−/−MM mice exhibited a remarkably similar histologic phenotype characterized by epithelial cysts (Figure S3H, I), a reduction in the number of NCAM-positive nephrogenic precursor structures (Figure 3A–3C), and glomeruli, which are characterized by expression of WT1 in podocytes (Figure 3D–I). As in previous analyses, quantitation of NCAM-positive and WT1-positive structures was performed in sagittal tissue sections generated starting at the mid-point of a kidney. The number of NCAM-positive structures in Kif3a−/−UB and Kif3a−/−MM mice was reduced by 25% and 32%, respectively, at E13.5 (Figure 3J, no. NCAM-positive structures/section, WT versus Kif3a−/−UB: 16.5±3.05 versus 12.33±2.36, p = 0.003; WT versus Kif3a−/−MM : 16.5±3.05 versus 11.16±2,47, p = 0.001, n = 4 mice/group). The number of WT1-positive structures in Kif3a−/−UB and Kif3a−/−MM mice was reduced by 24% and 34%, respectively, at E15.5 (Figures 3D–F and Figure 3K, no. WT1-positive structures/section: WT versus Kif3a−/−UB: 25±3.43 versus 18.8±2.71, p = 0.002; WT versus Kif3a−/−MM: 25±3.43 versus 15.71±1.25, p = 0.001, n = 4 mice/group), and by 25% and 35%, respectively, at P0 (Figure 3G–I and Figure 3K, no. WT1-positive structures/section: WT versus Kif3a−/−UB: 34.25±5.32 versus 25.12±3.68, p = 0.002; WT versus Kif3a−/−MM: 34.25±5.32 versus 21.6±5.69, p = 0.0001, n = 4 mice/group). Nephron number was also quantitated by counting the number of glomeruli, identified by their characteristic morphology in sagittal tissue sections generated in both directions from the mid-point of a kidney at 36 micrometer intervals to the outer limit of the organ [17]. At E15.5, this analysis demonstrated a reduction in glomerular number of 24% in Kif3a−/−UB kidneys and 33% in Kif3a−/−MM kidneys (Figure S3B and S3C versus S3A and Figure S3J; no. glomeruli/kidney, WT versus Kif3a−/−UB: 78.14±7.81 versus 62.85±4.29, p = 0.002; WT versus Kif3a−/−MM : 78.14±7.81 versus 54.85±7.20, p = 8.75E–05, n = 7 mice/group). Glomerular number was similarly reduced at E18.5 (Figure S3D–F, Figure S3J; no. glomeruli/kidney, WT versus Kif3a−/−UB: 1153±151 versus 887±74, p = 0.01; WT versus Kif3a−/−MM: 1153±151 versus 827±169, p = 0.003, n = 6 mice/group) and at P0 (Figure S3G–I, Figure S3J; no. glomeruli/kidney, WT versus Kif3a−/−UB: 2025±211 versus 1513±290, p = 0.01; WT versus Kif3a−/−MM: 2025±211 versus 1285±255, p = 0.001, n = mice/group). Cyst formation in glomeruli and tubules (Figure 3I and Figure S3H and S3I, arrowheads) was consistent with published analysis of KIF3A function in the kidney [26]. Together, these results demonstrate that Kif3a deficiency in either the ureteric or metanephric mesenchyme cell lineage causes nephron deficiency.
Figure 3. Nephron number is decreased in both Kif3a−/−UB and Kif3a−/−MM kidneys.

(A–C) Nephrogenic precursor structures, marked by expression of NCAM (red color, marked by arrows), in Kif3a−/−UB mice (B) and Kif3a−/−MM mice (C) versus control mice (A). (D–F) Identification of glomeruli via expression of WT1 (green color, marked by arrow). Collecting ducts are identified by expression of cytokeratin (red color). (G–I) Identification of WTI-positive structures (red color, marked by arrow) at P0. Proximal tubules are identified by expression of LTL (green color, marked by arrowhead). (J, K) Quantification of the number of NCAM–positive and WT1-positive structures demonstrates a decreased number in Kif3a−/−UB and Kif3a−/−MM mice. (***, P<0.001; **, P<0.01; *, P<0.05). Scale bars: 25 micrometer.
Kif3a Controls Branching Morphogenesis in a GLI3R-dependent Manner
Formation of nephrons is initiated by signals released from ureteric bud-derived cells adjacent to mesenchymal nephrogenic progenitor cells [15]. Since nephron number is directly related to the number of ureteric branches elaborated during branching morphogenesis, we analyzed the effect of Kif3a deficiency on formation of ureteric branches by quantitating the number of ureteric bud tips, marked by expression of either GFP or Cytokeratin. In Kif3a−/−UB mice, formation of the initial ‘T’ shape ureteric branch at E11.5 and the first two branch generations at E12.5 was normal compared with WT (Figure 4A, 4A’, 4B and 4B’). However, at E13.0 and E14.0, the number of ureteric bud tips was significantly reduced in Kif3a−/−UB mice (Figures 4C’ versus 4C, 4D’ versus 4D and Figure 4R; no. UB tips/kidney, WT versus Kif3a−/−UB at E13.0∶19.2±2.28 versus 16±1.41, p = 0.03; WT versus Kif3a−/−UB at E14.0∶39±2.59 versus 34±1.15, p = 0.03,n = 6 mice/group). Since ureteric branching is dependent, in part, on ureteric tip cell proliferation, we next identified ureteric tip cells undergoing mitosis using antibody specific for phospho-histone H3. Consistent with the decrease in ureteric branching, mitotic ureteric tip cell was decreased in Kif3a−/−UB mice (Figure 4K, 4L, and 4S; no. mitotic tip cells/tip, WT versus Kif3a−/−UB: 7.72±1.81,versus 3.23±0.54, p = 0.001, n = 5 mice/group). These results are consistent with decreased nephron number observed in Kif3a−/−UB mice (Figure 3B versus 3A, and Figure 3J).
Figure 4. Kif3a controls renal branching morphogenesis in a GLI3R-dependent manner.

(A–C’’) Cytokeratin immunofluorescence demonstrating ureteric branching at E11.5 and E12.5. The branch pattern is similar among WT (A,B), Kif3a−/−UB (A’,B’) and Kif3a−/−UB;Kif3aΔ699/+(constitutive expression of Gli3Δ699/+ in Kif3a−/−UB background, A’’, B’’) kidneys. (C–D’’) Ureteric branching in WT, Kif3a−/−UB, and Kif3a−/−UB;Gli3Δ699/+mice at E13.0 and E14.0. In Hoxb7-GFP-Cre mice, GFP expression in the kidney is restricted to the ureteric cell lineage. Ureteric branches expressing GFP are visualized in whole mount preparations of kidney explants generated from E13.0 (C, C’ and C’’) and E14.0 (D, D’ and D’’). Imaging suggests that branch number in Kif3a−/−UB mice (C’, D’) is less than that in WT (C, D) and Kif3a−/−UB;Gli3Δ699/+mice (C’’, D’’). (E–J) Wnt11 and Ret expression. In WT kidney, Wnt11 (E) and Ret (H) are strongly expressed in ureteric tip cells (arrows). Expression of Wnt11 (F) and Ret (I) is markedly decreased in Kif3a−/−UB mice. In Kif3a−/−UB;Gli3Δ699/+ mice, Wnt11 (G) and Ret (J) expression are rescued to levels similar to that observed in WT mice. (K–M) Phosho-Histone H3 (pHH3) is stained in mitotic cell of ureteric tip (red color) that is marked by cytokeratin (green color) at E13.5. (N–P) NCAM staining showing nephrogenic structures at E13.5. Decreased formation of NCAM-positive structures in Kif3a−/−UB mice (O versus N) is rescued in Kif3a−/−UB;Gli3Δ699/+ (P versus O) mice (Q) Schematic showing the Wnt11/Gdnf/Ret gene network that promotes ureteric branching morphogenesis. (R) Quantitation of ureteric branch tip number reveals a significant decrease in Kif3a−/−UB mice compared to WT at E13.0 and E14.0 but increased branching in Kif3a−/−UB;Gli3Δ699/+ mice compared to Kif3a−/−UB mice. (S) Quantitation of mitotic tip cells in K, L and M. pHH3 marked cells are decreased in Kif3a−/−UB mice (L) compared to WT (K). pHH3-positive cells is remarkably increased in in Kif3a−/−UB;Gli3Δ699/+ mice compared to Kif3a−/−UB mice (M versus L). (T) Quantitation of NCAM-positive structures in N, O and P reveals a significant decrease in Kif3a−/−UB mice compared to WT and Kif3a−/−UB;Gli3Δ699/+ mice. (*, P<0.05, **, P<0.01, *** P<0.001). Scale bars: 50 micrometer.
Ureteric branching is controlled by a Wnt11/Ret/Gdnf signaling axis [27], [28]. GDNF, an extracellular ligand expressed by metanephric mesenchyme cells, binds to RET on the surface of ureteric tip cells. In turn, GDNF/RET signaling controls ureteric tip cell expression of Wnt11, a positive regulator of ureteric branching (Figure 4Q). In Kif3a−/−UB mice, overall expression of Wnt11 and Ret was markedly reduced (Figures 4F’ and 4I’, inserts). Higher resolution imaging of ureteric tips revealed weak expression of Wnt11 and Ret in ureteric cells (Figure 4F and 4I, arrowheads).
Previously, we demonstrated that expression of Wnt11 and Ret in ureteric tip cells is controlled by GLI3R [17]. Suppression of GLI3R formation in Patched1 deficient mice decreases Wnt11 and Ret expression and lowers ureteric branch and nephron number. Obligate expression of GLI3R via the Gli3Δ699 allele, which expresses GLI3R in a constitutive manner [29], rescues these abnormalities [17]. Since defects in the primary cilium alter the ratio of GLI3 activator to GLI3R in favor of GLI3 activator [1], we hypothesized that GLI3R deficiency in Kif3a−/−UB mice could cause reduced branching morphogenesis. Thus, we determined whether constitutive expression of GLI3R in Kif3a−/−UB mice rescues ureteric branching. Analysis of Kif3a−/−UB;Gli3Δ699/+ mice at E13.0 and E14.0 revealed that the number of ureteric tips was significantly increased compared to Kif3a−/−UB mice (Figure 4C’’, 4D’’ and 4R; no. UB tips/kidney, Kif3a−/−UB;Gli3Δ699/+ versus Kif3a−/−UB, at E13.0∶19.2±2.28 versus 15.33±0.82, p = 0.01; at E14.0∶40.0±2.33 versus 33.33±2.5, p = 0.001, n = 6 mice/group). Further, expression of Wnt11 and Ret was markedly increased compared to that observed in Kif3a−/−UB mice (Figure 4G and 4J versus Figure 4F and 4I). Concomitant with a rescue of ureteric branching, the number of NCAM-positive nephrogenic precursors in Kif3a−/−UB;Gli3Δ699/+ mice was comparable to that observed in WT mice (Figure 4P versus 4O, and 4T; NCAM-positive structures/section - Kif3a−/−UB;Gli3Δ699/+ versus WT: 15.5±3.40 versus 14.7±4.24, P = 0.27, n = 4 mice/group). Taken together, these results demonstrate that Kif3a regulates branching morphogenesis in a GLI3R-dependent manner.
Kif3a Controls Metanephric Mesenchyme Cell Survival
Decreased nephron number in Kif3a−/−MM mice (Figure 3J, 3K and Figure S3J) demonstrated that Kif3a functions in a cell autonomous manner within metanephric mesenchyme. We investigated mechanisms underlying Kif3a-dependent functions by first analyzing the effect of Kif3a deficiency on metanephric mesenchyme cells that are progressively committed to a nephrogenic fate. Kif3aloxP/loxP [3] mice were intercrossed with R26RLacZ/LacZ [30] to generate Kif3aloxP/loxP;R26RLacz/+ mice, which were used as a reporter for Rarb2-Cre activity and to label Kif3a-deficient cells [31]. Analysis of lacZ expression at E11.0 demonstrated that the metanephric blastema was smaller in Kif3a−/−MM mice compared to controls (Figure 5A and 5B, area of lacZ-positive tissue (µm2): control –53806.86±662; Rarb2-Cre;R26R;Kif3aloxP/− –33486±2563; p = 3.27×10−5, n = 6 mice/group). Consistent with this finding, the number of SIX2-positive nephrogenic progenitor cells was decreased in Kif3a−/−MM mice (Figure S4D versus S4E, S3F; no. SIX2-positive cells/tissue section, WT versus Kif3a−/−MM : 48.5±2.65 versus 43.25±3.5, p = 0.008, n = 6 mice/group). This finding is consistent with the decreased number of NCAM-positive structures in Kif3a−/−MM mice (Figure 3A, 3C and 3J). Next, we determined whether these Kif3a-dependent effects on nephron number were associated with altered cell proliferation and/or apoptosis. We analyzed apoptosis at the stages when the process of nephron formation is well established (E12.5). The number of apoptotic nuclei, identified by the TUNEL assay, was significantly increased in metanephric mesenchyme cells in Kif3a−/−MM kidney tissue (Figure 5F versus 5E and 5H; no. TUNEL-positive cells/tissue section, WT versus Kif3a−/−MM: 3.25±0.96 versus 7.2±1.29, p = 0.0006, n = 4 mice/group). In contrast, the proportion of proliferating (BrdU-positive) cap mesenchyme cells did not differ in mutant mice (Figure S4A, S4B and S4C) (% BrdU-positive cap mesenchyme cells, WT versus Kif3a−/−MM: 89.6±2.88 versus 88.2±3.56, p = 0.51, n = 5 mice/group ). Previously, we demonstrated that deficiency of nephrogenic progenitors during early stages of murine kidney development can limit the number of nephrogenic cells available to participate in more advanced stages of nephron formation [31]. We investigated this possibility in Kif3a−/−MM mice using the R26R allele and examining cortical sections for lacZ expression in nephrogenic structures. In control mice, lacZ-marked cells comprised 95% of the total number of cells (4547 of 4613 cells) resident within nephrogenic structures. In contrast, in mutant mice, lacZ-marked cells constituted only 82% of the total number of cells in nephrogenic structures (2413 of 3251 cells) (p = 0.02; n = 6 cortical section counted in each of 6 mice/group, Figure 5C, 5D and 5G). These data suggest that depletion of Kif3a deficient nephrogenic progenitors may provide a selective advantage for Rarb2-Cre-negative cells with WT levels of KIF3A to participate in nephron formation. Together, these analyses of cell fate and cell survival indicate that Kif3a deficiency interferes with survival of nephrogenic progenitor cells and decreases the size of the cellular pool available to participate in nephron formation.
Figure 5. Kif3a controls survival of nephrogenic precursor cells.

(A–D) LacZ expression in kidney tissue. (A, B) LacZ-marked metanephric blastema at E11.0 is demarcated by red dotted lines. The position of the ureteric bud is marked by white dotted lines. The metanephric blastema is smaller in Kif3a−/−MM mice (B) compared to control (A). (C, D) Incorporation of pink (lacZ-negative; Kif3a+/+ cells) into nephrogenic structures (arrows) is greater in Kif3a−/−MM mice (D) compared to controls (C). (E, F) TUNEL assay identify the apoptotic cells (arrows) in the metanephric mesenchymal cells at E12.5. The number of TUNEL-positive cells is greater in Kif3a−/−MM mice (F, arrows) compared to control (E, arrows). (G, H) Quantitation of lacZ-positive and –negative cells (G) and TUNEL-positive cells (H). Kif3a−/−MM mice exhibit a significantly larger contribution of lacZ-negative cells to nephrogenic structures (D), and increased apoptosis in metanephric mesenchymal cells (F). (***, P<0.001; **, P<0.01; *, P<0.05). Scale bars: 50 micrometer.
Kif3a Deficiency Leads to Fgf8 Deficiency in Nephrogenic Mesenchyme
Nephron formation is dependent on a signaling axis in which Fgf8 functions upstream of Wnt4 and Lim1 (Figure 6A). In the absence of Fgf8, neither Wnt4 nor Lim1 is expressed and nephron formation largely fails to progress to the stage of the S-shaped body [22]. Since FGF receptors have been localized to the cilium in nonrenal tissues [12], we investigated the possibility that Kif3a deficiency and loss of primary cilia interfere with FGF8-mediated signaling during nephron formation. In support of this possibility, analysis of kidney tissue derived from Kif3a−/−MM mice at P0 revealed a 28% reduction in the number of S-shaped bodies (Figure 6H and 6I, Kif3a−/−MM versus control – no. S-shape bodies/kidney: 6.16±0.5 versus 3.83±1.73; p = 0.0003, n = 5 mice/group). While expression of Fgf8 mRNA was only mildly decreased at E13.5 (Figure S5), by E15.5, Fgf8 expression was almost undetectable (Figure 6C versus 6B, insert and Figure 7G, lane 3). Further, expression of Wnt4 and Lim1 was markedly reduced with only a focal pattern of expression within some nephrogenic structures (Figure 6E versus 6D and 6G versus 6F, insert). Together, these results indicate that Kif3a deficiency decreases Fgf8-dependent signaling during renal development.
Figure 6. Kif3a controls expression of a Fgf8-mediated signaling pathway during nephron formation.

A. Schematic of Fgf8/Wnt4/Lim1 signaling pathway required during nephron formation. (B–G) In situ RNA hybridization in kidney tissue sections generated from E15.5 embryos. Fgf8 (B, C), Wnt4 (D, E) and Lim1 (F, G) are expressed in nephrogenic progenitors in WT mice. Fgf8 expression is barely detectable in Kif3a−/−MM mice (C, C’). Expression of Wnt4 (E) is markedly decreased as is expression of Lim1 (G). (H, I) Histological sections from P0 kidney tissue. There are fewer S-shaped bodies (arrows) in Kif3a−/−MM mice (I) than in control mice (H). Scale bars: 25 micrometer.
Figure 7. Kif3a acts upstream of Fgf8.

(A–D) Analysis of primary cilia (arrows) in metanephric mesenchyme cells derived from WT and Kif3a−/−MM metanephroi dissected free of ureteric bud. Cilia are identified by expression of α-AcT. (A’–D’) Higher magnification of images in A–D, respectively. WT and Kif3a-deficient cells were transfected with a plasmid encoding Kif3a fused to GFP. Cilia in Kif3a-deficient mesenchyme cells (B, B’) are vestigial in comparison to cilia on WT cells (A, A’). Transfection with Kif3a results in localization of GFP to the cilium in each treatment group (C, D) and lengthening of the cilium in Kif3a−/−MM cells (D versus B). (E) Quantitation of cilia length in untransfected and transfected WT and Kif3a−/−MM cells. Expression of Kif3a partially rescues cilia length in Kif3a-deficient cells. (F) Quantitation of Ptc1 and Gli1 mRNA expression, measured by quantitative RT-PCR in untransfected and transfected WT and Kif3a−/−MM cells. Ptc1 and Gli1 mRNA expression is not affected by Kif3a deficiency or transfection with Kif3a. (G) Expression of Fgf8, Ptc1, and Gli1 mRNA, measured by real time RT-PCR using RNA isolated from kidney explants and from untransfected and transfected cultured metanephric mesenchyme cells. (H) Quantitation of Fgf8 mRNA levels measured by quantitiative RT-PCR as in panel G. MM, metanephric mesenchyme; UB, ureteric bud; WT, wild-type. (***, P<0.001; **, P<0.01; *, P<0.05), Scale bars: (A–D) 25 micrometer, (I–L) 50 micrometer.
Kif3a Controls Expression of Fgf8 by Metanephric Mesenchyme Cells
Previous studies have demonstrated that Fgf8 signalling regulates cilia length via the IFT pathway in diverse epithelia [11]. Yet, a role for Kif3a in controlling Fgf8 expression has not been previously elucidated. Our results demonstrating that Kif3a deficiency precedes Fgf8 deficiency in metanephric mesenchyme suggested the possibility that Kif3a controls Fgf8 expression in metanephric mesenchyme cells. We tested our hypothesis in cultured metanephric mesenchyme cells isolated from WT and Kif3a−/−MM mice and transfected with a DNA construct encoding a Kif3a-GFP fusion protein (Figure 7). Analysis of primary cilia length, identified by α-AcT expression and imaged by confocal microscopy in cultured primary metanephric mesenchyme cells, revealed that cilia were markedly shorter in cells isolated from Kif3a−/−MM mice (Figure 7B versus 7A and Figure 7E, cilium length, WT versus Kif3a−/−MM MM cells: 4.585±1.523 micrometer versus 1.576±0.449 micrometer; p = 3.5E–10, n = 10 culture wells/group). Transfection of Kif3a fused with GFP in metanephric mesenchyme cell cultures caused localization of KIF3A and GFP to primary cilia and a significant increase in cilia length in metanephric mesenchyme cells isolated from Kif3a−/−MM mice (Figure 7C and 7D and Figure 7E, cilium length, untransfected versus transfected Kif3a−/−MM metanephric mesenchyme cells: 1.576±0.449 micrometer versus 1.962±0.360 micrometer; p = 0.03, n = 10 culture wells/group). Next, we analyzed the effect of Kif3a on expression of Hh signaling effectors, the expression of which has been shown to be dependent on Kif3a in nonrenal tissues. Surprisingly, neither loss of Kif3a expression in Kif3a−/−MM metanephric mesenchyme tissue, cultured metanephric mesenchyme cells nor transfection-mediated Kif3a expression in these cells was associated with a detectable change in Ptc1 or Gli1 mRNA levels assayed by quantitative PCR (Figure 7G and 7F). In contrast, Fgf8 mRNA expression was significantly lower in cultured Kif3a−/−MM metanephric mesenchyme cells compared to controls (Figure 7G and 7H). Moreover, Kif3a transfection significantly increased expression of Fgf8 mRNA in both WT and Kif3a-deficient metanephric mesenchyme cells (Figure 7G and 7H). Together, these data indicate that Kif3a controls Fgf8 expression in metanephric mesenchyme cells.
Discussion
Cilia proteins KIF3A, IFT88 and IFT20, which are involved in IFT [2], [32], [33], are required for renal ciliogenesis; inactivation of each is known to cause cystic kidney disease [3], [14], [34], [35]. To our knowledge this is the first study demonstrating a role for the primary cilium in the regulation of nephron number. Our data show that Kif3a expression and primary cilia are found in both ureteric cells and metanephric mesenchyme cells from the onset of murine kidney development. CRE-mediated recombination using a Kif3aloxP allele results in near total loss of Kif3a in CRE-expressing cells. Loss of cilia occurs with slower kinetics. Yet, Kif3a deficiency and a decrease in the number of primary cilia reduce the number of nephron precursor structures formed and the final number of mature nephrons. Experiments that investigated the mechanisms underlying this phenotype support a model of Kif3a function during murine renal development (Figure 8). Our model suggests that the functions of Kif3a in controlling nephron formation are specific to the ureteric and metanephric mesenchyme cell lineages. In the ureteric lineage, Kif3a controls the number of ureteric branches formed in a GLI3R-dependent manner. Control of branch number is a critical determinant of nephron number. In metanephric mesenchyme cells, Kif3a exerts two major distinct effects. First, Kif3a controls cell survival such that the metanephric blastema that gives rise to nephrons is smaller in Kif3a deficient mice and that surviving Kif3a deficient metanephric mesenchyme cells are less able to take part in forming nephrogenic structures compared to their wild type counterparts. Second, Kif3a controls expression of genes required during nephron formation in metanephric mesenchyme cells.
Figure 8. Model of Kif3a-mediated regulation of nephron number.
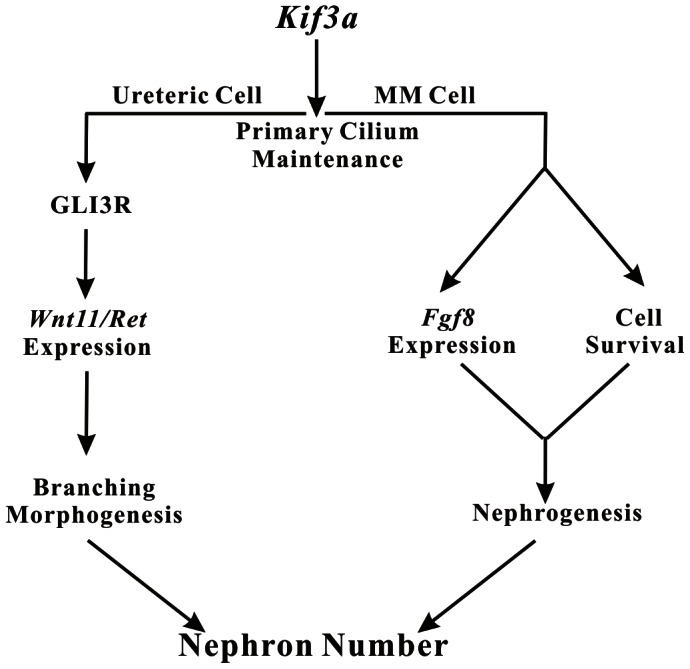
A Kif3a control is required in both ureteric and metanephric mesenchyme cells for processes that control nephron number. In ureteric cells Kif3a acts in a GLI3R-dependent manner to control expression of Wnt11/Ret and ureteric branching. In metanephric mesenchyme cells, Kif3a functions to control cell survival and expression of Fgf8, which is required for maturation of nephrogenic progenitors. The lineage-specific functions of Kif3a in ureteric and metanephric mesenchyme cells converge to control nephron number.
Our results demonstrate a central role for KIF3A in controlling ureteric and mesenchyme cell function. Lineage-specific deletion of Kif3a is efficient with little residual Kif3a mRNA expression in either the ureteric or metanephric mesenchyme cell populations by E11.5, the stage at which these cell populations were separated and analyzed in Kif3a−/−UB and Kif3a−/MM mice (Figure S2). Our results also strongly suggest that the functions of KIF3A protein are related to its specific expression in the primary cilium. Analysis of KIF3A protein expression in Kif3a-deficient mouse strains demonstrated a specific KIF3A signal in the primary cilium (Figure 2A, 2A’, 2A’’). Interestingly, primary cilia exhibit a comparatively slower turnover rate, compared to KIF3A protein, in ureteric and mesenchyme cells. While we could detect very little KIF3A protein by E13.5, in the ureteric or metanephric mesenchyme cells of Kif3a−/−UB and Kif3a−/MM mice, respectively, primary cilia could be detected at E15.5, albeit in reduced numbers. These observations suggest that primary cilium structure can be maintained in non-dividing cells in the face of KIF3A depletion. However, our studies do not provide information as to whether the function of KIF3A-deficient cilia is normal. Our data are also consistent with the rather modest effect of KIF3A deficiency on nephron number. Given the slow kinetics of KIF3A depletion and loss of cilia, it is likely that remaining number of cilia during early critical stages of nephron formation are sufficient to support this process.
Our results demonstrate a critical role for GLI3R in primary cilium function in ureteric cells and are consistent with our previous work related to GLI3R and ureteric branching [17]. The initial stage of ureteric bud invasion into the mesenchyme appears normal in Kif3a−/−UB kidneys. However, the expression of Wnt11 and Ret, both of which are required for ureteric branching [28], [36], is decreased in mutant kidneys at E13.5 (Figure 4F,G and 4H, I). Primary cilia are present on only a subset of UB cells at this time point as primary cilia are gradually lost from E13.5 to P0. Concomitantly, the expression of the ureteric tip markers, and the number of ureteric tips is significantly reduced at E14.0.
In our previous published work, analysis of Hh signaling activity, using a Ptc1-lacZ reporter, demonstrated that ureteric tips are characterized by low Hh activity [17]. Activation of Hh signaling activity in ureteric cells in mice with Ptc1 deficiency causes decreased Ret and Wnt11 expression, decreased ureteric branching and low nephron number. But, constitutive expression of GLI3R (via the Gli3 Δ699 allele) rescues these abnormalities [17] and suggests that GLI3R, rather than GLI activators, is the regulatory target of SHH signaling during formation of nephrons. Our prior analyses in Shh deficient mice also support the concept that regulation of GLI3R is the critical event during kidney development. Mice with homozygous deficiency of Shh are characterized by disruption of initial ureteric-metanephric mesenchyme tissue interactions and an elevated ratio of GLI3R to GLI activator proteins in Shh deficient renal tissue. Remarkably, these abnormalities are rescued by homozygous deficiency of Gli3 in Shh deficient mice, thus implicating regulation of GLI3R formation as a critical event during renal development [16]. Results here suggest that the primary cilium plays a critical role in GLI3R expression in ureteric cells.
In contrast to ureteric cells, our results do not invoke Hh signaling and GLI3R in regulating metanephric mesenchyme cell survival and nephron formation. Our data, are consistent with our published analysis of kidney development in mice with conditional inactivation of Smo in metanephric mesenchyme cells [37]. In these mice (Rarb2-Cre;SmoloxP/−), genetic inactivation of Smo was mediated by CRE recombinase, the expression of which was driven by a Rarb2 promoter element which directs expression in the intermediate and metanephric mesenchyme [31]. In Rarb2-Cre;SmoloxP/− mice, renal development is normal until E15.5 when pelvic dilatation arises due to ureteric dyskinesia and abnormal pacemaker cell function, demonstrating that loss of Hh signaling in intermediate and metanephric does not disrupt the mass of cells available to take part in nephron formation. Our results in cultured metanephric mesenchyme cells (Figure 7) are consistent with these findings in Rarb2-Cre;SmoloxP/− mice since Ptc1 and Gli1 are expressed in Kif3a deficient metanephric mesenchyme cells isolated at E11.5. Moreover, transfection of Kif3a in Kif3a-deficient cells has no significant effect on Ptc1 and Gli1 expression.
Our analyses in Kif3a−/−MM mice and in cultured metanephric mesenchyme cells suggest a role for KIF3A upstream of FGF8. Kif3a−/−MM mice are characterized by cilia in developing nephron structures and intact nephron formation before E13.5. However, the total pool of Kif3a-negative mesenchymal precursor cells is decreased in the kidney blastema at E11.0. Surviving Kif3a-negative mesenchyme cells exhibit the ability to undergo a mesenchymal to epithelial transition probably because a sufficient level of Fgf8, Wnt4 and Lim1 mRNA is present to support this process (Figure S5). However by E15.5, Fgf8 expression is lost – the same stage at which the number of cilia is significantly reduced in mesenchyme- derived cells. By E15.5, Wnt4 and Lim1 expression is markedly reduced consistent with loss of Fgf8. Yet, our results related to FGF8 are distinct from previous published analyses of Fgf8 activity during renal development. Mice with total loss of Fgf8 are able to initiate formation of nephrons but development of nephron precursors does not progress to the S-shape stage. While Kif3a−/−MM mice similarly exhibit a lower number of S-shape bodies compared to controls, S-shaped bodies are formed [22]. In FGF8-deficient mice, cells within nascent nephrons undergo high rates of apoptosis [22], a finding that is similar to that we observed in Kif3a−/−MM mice. Thus, decreased expression of Fgf8 may be the cause of increased cell death in Kif3a−/−MM null kidneys. Our studies in cultured WT and Kif3a-deficient metanephric mesenchyme cells further link Kif3a to Fgf8. Previous studies demonstrated that Fgf signaling regulates cilia length through Fgf8-Fgfr1 and the IFT pathway [11]. Our data show that Fgf8 expression is decreased in Kif3a-deficient cells and that Fgf8 expression is partially restored to WT levels by transfection of these cells with a plasmid encoding Kif3a.
Materials and Methods
Mouse Strains and Genotyping
The following mouse strains were used in these studies: Kif3aloxP/loxP, Kif3a+/−, Cre-ER™, Cre-ER™;Kif3aloxP/− (termed Kif3a™), Hoxb.7-CreEGFP;Kif3a+/−, Rarb2-Cre;Kif3a+/−, Hoxb.7-CreEGFP;Kif3aloxP/− (termed Kif3a−/− UB), Rarb2-Cre;Kif3aloxP/− (termed Kif3a−/− MM), Hoxb.7-CreEGFP;Kif3aloxP/+ (termed Kif3a+/− UB); Rarb2-Cre;Kif3aloxP/+ (termed Kif3a+/− MM), Hoxb.7-CreEGFP;Kif3aloxP/−;Gli3Δ699/+(termed Kif3a−/− UB;Gli3Δ699/+) and R26RLacZ/LacZ.;Kif3aflox/flox [3] mice were maintained on the C57BL/6 (B6) genetic background. Cre-ER™ [23], Hoxb7-CreEGFP [24] and Rarb2-Cre [25] mice were maintained on the CD1 inbred genetic background. Cre-ER™;Kif3a+/−, Cre-ER™;Kif3aflox/−(Kif3a™); Hoxb7-CreEGFP;Kif3a+/−; Hoxb7-CreEGFP;Kif3aflox/− (Kif3a−/−UB) and Rarb2-Cre;Kif3a+/−, Rarb2-Cre;Kif3aflox/− (Kif3a−/−MM) mice were maintained on a mixed background. R26R reporter mice [30] were maintained on a B6 x129/SV mixed genetic background. Genotyping was performed by PCR using genomic DNA isolated from ear clips or tails. PCR primers are listed in Table S1 in File S1.
Tamoxifen-induced CRE Recombinase Expression
Tamoxifen (TM, T5648, Sigma) was dissolved in sesame oil at a concentration of 20 mg/ml. TM was injected at a dose of 3mg/40g body weight intraperitoneally into pregnant mice at E10.5.
Tissue-based Assays
Non-radioactive section in situ hybridization was performed as per published methods using 5µm paraffin-embedded tissue sections [38]. Digoxigenin-labeled antisense probes were generated from linearized plasmids containing mouse Wnt4 [39], Wnt11 [40], Wnt9b [20], Ret [41], Foxd1 [42], Lim1 [25], Fgf8 [43], [44]. To detect LacZ activity, tissues were fixed in 2% paraformaldehyde (PFA), embedded in OCT (Tissue-TeK 4583), and cryosectioned at 10 micrometer. Sections were dried at RT for 30 minutes, rinsed in PBS, and stained with standard X-gal staining solution overnight at 37°C before counterstaining with Eosin. The number of NCAM-positive structures, WT1-positive structures and S-shaped bodies was estimated by counting the number of structures in five tissue sections - a mid-sagittal section and two sections generated 40 micrometer and 80 micrometer in both directions from the mid-sagittal section - in each kidney analyzed. Quantitation was expressed the number of structures per section [43]. The average number of mitotic cell of ureteric tip (mitotic cell number/tip) was quantified by counting in five randomly chosen ureteric tips per section; five sections in each kidney were analyzed. The terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) method was used to detect apoptosis. Paraffin wax was removed from 5 µm tissue sections with xylene and tissues were rehydrated and incubated with 20 mg/ml of Proteinase K for 15 minutes at 37°C. Non-specific binding was reduced with a hydrogen peroxide/methanol solution. Fragmented DNA was labelled using a reaction mixture, according to the manufacturer’s instructions (Roche). Bound probes were detected using 3,3′-diaminiobenzidine (DAB) as a substrate (Vector Laboratories). An in situ BrdU-uptake assay was used as a surrogate measure of cell proliferation. Pregnant mice were injected intraperitoneally with a solution containing bromodeoxyuridine (BrdU, Sigma B-5002) at a dose of 10 micrograms/g body weight. Mice were sacrificed at two hours post-injection. Kidneys were fixed with 4% PFA overnight at 4°C, dehydrated and embedded in paraffin. BrdU incorporation was detected in 5 micrometer sections using anti-BrdU horseradish peroxidase (1∶50, Roche, USA).
Transient Transfection of Metanephric Mesenchymal Cells
Metanephric mesenchyme cells were isolated at E11.5 and cultured in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) supplemented with 15% fetal bovine serum, 100 U/ml penicillin and 100 micrograms/ml streptomycin [45]. Isolated cells were cultured overnight then passaged to a new dish and cultured for 16 hours after which cells were transfected with a pEGFP-N1 vector containing full length Kif3a cDNA (0.8 micrograms/well) using Lipofectamine™ 2000 (Invitrogen) according to the manufacturers’ instructions. Cells were harvested 36 hours after transfection.
Antibodies
Immunofluorescence analyses of kidney tissue and cultured cells was performed using published methods [44], [46], [47] using antibodies directed against: KIF3A (1∶100, K3513, Sigma), PAX2 (1∶500, PRB-276P, Covance), α-AcT (1∶1000, T6793, Sigma), Cytokeratin (1∶200, C2562, Sigma), NCAM (Neural Cell Adhesion Molecule) (1∶50, C9672, Sigma), WT1 (1∶500, sc-192, SANTA CRUZ), LTL (Lotus Tetragonolobus Lectin) (1∶100, FITC, FL-1321, Vector Laboratories, Inc.), SIX2 (1∶200, ab68908, Abcam), Phospho-Histone H3 (Ser28) (1∶200, Cell Signaling) and GFP (1∶1000, Ab16901, Millipore). Secondary antibodies used were Alexa 488 anti-rabbit IgG, anti-rat IgG and anti-chicken IgG, as well as Alexa 546 anti-rat IgG and anti-rabbit IgG, (1∶1000, Molecular Probes, Invitrogen Detection Technologies). DAPI (1∶1000, D9564, Sigma) was used for nuclear staining.
RNA Isolation and Real-Time PCR
Total RNA was purified from isolated E11.5 ureteric bud and metanephric mesenchyme or whole embryonic kidneys using the RNeasy Mini Kit (QIAGEN). cDNA was synthesized using a first strand cDNA synthesis kit (Invitrogen). Real-time RT-PCR [46] was performed to determine the expression of Kif3a, Ptc1, Gli1 and Fgf8. Gapdh served as an endogenous control. The primers used, the fragments amplified, and the annealing temperatures are detailed in Table S2 in File S1. Quantitative RT-PCR (qRT-PCR) was performed using an Applied Biosystems 7900 HT fast RT-PCR system with TaqMan® Universal PCR Master Mix and TaqMan® probes for Kif3a (Mm00492876_m1) or Fgf8 (Mm00438922_m1). Mouse Gapdh was used as an endogenous control (Mm03302249_g1, Applied Biosystems). Primers for Ptc1 and Gli1 were designed using Primer 3 software. Relative mRNA expression was determined using the standard curve method. Samples were analysed in triplicates.
Image Capture and Data Analysis
Kidneys for SEM were perfused with 4% PFA and 2% glutaraldehyde in PBS, prepared as described previously [14], and visualized with a FEI XL30 Scanning Electron Microscope at the Advanced Bioimaging Center of Mount Sinai Hospital, University of Toronto. Microscopy was also performed using a spinning disk confocal laser scanning microscope or Zeiss Axiovision4 light microscope. A minimum of four mice (derived from different litters) were analysed for each developmental stage, gene, antigen and genotype. Student’s t-test (two-tailed) was used to analyze the mean differences between groups. The statistical significance was taken at a value of P<0.05. Images were combined using Adobe Photoshop CS2 and CorelDRAW 14 software.
Ethics Statement
All experiments using animals have been conducted according to the guidelines adopted by the Toronto Centre for Phenogenomics and which are in accord with national and international guidelines. The experiments, the results of which are reported here, were approved by the Institutional Animal Care and Use Committee (IACUC) of the Toronto Centre for Phenogenomics. Animals were sacrificed via inhalation of CO2.
Supporting Information
Primary cilia are present in both ureteric epithelial and metanephric mesenchyme cells in the developing murine kidney. (A–D) Schematic of ureteric bud epithelial cells (black), metanephric mesenchyme cells and metanephric-derived nephrogenic stuctures during progressive stages of kidney development (green). (A’–D’’) Primary cilia (acetylated α-tubulin, red, arrows) are present in both ureteric (arrows), metanephric mesenchyme cells and their derivatives (Pax2, green, arrow heads) in E11.5 (A’), E13.5 (B’) and E15.5 (C’) kidneys. (A’’–D’’) Single color shows primary cilia in the developing kidney. CM, Condensate Mesenchyme; CSB, Comma-Shape Body; UB, Ureteric Bud; RV, Renal vesical; WT, wild type. Scale bar: C’–F’’, 25 micrometer.
(TIF)
Expression of Kif3a in kidney tissue. Ureteric bud was dissected free of metanephric mesenchyme in E11.5 kidney tissue of WT, Kif3a−/−UB, and Kif3a−/−MM mice. Kif3a mRNA expression was analyzed by quantitative RT-PCR and quantified. Kif3a is not expressed in the ureteric bud of Kif3a−/−UB mice but is expressed in metanephric mesenchyme. Kif3a is not expressed in the metanephric mesenchyme of Kif3a−/−MM mice but is expressed in ureteric bud. (***, P<0.001).
(TIF)
Decreased nephron number in both Kif3a−/−UB and Kif3a−/−MM kidneys. (A–F) Histological sections, stained with hematoxylin and eosin demonstrate a qualitative decrease in the number of glomeruli (arrows) at E15.5 (A,B,C) and E18.5 (D,E,F) in both Kif3a−/−UB (B, E) and Kif3a−/−MM (C, F) mice compared to WT (A, D). (G–I) The decrease in mature glomeruli (arrows) in both Kif3a−/−UB (H) and Kif3a−/−MM (I) mice is greater at P0. Cysts are present in collecting duct (H, arrowheads) and tubules (I, arrowheads) in both mutant mouse strains. (J) Quantification of the number of mature glomeruli demonstrates a decrease in Kif3a−/−UB and Kif3a−/−MM mice at E15.5, E18.5 and P0 compared to controls. (***, P<0.001; **, P<0.01; *, P<0.05). Scale bars: 50 micrometer.
(TIF)
Cell proliferation and SIX2-positive nephrogenic progenitor cells in Kif3a−/−MM kidney tissue. (A, B) In situ BrdU incorporation assay in E13.5 kidney tissue. Ureteric bud tip is demarcated by the yellow dotted line. (C) Quantification of BrdU-positive cap mesenchyme cells reveals no significant difference between Kif3a−/−MM and WT mice. (D, D’) SIX2-positive cells (nephrogenic precursors) are organized in a tightly packed layer around the ureteric bud tip at E13.5 in WT mice. (E, E’) The SIX2-positive cells are disorganized surrounding the ureteric tip in Kif3a−/−MM mice. (F) Quantification of the SIX2-positive cells demonstrates a significant decrease in Kif3a−/−MM mice versus WT control mice. (**, P<0.01). Scale bars: 50 micrometer.
(TIF)
Expression of Fgf8, Wnt4, Lim1 and Wnt9b mRNAs in E13.5 Kif3a−/−MM mice. Expression was determined by in situ hybridization. Expression of Fgf8 is mildly decreased in Kif3a−/−MM mice (A’) compared to WT (A) but expression of Wnt4. (B, B’), Lim1 (C, C’) and Wnt9b (D, D’) is unchanged. Scale bars: 50 micrometer.
(TIF)
Table S1, Primers used to genotype the various mutant mouse lines. Table S2, The primers and their RT-PCR products used to estimate the mRNA expression in kidneys and in MM cells.
(DOC)
Acknowledgments
We thank Dr. Cathy L. Mendelsohn for the Rarb2-Cre mouse strain and Dr. Lawrence S. B. Goldstein for Kif3a flox/flox mice.
Funding Statement
This work was supported by grants from the Canadian Institutes of Health Research, the Kidney Foundation of Canada and the Canada Research Chairs Program (to NDR) and operating grants from the Canadian Cancer Society Research Institute (to CCH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scholey JM, Anderson KV (2006) Intraflagellar transport and cilium-based signaling. Cell 125: 439–442. [DOI] [PubMed] [Google Scholar]
- 3. Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS (1999) Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A 96: 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, et al. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426: 83–87. [DOI] [PubMed] [Google Scholar]
- 5. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, et al. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437: 1018–1021. [DOI] [PubMed] [Google Scholar]
- 6. Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, et al. (2005) Gli2 and gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317: 372–376. [DOI] [PubMed] [Google Scholar]
- 8. Huangfu D, Anderson KV (2005) Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A 102: 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, et al. (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37: 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, et al. (2009) Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med 15: 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ (2009) FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature 458: 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Evans MJ, Fanucchi MV, Van Winkle LS, Baker GL, Murphy AE, et al. (2002) Fibroblast growth factor-2 during postnatal development of the tracheal basement membrane zone. Am J Physiol Lung Cell Mol Physiol 283: L1263–1270. [DOI] [PubMed] [Google Scholar]
- 13. Yoder BK (2007) Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol 18: 1381–1388. [DOI] [PubMed] [Google Scholar]
- 14. Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, et al. (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A 100: 5286–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saxen L (1987) Organogenesis of the kidney. Cambridge: Cambridge University Press.
- 16. Hu MC, Mo R, Bhella S, Wilson CW, Chuang PT, et al. (2006) GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development 133: 569–578. [DOI] [PubMed] [Google Scholar]
- 17. Cain JE, Islam E, Haxho F, Chen L, Bridgewater D, et al. (2009) GLI3 Repressor Controls Nephron Number Via Regulation of Wnt11 and Ret in Ureteric Tip Cells. PLoS ONE 4: e7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bridgewater D, Cox B, Cain J, Lau A, Althaide V, et al. (2008) Canonical WNT/beta-catenin signaling is required for ureteric branching. Dev Biol 317: 83–94. [DOI] [PubMed] [Google Scholar]
- 19. Park JS, Valerius MT, McMahon AP (2007) Wnt/{beta}-catenin signaling regulates nephron induction during mouse kidney development. Development 134: 2533–2539. [DOI] [PubMed] [Google Scholar]
- 20. Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP (2005) Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell 9: 283–292. [DOI] [PubMed] [Google Scholar]
- 21. Stark K, Vainio S, Vassileva G, McMahon AP (1994) Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4 . Nature 372: 679–683. [DOI] [PubMed] [Google Scholar]
- 22. Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, et al. (2005) FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132: 3847–3857. [DOI] [PubMed] [Google Scholar]
- 23. Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244: 305–318. [DOI] [PubMed] [Google Scholar]
- 24. Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, et al. (2004) Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276: 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobayashi A, Kwan KM, Carroll TJ, McMahon AP, Mendelsohn CL, et al. (2005) Distinct and sequential tissue-specific activities of the LIM-class homeobox gene Lim1 for tubular morphogenesis during kidney development. Development 132: 2809–2823. [DOI] [PubMed] [Google Scholar]
- 26. Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, et al. (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100: 5286–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Majumdar A, Vainio S, Kispert A, McMahon J, McMahon AP (2003) Wnt11 and Ret/Gdnf pathways cooperate in regulating ureteric branching during metanephric kidney development. Development 130: 3175–3185. [DOI] [PubMed] [Google Scholar]
- 28. Michos O, Cebrian C, Hyink D, Grieshammer U, Williams L, et al. (2010) Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet 6: e1000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bose J, Grotewold L, Ruther U (2002) Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet 11: 1129–1135. [DOI] [PubMed] [Google Scholar]
- 30. Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70–71. [DOI] [PubMed] [Google Scholar]
- 31. Di Giovanni V, Alday A, Chi L, Mishina Y, Rosenblum ND (2011) Alk3 controls nephron number and androgen production via lineage-specific effects in intermediate mesoderm. Development 138: 2717–2727. [DOI] [PubMed] [Google Scholar]
- 32. Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3: 813–825. [DOI] [PubMed] [Google Scholar]
- 33. Gerdes JM, Davis EE, Katsanis N (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137: 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jonassen JA, San Agustin J, Follit JA, Pazour GJ (2008) Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol 183: 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, et al. (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 151: 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuchardt A, D'Agati V, Pachnis V, Costantini F (1996) Renal agenesis and hypodysplasia in ret-k - mutant mice result from defects in ureteric bud development. Development 122: 1919–1929. [DOI] [PubMed] [Google Scholar]
- 37. Cain JE, Islam E, Haxho F, Blake J, Rosenblum ND (2011) GLI3 Repressor controls functional development of the mouse ureter. Journal of Clinical Investigation 121: 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, et al. (1998) Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125: 2533–2543. [DOI] [PubMed] [Google Scholar]
- 39. Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP (1999) Female development in mammals is regulated by Wnt-4 signalling. Nature 397: 405–409. [DOI] [PubMed] [Google Scholar]
- 40. Kispert A, Vainio S, Shen L, Rowitch DH, McMahon AP (1996) Proteoglycans are required for maintenance of Wnt-11 expression in the ureter tips. Development 122: 3627–3637. [DOI] [PubMed] [Google Scholar]
- 41. Sainio K, Suvanto P, Davies J, Wartiovaara J, Wartiovaara K, et al. (1997) Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development 124: 4077–4087. [DOI] [PubMed] [Google Scholar]
- 42. Hatini V, Huh SO, Herzlinger D, Soares VC, Lai E (1996) Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev 10: 1467–1478. [DOI] [PubMed] [Google Scholar]
- 43. Chi L, Zhang S, Lin Y, Prunskaite-Hyyrylainen R, Vuolteenaho R, et al. (2004) Sprouty proteins regulate ureteric branching by coordinating reciprocal epithelial Wnt11, mesenchymal Gdnf and stromal Fgf7 signalling during kidney development. Development 131: 3345–3356. [DOI] [PubMed] [Google Scholar]
- 44. Rosenquist TA, Martin GR (1996) Fibroblast growth factor signalling in the hair growth cycle: expression of the fibroblast growth factor receptor and ligand genes in the murine hair follicle. Dev Dyn 205: 379–386. [DOI] [PubMed] [Google Scholar]
- 45. Chi L, Rosenblum N (2012) Investigating primary cilia in cultured metanephric mesenchymal cells. Methods Mol Biol 886: 157–163. [DOI] [PubMed] [Google Scholar]
- 46. Chi L, Itaranta P, Zhang S, Vainio S (2006) Sprouty2 is involved in male sex organogenesis by controlling fibroblast growth factor 9-induced mesonephric cell migration to the developing testis. Endocrinology 147: 3777–3788. [DOI] [PubMed] [Google Scholar]
- 47. Hartwig S, Bridgewater D, Di Giovanni V, Cain J, Mishina Y, et al. (2008) BMP receptor ALK3 controls collecting system development. J Am Soc Nephrol 19: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primary cilia are present in both ureteric epithelial and metanephric mesenchyme cells in the developing murine kidney. (A–D) Schematic of ureteric bud epithelial cells (black), metanephric mesenchyme cells and metanephric-derived nephrogenic stuctures during progressive stages of kidney development (green). (A’–D’’) Primary cilia (acetylated α-tubulin, red, arrows) are present in both ureteric (arrows), metanephric mesenchyme cells and their derivatives (Pax2, green, arrow heads) in E11.5 (A’), E13.5 (B’) and E15.5 (C’) kidneys. (A’’–D’’) Single color shows primary cilia in the developing kidney. CM, Condensate Mesenchyme; CSB, Comma-Shape Body; UB, Ureteric Bud; RV, Renal vesical; WT, wild type. Scale bar: C’–F’’, 25 micrometer.
(TIF)
Expression of Kif3a in kidney tissue. Ureteric bud was dissected free of metanephric mesenchyme in E11.5 kidney tissue of WT, Kif3a−/−UB, and Kif3a−/−MM mice. Kif3a mRNA expression was analyzed by quantitative RT-PCR and quantified. Kif3a is not expressed in the ureteric bud of Kif3a−/−UB mice but is expressed in metanephric mesenchyme. Kif3a is not expressed in the metanephric mesenchyme of Kif3a−/−MM mice but is expressed in ureteric bud. (***, P<0.001).
(TIF)
Decreased nephron number in both Kif3a−/−UB and Kif3a−/−MM kidneys. (A–F) Histological sections, stained with hematoxylin and eosin demonstrate a qualitative decrease in the number of glomeruli (arrows) at E15.5 (A,B,C) and E18.5 (D,E,F) in both Kif3a−/−UB (B, E) and Kif3a−/−MM (C, F) mice compared to WT (A, D). (G–I) The decrease in mature glomeruli (arrows) in both Kif3a−/−UB (H) and Kif3a−/−MM (I) mice is greater at P0. Cysts are present in collecting duct (H, arrowheads) and tubules (I, arrowheads) in both mutant mouse strains. (J) Quantification of the number of mature glomeruli demonstrates a decrease in Kif3a−/−UB and Kif3a−/−MM mice at E15.5, E18.5 and P0 compared to controls. (***, P<0.001; **, P<0.01; *, P<0.05). Scale bars: 50 micrometer.
(TIF)
Cell proliferation and SIX2-positive nephrogenic progenitor cells in Kif3a−/−MM kidney tissue. (A, B) In situ BrdU incorporation assay in E13.5 kidney tissue. Ureteric bud tip is demarcated by the yellow dotted line. (C) Quantification of BrdU-positive cap mesenchyme cells reveals no significant difference between Kif3a−/−MM and WT mice. (D, D’) SIX2-positive cells (nephrogenic precursors) are organized in a tightly packed layer around the ureteric bud tip at E13.5 in WT mice. (E, E’) The SIX2-positive cells are disorganized surrounding the ureteric tip in Kif3a−/−MM mice. (F) Quantification of the SIX2-positive cells demonstrates a significant decrease in Kif3a−/−MM mice versus WT control mice. (**, P<0.01). Scale bars: 50 micrometer.
(TIF)
Expression of Fgf8, Wnt4, Lim1 and Wnt9b mRNAs in E13.5 Kif3a−/−MM mice. Expression was determined by in situ hybridization. Expression of Fgf8 is mildly decreased in Kif3a−/−MM mice (A’) compared to WT (A) but expression of Wnt4. (B, B’), Lim1 (C, C’) and Wnt9b (D, D’) is unchanged. Scale bars: 50 micrometer.
(TIF)
Table S1, Primers used to genotype the various mutant mouse lines. Table S2, The primers and their RT-PCR products used to estimate the mRNA expression in kidneys and in MM cells.
(DOC)