

Abstract
To identify rate-limiting steps in T cell-independent type 2 (TI-2) antibody production against polysaccharide antigens, we performed a genome-wide screen by immunizing several hundred pedigrees of C57BL/6 mice segregating ENU-induced mis-sense mutations. Two independent mutations, Tilcara and Untied, were isolated that semi-dominantly diminished antibody against polysaccharide but not protein antigens. Both mutations resulted from single amino acid substitutions within the kinase domain of Protein Kinase C Beta (PKCβ). In Tilcara, a Ser552>Pro mutation occurred in helix G, in close proximity to a docking site for the inhibitory N-terminal pseudosubstrate domain of the enzyme, resulting in almost complete loss of active, autophosphorylated PKCβI whereas the amount of alternatively spliced PKCβII protein was not markedly reduced. Circulating B cell subsets were normal and acute responses to BCR-stimulation such as CD25 induction and initiation of DNA synthesis were only measurably diminished in Tilcara homozygotes, whereas the fraction of cells that had divided multiple times was decreased to an intermediate degree in heterozygotes. These results, coupled with evidence of numerous mis-sense PRKCB mutations in the human genome, identify Prkcb as a genetically sensitive step likely to contribute substantially to population variability in anti-polysaccharide antibody levels.
Keywords: Protein kinase c beta, T independent antibody response, mis-sense mutation
Introduction
A hallmark of the adaptive immune system is the production of antibodies by B cells targeting varying types of antigens. Genetic studies in mice have distinguished different routes for antibody production depending on the nature of the antigen. Firstly, antigens have been classified as eliciting either T cell-dependent or T cell-independent (TI) responses based on the ability to induce antibodies to a given antigen in athymic nude mice1. Antigens that elicit T-independent antibody responses have been further sub-divided into TI-1 or TI-2 antigens based on their ability to stimulate responses in the X-linked immunodeficiency (Xid) mouse strain, which has a crippling Btk mutation that affects some intracellular signals elicited by antigen binding to the B cell antigen receptors (BCR). Xid mice are capable of making antibodies against TI-1 antigens, such as bacterial lipopolysaccharide or flagellin, because these signal B cell proliferation through Toll-like receptors2. By contrast, Xid mice fail to make antibodies to TI-2 antigens such as the polysaccharide envelopes of some bacteria or synthetic polysaccharides such as Ficoll, because these antigens signal B cell proliferation by extensively cross-linking the BCR into large aggregates that persist on the cell surface1, 3, 4. TI-2 responses are mediated primarily by the secretion of short-lived IgM isotype antibodies by marginal zone (MZ) B cells in the spleen5 and the B-1 cells in the serous cavities6, 7.
In a T-independent antibody response, the stimulation of the B cell receptor (BCR) leads to the phosphorylation of immunoreceptor tyrosine-based activation motifs in the cytoplasmic tails of the signal transducing molecules CD79a (Igα) and CD79b (Igβ). This allows the phosphorylation of multiple signalling kinases such as Bruton’s tyrosine kinase (Btk) and the phospholipase c gamma-2 (PLCγ2)-mediated elevation of intracellular calcium. These kinases activate protein kinase c beta (PKCb) which starts a phosphorylation cascade activating the Card11/Malt1/Bcl-10 complex. This results in the recruitment of the nuclear factor kappa B transcription factor to inducible genes that stimulate B cell proliferation and TI-2 antibody production.
Defects in TI-2 antibody responses result in increased susceptibility to encapsulated bacterial infections such as Haemophilus influenza type b, Neisseria meningitides and Streptococcus pneumoniae8. These infections severely impact young children particularly in the developing world. Chronic infection and poor disease management can lead to mortality. Lower antibody production against these antigens is correlated with recurrent infection9–11 but the underlying basis for population variation is not known. Further understanding of the underlying genetic alterations that make individuals more susceptible to these diseases are important for the identification of therapeutic targets for the disease and to enhance vaccination and immunisation strategies.
A unique feature of modern humans is the massive degree of population expansion that has occurred in the last several hundred generations. Deep sequencing efforts have recently revealed that this growth has resulted in an equally massive increase in the frequency of heterozygous deleterious mutations12–16 with each person carrying on average 100 deleterious mutations in the genome with approximately 20 being complete loss of function variants17. Each of these mutations are individually too recently arising and rare in the population at large to have yet been removed by purifying selection, yet cumulatively they amount to a large genetic burden. Rare variants account for 95% of the protein-altering mutations predicted to be deleterious in the current generation of humans, and account for ~25% of the deleterious mutations in any individual’s genome. Based on the de-novo mutation rate in human reproduction (1.4×10−8/bp), the current generation of humans is now large enough (6×109 individuals) to saturate every nucleotide of the genome with de-novo mutations12. Among this burden of recent mutations will be many unique heterozygous mutations in the large network of genes that control antibody production. Heterozygous mutations that affect rate-limiting steps in antibody production will account for a substantial amount of inherited variability in antibody formation, but no systematic efforts have yet been made to identify these rate-limiting steps.
In this study, we have taken an experimental approach in mice to evaluate the impact of increasing the genome-wide burden of rare variants on TI-2 antibody formation against polysaccharide antigens. We used N-ethyl-N-nitrosurea (ENU) mutagenesis of inbred C57BL/6 mice to increase the frequency of single nucleotide variants, coupled with systematic screening for variability in the antibody response following immunization with a haptenated polysaccharide, NP-Ficoll18. Two semi-dominant mutations were isolated causing selective deficits in TI-2 antibody response, and both were due substitutions in conserved residues in the catalytic domain of Protein kinase c beta (Prkcb). These results identify PRKCB as a rate-limiting point in the network of genes controlling TI-2 antibody, and provide novel animal models to analyze the biochemistry of the two PKCβ enzyme isoforms.
Results
Generation of the Tilcara and Untied mutant mice and identification of the causative mutation in Prkcb
A genetic screen was performed to identify genes that are critical for T-cell independent antibody production. First generation offspring (G1) from ENU-treated inbred C57BL/6 male mice were used as founders for separate inbreeding pedigrees by breeding with wild-type females, and subsequently intercrossing their G2 progeny to generate a G3 generation. Twelve-week-old G3 offspring from 130 pedigrees were screened by immunization with a panel of antigens that differentially elicit T-cell independent and T-dependent antibody responses. T cell-dependent antibody response were measured by immunizing each G3 animal with alum-precipitated chicken gamma globulin coupled to hapten arsonate (ABA-CGG) and heat killed Bordetella pertussis (BP). After 5 weeks, the mice were re-immunized with ABA-CGG to test T-cell dependent memory and affinity maturation, and immunized at the same time with NP-Ficoll, which elicits a T-cell independent type 2 (TI-2) antibody response18, 19. Three of five mice tested in the Tilcara pedigree exhibited lower TI-2 responses compared to littermate counterparts (Figure 1a and b), but had normal T-dependent responses indicating that these mice have no general problem in the generation of specific antibodies.
Figure 1.

(a) Tilcara strain pedigree and TI-2 antibody response. Open and black-filled shapes in pedigree denote third generation (G3) mice with normal or low TI-2 responses, respectively, based on ELISA measurement shown on right. NP-specific IgM antibodies were measured in 1:150 diluted serum from individual G3 mice in the Tilcara pedigree and other ENU6 pedigrees screened in parallel, 6 days after immunization with NP-Ficoll. (b) Semi-dominant low TI-2 antibody following immunization in G3 offspring from the Tilcara pedigree. (c) Informative recombinant Ch7 haplotypes in (CBA×B6til/+)F1×B6+/+ mice with low (affected) or normal antibody response to NP-Ficoll. Black, homozygous B6 genotype; white, heterozygous CBA/B6 genotype. Maximum non-recombinant interval is shown. Map positions refer to Build GRCm38 of the mouse genome assembly (http://mouse.ensembl.org). (d) Sequencing chromatograms from amplified Prkcb splenic cDNA from mice of the indicated genotypes, centred on nucleotide 1654. (e) Semi-dominant low TI-2 antibody following immunisation with 2,4,6-trinitrophenyl (TNP)-Ficoll in G3 offspring from the Untied pedigree. (f) Location of Tilcara and Untied mutations within the human PKCβII kinase domain crystal structure (PDB ID: 210E). (g) Location of the S552 residue, Y417 residue and the series of acidic residues in helix G that interact with the N-terminal pseudosubstrate domain. The ATP-binding pocket within the catalytic cleft is also shown. (h) Location of Tilcara and Untied mutations with respect to PKCβ protein domains: ps, pseudosubstrate inhibitory domain; C1A & C1B, diacylgycerol and phospholipid binding domains; C2, calcium binding domain. The green box represents the region that differs in PKCβI PKCβII. (i) Alignment of amino acid sequences of PKCβ from the indicated species (top) and with other murine PKC family members (bottom).
To link the mutation responsible for low TI-2 antibody to a single chromosome, an F2 intercross was performed between offspring from an affected C57BL/6 (B6) G3 mouse and a wild-type C57BL/10 (B10) mouse. Immunization of the F2 offspring with NP-Ficoll identified three groups of animals. The first group had a very low TI-2 antibody response (“affected”), a second group had normal levels of antibodies (“unaffected”) and a third group showed an intermediate response. Pooled DNA samples from affected and unaffected mice was tested using a panel of single nucleotide polymorphisms (SNPs) between the B6 and B10 strains, linking the Tilcara trait to chromosome 7 distal to SNP rs1347954 at 109 Mb. Because of the semi-dominant inheritance the location of the mutation was further refined by immunizing offspring from a cross between (C57BL/6×CBA)F1 animals with an intermediate response and wild-type B6 mice. Genotyping of intermediate responders and unaffected animals localised the mutation to an 8.3 Mb interval (Figure 1c) containing 199 predicted genes, including 16 genes with known immunological function. Three of these, Cd19, Cd11b and Prkcb, were selected as candidate genes for sequencing of splenic mRNA, revealing a single T-to-C point coding sequence mutation in Prkcb at base pair 1654 (Figure 1d).
A second screen of another cohort of ENU mutagenized mice revealed another mouse strain, Untied, that also displayed a semi-dominant defect in IgM antibody selectively against TI-2 antigens (Figure 1e). Sequencing of Prkcb revealed an independent mutation in this strain: in this case a T to C transition at position 1469 of the Prkcb transcript. The Tilcara and Untied mutations both result in single amino acid substitutions of highly conserved residues in the PKCβ kinase domain. Untied results from a tyrosine to histidine substitution at amino acid 417. This alters a residue that is absolutely conserved from mammals to invertebrates, in the middle of beta sheet 5 near the conserved gatekeeper residue (Met420) that controls access to the ATP binding pocket (Figure 1f, g and h). In Tilcara, Ser552 in the middle of alpha helix G on the C-lobe of the kinase domain, on the lower lip of the substrate binding cleft (Figure 1f, g and h), is mutated to a proline that would be predicted to break the helical structure. The mutated S552 is absolutely conserved from mammals to fish and highly conserved between different murine PKC family members (Figure 1i). The genetic evidence that these are causal is thus supported by three independent mutations, since a comparable semi-dominant loss of TI2 antibody responses was also observed in Prkcb knockout mice20.
Two isoforms of protein kinase C beta, PKCβI and PKCβII are produced in mice, humans and other mammals by alternative mRNA splicing of exons encoding PKCβI residues 622–671 (622–673 in PKCβII). Hence the S552P Tilcara mutation affects the part of the protein that is common to both isoforms. Western blotting with antibodies specific for one or other isoform revealed a consistent reduction in the PKCβI isoform in purified B cell lysates from Prkcbtil/til mice compared to wild-type controls, while Tilcara heterozygous mice had intermediate protein expression (Figure 2a). By contrast, there was no consistent decrease in the PKCβII isoform (Figure 2a).
Figure 2.

(a) Western blot for PKCβI, PKCβII in unstimulated purified B cell lysates from wild-type (Prkcbwt/wt), Tilcara heterozygous (Prkcbwt/til) and Tilcara homozygous (Prkcbtl/til) mice. (b) Western blot of total NP-40 cell lysates and the pellet or supernatant fractions from 293 cells transfected with expression vectors encoding V5-tagged wild-type or S552P mutant PKCβI or PKCβII. (c,d) Western blot of lysates from unstimulated B cells of the indicated genotypes, probed with (c) antibody to phosphorylated threonine 500 or (d) phophorylated threonine 642 in PKCβI. Blots in a, c and d were reprobed with β-actin to ensure equal loading. The results are representative of two independent experiments analyzing 3–4 mice of each genotype.
Following translation, both PKCβI and PKCβII undergo sequential phosphorylation at threonine and serine residues to become catalytically active, and these modifications can be revealed indirectly by slower mobility in SDS-PAGE and directly by phospho-specific antibodies21. When the mutant or wild-type PKCβI isoform was expressed in 293T cells, the Tilcara S552P mutation caused almost complete absence of the slower migrating PKCβI form although the more rapidly migrating species was present at comparable levels in soluble cytoplasmic component of the NP-40 lysed-transfected with mutant or wild-type vectors (Figure 2b). The S552P mutation had a similar effect when the PKCβII isoform was expressed in transfected 293T cells, although a small amount of slowly migrating protein was present in this case. The first post-translational modification step in the maturation of PKCβI and II, involving Thr500 phosphorylation in the kinase domain activation loop by phosphoinositide-dependent kinase-1 (PDK-1)22 was tested by Western blotting B cell lysates with an antibody to phospho-T500 (Figure 2c). Comparable amounts of phospho-T500 PKCβ were present in wild-type, heterozygous and Prkcbtil/til B cell lysates. This activation loop phosphorylation in turn allows PKCβI to auto-phosphorylate T642 in the turn motif and S660 in the hydrophobic motif of the alternatively spliced C-terminal tail, releasing the fully active enzyme into the cytosol23. Western blotting with phospho-T642 PKCβI antibody revealed a profound decrease in autophophorylated protein in B cells from homozygous Prkcbtil/til mutant mice and an intermediate deficit in heterozygotes (Figure 2d). Thus, the S552P mutation almost completely eliminates the fully active species of PKCβI in B cells, establishing that the Prkcbtil/til mutation diminishes PKCβ function both in situ in primary B cells and when the mutant protein is expressed on its own in a heterologous cell line.
Diminished basal serum IgM and IgG3 and T-independent antibody responses in Tilcara mouse
The semi-dominant biochemical effects of the Tilcara S552P PKCβ mutation defined above were accompanied by semi-dominant effects on antibody production to the TI-2 antigen, NP-Ficoll (Figure 3a). When NP-specific IgM and IgG3 were measured in a large cohort of animals seven days post immunization, Tilcara heterozygous mice made significantly less antibody than wild-type controls while Prkcbtil/til homozygotes made lower responses than heterozygotes. Likewise, in unimmunised age-matched mice there was a semi-dominant decrease in natural serum IgM antibody (Figure 3b), which decreased ~50% and ~75% in Tilcara heterozygous and homozygous mice, respectively, compared to wild-type controls. Basal serum IgG3 was also decreased ~75% in Tilcara heterozygous and homozygous mice.
Figure 3. Semi-dominant defects in TI-2 antibody but normal TD antibody responses in Tilcara.
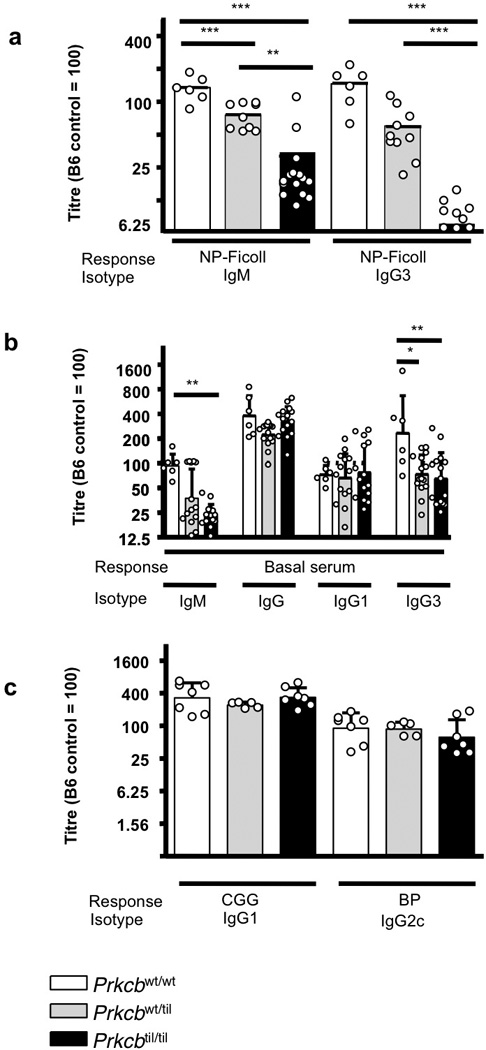
(a) NP-binding IgM and IgG3 antibody titres measured 6 days after immunization with the TI-2 antigen, NP-Ficoll. (b) Relative concentration of basal serum antibodies of the IgM, IgG, IgG1 and IgG3 isotypes. (c) IgG2ab and IgG1 isotypes TD antibody titres measured 15 days after primary immunization with Bordetella pertussis (BP) and alum precipitated chicken gamma globulin coupled to hapten arsonate (ABA-CGG), respectively. For a–c, relative antibody titres are shown, compared to the mean value in C57BL/6 control sera set at an arbitrary value of 100. Each dot represents one mouse, the bars represent the means for groups of Tilcara mice of the indicated genotypes, and error bars represent the standard deviations. *, ** and *** indicates a significant difference of P<0.05, P<0.01, P<0.001, respectively, calculated by The One-way ANOVA statistical test and Bonferroni post-test. The results are representative of three independent experiments.
Serum IgG1 and total IgG were nevertheless comparable between mutant and wild-type animals (Figure 3b). Likewise, there were normal T cell dependent antibody responses following immunization with alum precipitated chicken gamma globulin coupled to hapten arsonate (ABA-CGG) and heat killed Bordetella pertussis (BP), which depend on TH2 and TH1 cells to elicit CGG-specific IgG1 and BP-specific IgG2c response24. There were no significant differences in these antibody responses between wild-type mice and Tilcara heterozygous or homozygous animals (Figure 3c). The S552P mutation in Prkcb thus causes a selective defect in T-independent antibody responses.
Lymphocyte development and decreased B-1a cells in Tilcara mouse
B lymphocyte development and peripheral subsets were assessed in Tilcara mice by flow cytometry, with particular focus on B cells that are responsible for TI-2 antibody responses – marginal zone and B-1 cells. In the bone marrow, no significant differences in percentages of early pro-B/pre-B (B220loIgMlo), immature (B220loIgMhi) and mature (B220hiIgMhi) B cells could be detected between wild-type, Prkcbwt/til, Prkcbtil/ti mice (Figure 4a). Similarly, there was no discernable effect of the mutation on transitional 1 (CD93hiIgMloCD23lo), transitional 2 (CD93hiIgMhiCD23hi), transitional 3 (CD93hiIgMloCD23hi), follicular (CD93loCD23hiCD21int) and marginal zone (CD93loCD23loCD21int) B cell subpopulations in the spleen (Figure 4b). Cell surface IgM was downregulated normally during maturation of follicular B cells in Tilcara homozygotes: a result that stands in contrast to mutations in BTK25 or PLCγ226 immediately upstream of PKCβ or with mutations in CARD1127 immediately downstream, which cause surface IgM to remain high on follicular B cells. T cell subsets in the thymus, lymph nodes and spleen were also comparable (data not shown). The only striking abnormality in lymphocyte development was in the peritoneal cavity, where CD11bhiIgMhi B-1 cells were markedly decreased in frequency selectively in Prkcbtil/til homozygous mice compared with wild-type and Prkcbwt/til heterozygotes (Figure 4c). Sub-division of the B-1 cells into CD5+ B-1a and CD5− B-1b subpopulation revealed that this difference was attributed largely to the near absence of the B-1a population in homozygous mutant animals (Figure 4c).
Figure 4.

A representation of flow cytometric plots and percentages from Prkcbwt/wt, Prkcbwt/tiland Prkcbtil/til mice for (a) Pro-B/pre-B (B220intIgMint), immature (B220intIgMhi) and mature (B220hiIgMhi) B cell populations from total lymphocytes for bone marrow cell suspensions. (b) Transitional 1 (B220hiCD93hiIgMloCD23lo), transitional 2 (B220hiCD93hiIgMhiCD23hi), transitional 3 (B220hiCD93hiIgMloCD23hi), follicular (B220hiCD93loCD23hiCD21int) and marginal zone (B220hiCD93loCD23loCD21int) splenocytes. (c) B-1 cell (IgMhiCD11bhi) population in the peritoneal cavity. The histograms show the B-1a (CD5hi) and B-1b (CD5lo) cell populations The graphs show the percentages of cells from total lymphocytes. *, ** and *** indicates a significant difference of P<0.05, P<0.01, P<0.001, respectively, calculated by The One-way ANOVA statistical test and Bonferroni post-test. Results from a–c representative of three independent experiments.
Competitive mixed bone marrow chimeras were produced by transplanting irradiated mice with equal amounts of bone marrow from CD45.1 wild-type and CD45.2 Prkcbtil/til donors. Analysis of the reconstituted chimeric animals showed that the Prkcbtil/til mutation did not affect the numbers of B and T cell subsets in central and peripheral lymphoid tissue, which were drawn equally from mutant and wild-type lymphocytes. By contrast, peritoneal B-1a cells and to a lesser extend marginal zone cells were preferentially derived from the wild-type lymphocytes and not from those harbouring the Prkcbtil/til mutation, demonstrating an intrinsic, cell-autonomous requirement for fully active PKCβ selectively in the formation of B1 B cells (Supplementary Table 1 and supplementary Figure 1).
Recessive deficits in the acute BCR-response
We next analysed the consequences of the S552P Prkcbtil mutation on B cell responses to receptor stimulation in vitro. Increased intracellular calcium after BCR stimulation was monitored using the calcium-sensing dye, Indo-1, in co-cultured CD45.1 wild-type and CD45.2 Prkcbtil/til cells stimulated with sub-optimal and optimal concentrations of anti-IgM antibody (Figure 5a). By overlaying the paired responses from mutant and wild-type cells stimulated together, there was no discernable difference in the intracellular calcium response between wild-type and Prkcbtil/til cells. Thus, the early steps in BCR signalling leading to Plcγ activation, which lies immediately upstream of PKCβ, appeared unaffected by the Prkcbtil mutation.
Figure 5. Cell autonomous effects of the Tilcara mutation on acute B cell response to BCR crosslinking.

(a) Intracellular calcium response in mixtures of B cells from Prkcbwt/wt CD45.1 and Prkcbtil/til CD45.2 mice in response to optimal (50 µg/mL) and sub-optimal (5 and 0.5 µg/mL) concentrations of anti-IgM antibody introduced at t=30 s. The vertical axis shows the geometric mean fluorescence ratio of calcium-bound Indo-1 (395 nm, FL5) over un-bound Indo-1 (495 nm, FL4). Paired responses of CD45.1 and CD45.2 cells are overlaid for each anti-IgM dose. The results are representative of three independent experiments. (b and c) Induction of CD25 on homozygous or heterozygous CD45.2+B220+ cells (open histograms) co-cultured with CD45.1 wild-type B cells (shaded histograms) for 16 hrs in the presence of the indicated stimuli. The mean % positive cells in each culture are shown. The results are representative of two independent experiments with n = 5 per group.
BCR, CD40 or TLR signalling induces an early wave of gene transcription in B cells that includes the cell surface protein, CD25. CD25 induction by the BCR but not CD40 or TLR4 is compromised in B cells with mutations in Card1127, a known immediate substrate of PKCβ28, 29. Flow cytometric analysis showed that CD25 was induced by anti-IgM stimulation of wild-type B cells but poorly induced in homozygous mutant Prkcbtil/til B cells stimulated in the same culture (Figure 5b and c). By contrast, CD25 was induced equivalently in mutant and wild-type B cells stimulated with 1 µg/ml IgM + 10µg/ml CD40L or the TLR4 agonist, LPS. Unlike the homozygous mutant B cells, Tilcara heterozygous B cells exhibited no discernable decrease in CD25 induction following anti-IgM stimulation (Figure 5b and c).
Entry of B cells into cell cycle was measured by incorporation of radioactive thymidine after 2 days in culture. Spleen cells from individual homozygous Prkcbtil/til mice had markedly diminished thymidine incorporation in response to BCR-stimulation, whereas their response to LPS (TLR4), CpG (TLR9) or antibody to CD3 was normal (Figure 6a–d). Heterozygous Prkcbtil/+ B cells did not have significantly reduced DNA incorporation upon anti-IgM stimulation. The combination of anti-IgM and anti-CD40 stimulation partially restored the initiation of proliferation in Prkcbtil/til cells (Figure 6e). Measurement of B cell division by dilution of the intracellular dye, CFSE, yielded similar results and enabled cell autonomous effects of the Prkcb mutation to be confirmed by comparing CFSE dilution in co-cultured CD45.1 normal B cells 5 days post stimulation. Few homozygous mutant B cells had divided multiple times in response to anti-IgM, while the percentage of divided Tilcara heterozygous B cells was significantly lower than in wild-type controls (Figure 6f and g). Stimulation with anti-IgM and CD40 greatly enhanced the fraction of wild-type B cells that had divided multiple times, but only modestly increased the response of homozygous mutant B cells, while the response of heterozygous mutant B cells was clearly intermediate. Collectively, these results indicate that the semi-dominant effects of Prkcb mutation on BCR-induced proliferation are difficult to discern during the entry into cycle but become more apparent when measured after several cell divisions. This may reflect either less re-initiation of subsequent cell divisions in heterozygous mutants or diminished survival of their divided progeny compared to the wild-type B cells in the same culture.
Figure 6. Effects of Tilcara mutation on induction of B cell proliferation.

(a–e) Mean (± s.d.) 3H-thymidine incorporation in triplicate cultures of spleen cells from five individual mice of the indicated genotypes, 48 hrs after stimulation with the indicated agonists. Representative of two independent experiments. (f) Cell division measured by dilution of CFSE in wild-type, heterozygous and homozygous CD45.2 Prkcbtil/til B cells (open histograms) co-cultured with CD45.1 wild-type B cells (shaded histograms) for 5 days with the indicated stimuli. Histograms are gated on B220+ CD45.2 or CD45.1 cells. (g) The mean percentage of divided CD45.2 cells. The mean±s.d. for the percentage of all B cells that were CD45.2+ in unstimulated cultures are: 70.8±7.7% and 41.8±17.2% for for Prkcbwt/til (n = 5) and Prkcbtil/til (n = 5) respectively; for 10µg/mL anti-IgM: 66.6±10.5% and 29.4±15.0%; and for 1µg/mL anti-IgM+10 µg/mL CD40L 89.6±3.7% and 65.9±8.7%.
Discussion
The results above represent a genome-wide experimental approach to identify rate-limiting steps in TI-2 antibody production that are particularly sensitive to heterozygous mis-sense mutations. Two independent, semi-dominant mutations were revealed, Tilcara and Untied, both changing individual amino acids within the catalytic domain of PKCβ. Semi-dominant loss of TI-2 antibody was also reported in Prkcb knockout mice20. The findings identify Prkcb as a key genetic node with a gene-dosage sensitive role selectively in BCR-induced cell division and TI-2 antibody responses. In contrast with these semi-dominant effects on TI-2 antibody responses, formation of B1a B cells in the peritoneal cavity and basal production of serum IgM and IgG3 were only diminished in Tilcara homozygous mutants. Likewise, early responses to BCR-stimulation such as CD25 induction and initiation of DNA synthesis were only measurably diminished in homozygotes, whereas the fraction of cells that had divided multiple times was decreased to an intermediate degree in heterozygotes.
In Tilcara, the Ser552>Pro mutation differential affected the alternatively spliced PKCβI and PKCβII isoforms. The active, autophosphorylated PKCβI species was preferentially diminished and the overall amount of PKCβI protein was greatly decreased in homozygotes and markedly decreased in heterozygotes, whereas the overall amount of PKCβII protein was not markedly reduced. More complete loss of autophosphorylated PKCβI than PKCβII was also observed when tagged versions of the proteins were transiently expressed in 293 cells. Preferential loss of autophosphorylated PKCβI has several possible explanations. Ser552 lies in helix G of the catalytic domain, which forms a lower lip on the catalytic site and contains a series of acidic residues, EDEDELFQS552, that interact with basic Arg or Lys residues in the pseudosubstrate inhibitory domain that lies at the PKCβ N-terminus30–32. Serine552 is absolutely conserved in vertebrates, and its substitution by proline would be predicted to break the alpha-helical structure and diminish binding to the pseudosubstrate inhibitory domain and to kinase substrates. Decreased substrate binding may diminish autophosphorylation to generate the fully active species, while decreased binding to the pseudosubstrate domain may accelerate dephosphorylation and degradation of the kinase. When the pseudosubstrate domain is allosterically dislodged from Helix G upon PKCβ binding to membrane lipids, this opening up is rapidly followed by dephosphorylation of the kinase and, in the case of PKCβI, rapid protein degradation33. By contrast, PKCβII degradation after dephosphorylation occurs more slowly, and this may explain the selective loss of PKCβI in Tilcara B cells.
The resulting semi-dominant immunological effects of the Tilcara mutation favour the view that catalytically active PKCβI is the critical enzyme isoform for progressive B cell division in response to BCR-crosslinking and for TI-2 antibody formation. This genetic inference is supported by biochemical evidence that PKCβI preferentially associates with other essential proteins for BCR-induced NFκB signalling and proliferation, in particular BTK and CARD1129, 34.
The immune phenotype of Tilcara homozygotes had a number of significant differences compared to knockout mice that completely lack both PKCβ isoforms. PKCβ-null mice had defects in both TI-2 and TD antibody responses20, whereas Tilcara homozygous mice had low TI-2 antibody responses but normal TD antibody responses. PKCβ-null mice also had fewer mature IgMloIgDhi follicular B cells in the spleen35 and fewer mature follicular B cells that had recirculated to the bone marrow36, whereas the numbers of both subsets were normal in Tilcara homozygotes. There are two plausible explanations for the different phenotypes. Firstly, the Prkcbtil/til mutation may result in the expression of small amounts of catalytically competent PKCβI kinase with residual activity. This activity may be insufficient for the stimulation of TI-2 antibody responses but sufficient for TD responses and mature B cell survival. TI-2 antibody responses are highly dependent on BCR cross-linking and stimulation via the BCR receptor8. In contrast, the TD antibody response involves other mitogenic signals such as CD40 ligand37, that may activate complementary signalling pathways and kinases that counteract the partial defect in PKCβI kinase activity. This hypothesis is also supported by the observation that addition of CD40L to the anti-IgM stimulation was able to normalize upregulation of CD25 and helped at least partially to overcome the defective proliferation in response to anti-IgM alone. Alternatively, there may be a division of labor between the PKCβI and PKCβII isoforms, where the PKCβI isoform is involved in TI-2 antibody response, whereas the PKCβII isoform that was less compromised in Tilcara is involved in TD antibody production and mature B cell survival. Support for this possibility comes from the observation that PKCβI and PKCβII play different roles in the insulin receptor signalling pathway38. It would be interesting in future studies to engineer mice that selectively lack PKCβI protein entirely while retaining normal PKCβII.
Heterozygous mis-sense mutations, like those in Tilcara and Untied, have accumulated in the current human population at a massive scale because of exponential population growth and outbreeding12–16. Whereas offspring of ENU-mutagenized mice inherit ~40 novel mis-sense variants, a “normal” current human genome inherits more than 5 000 mis-sense variants including 300–600 that are predicted to be deleterious to protein function and 75–150 that represent rare, recent deleterious mutations yet to be subject to purifying selection16. Hence, the finding of two heterozygous missense Prkcb mutations that reduce TI-2 antibody responses amongst several hundred ENU mouse pedigrees can be extrapolated to predict at least a similar ~1% frequency in human families. Indeed, recent exome sequence data from 2 440 individuals16 annotates 21 independent mis-sense mutations in PRKCB including many in the kinase domain (http://evs.gs.washington.edu/EVS/). It will important to test if PRKCB is equally gene-dosage sensitive in human B cells. Extrapolating from the mouse experiments here, and from evidence that ~20% of missense mutations are profoundly crippling39, of the 1% of people carrying unique PRKCB variants at least 20% of these individuals (0.2% of the population) would be expected to make less antibody to polysaccharide antigens of enveloped bacteria but exhibit normal circulating B cell subsets, serum IgG, TD antibody to tetanus toxoid and MMR vaccination. One could readily envisage additive or epistatic interactions between heterozygous mutations in PRKCB and in other rate-limiting genes for BCR-induced proliferation, as we have shown occurs between heterozygous mutations in Lyn, Cd22 and Ptpn6 encoding interacting negative regulators of BCR signalling40. These experimental results, coupled with human exome sequencing, thus open up a new way to analyse variability in humoral immunity in the wider population, among individuals that would not be defined as having a primary immunodeficiency.
Materials and methods
ENU-treatment of mice
8–15 week old male C57BL/6 mice were treated with three doses of 100 mg/kg N-ethyl-N-nitrosurea (Sigma) prepared in 10% ethanol, citrate buffer (pH5.0), one week apart, as previously described 41. The mice were housed in specific pathogen free conditions at the Australian National University Bioscience Facility or the Scripps Research Institute vivarium. All animal procedures were approved by the Australian National University Animal Ethics and Experimentation Committee or the Institutional Animal Care and Use Committee of the Scripps Research Institute.
Immunization and enzyme-linked immunosorbent assay
Mice were immunized by intraperitoneal injections. For TI-2 immune response, the mice received 25 µg NP-Ficoll (Biosearch Technology) in sterile PBS. For TD responses, mice received 50 µg CGG with hapten arsonate (Biosearch Technology) and 5×108 whole killed Bordetella pertussis (Lee Labs, Becton Dickinson). Total and antigen specific antibodies in serially diluted sera were detected by enzyme-linked immunosorbent assay on Maxisord plates coated with CGG, sonicated B. pertussis or NP-Ficoll. Bound antibodies were detected with alkaline phosphate antibodies to specific mouse isotypes using the phosphatase substrate system. Plates were read at 405 to 650 nm using a Thermomax microplate reader. The titres for serum samples were calculated as the difference of serum concentrations required to achieve 50% maximum optical density compared to a standard control ran on each plate.
Typing of mutation
Candidate mRNAs were amplified in 300–800 bp fragments using polymerase chain reactions from spleen cDNA and resequenced by the Biomolecular Resource Facility (JCSMR, ANU) using the ABI 3730 capillary sequencer and analysed using the Sequencher 4.2 software. The primers for Prkbc were:
PrkcbA forward primer: 5' CGCGCAAGATGGCTGACC 3'
PrkcbA reverse primer: 5' CGATGTGGGCCTGGATATAG 3'
PrkcbB forward primer: 5' AAGCGCTGCGTGATGAAC 3'
PrkcbB reverse primer: 5' TGGCCAATCTTGGCTCTC 3'
PrkcbC forward primer: 5' TGTGGATGGCTGGTTCAA 3'
PrkcbC reverse primer: 5' AATACAGCATGGGGCTCCT 3'
PrkcbD forward primer: 5' GCTCCATTCCTGCTTCCA 3'
PrkcbD reverse primer: 5' CTTGCCTGGGTGTTTGGT 3'
PrkcbE forward primer: 5' CCGTTTGAAGGGGAGGAT 3'
PrkcbE reverse primer: 5' CATGGCCACTCTAGCCTCA 3'
Cell isolation and flow cytometry
Spleens and thymi were harvested from mice and single cells suspensions were prepared by sieving and gentle pipeting through a 70 µm nylon mesh filter. Bone marrow cells were flushed out from tibias and femurs, which were aseptically cut at both ends. Peritoneal cells were collected by abdominal lavage. Blood was collected by retro-orbital bleeds from live mice. For surface straining, samples were stained with the appropriate primary and secondary antibodies. A BD LSRII Benchtop Flow Cytometer with FACS Diva was used for acquisition of flow cytometric data, and Flowjo (Treestar, Inc.) was used for analysis.
Antibodies
Antibodies used for flow cytometry were from BD unless other wise indicated: 7-Amino-actinomycin D (Calbiochem), anti-mouse CD11b-APC/FITC (eBioscience), anti-mouse CD16/CD32 (Stemcell Technologies), anti-mouse CD1d-PE, anti-mouse CD21/CD35-FITC/PE, anti-mouse CD23-PE Cy7 (eBioscience), anti-mouse CD25-APC, anti-mouse CD4-FITC/PE Cy7, anti-mouse CD43-PE, anti-mouse CD45.1-biotin/FITC, anti-mouse CD45.1-Alexa Flour 700 (BioLegend), anti-mouse CD45.2-Biotin/PE, anti-mouse CD45.2-Pecific blue (BioLegend), anti-mouse CD45R-Alexa Fluor 405/APC-Cy7/FITC/PE-Cy7/PerCP, anti-mouse CD5-PE, anti-mouse CD62L-PE, anti-mouse CD69-FITC/PE Cy7, anti-mouse CD8- Alexa Fluor 700, PerCP Cy5.5 (eBioscience), anti-mouse CD86-APC/PE, anti-mouse CD93-APC (eBioscience), anti-mouse IgM-APC/FITC/PE Cy5.5, anti-mouse TCR beta- APC-Alexa Fluor 750 (eBioscience), streptavidin-PE/ PerCP/APC and streptavidin-Qdot 605 (Invitrogen). Antibodies used for ELISA: goat anti-mouse IgG/IgG1/IgG3/IgM-alkaline phosphate (Southern Biotechnology), goat anti-mouse IgG2ab-biotin (Southern Biotechnology) and strepavidin-alkaline phosphate (Vector).
Bone marrow chimeras
All recipients were sub-lethally irradiated with two doses of 450 rads and reconstituted via intravenous injections with 2×106 bone marrow cells.
In vitro cell stimulation assays
Goat anti-IgM F(ab')2 (Jackson Immunoresearch Laboratories), anti-CD40 (Clone 1C10), anti-TCR (Clone H57–597), bacterial DNA (CpG) (Geneworks) and lipopolysaccharide, from E.coli (Sigma) were used as agonist for stimulation. All cell stimulations were done in RPMI supplemented with 10% FCS, 50 units/mL penicillin/streptomycin, 10mM HEPES, 10mM sodium pyruvate, 10mM non-essential MEM and 55 µM β-mercaptoethanol. For DNA synthesis assay, 2×106 cells were cultured for 48 hours in 96-well flat bottom plates, before pulsing with 1 µCi [3H]thymidine for 8 hours. Intracellular calcium was measured on an LSR flow cytometer following labelling with Indo-1 (Molecular Probes). For labelling with CFSE, cells were diluted to a concentration of 20×106 cells/mL in PBS and stained with a final concentration of 2.5 µM CFSE at 37°C for 10 minutes. Following two washed with cold RPMI, 2×106 cells were cultured for 48 hours in 96-well flat bottom plates with appropriate agonist for 5 days.
Western blotting
For B cell purifications, red-cell depleted splenocytes were incubated with biotinylated antibodies to CD43 (BD) and B cells were negatively selected using strepavidin MACS bead separation (Miltenyi Biotech). Cell lysates were subjected to SDS-PAGE with a 12.5% gel using the BioRad Mini-Protean II system. Nitrocellulose membranes (Trans-blot BioRad) and the BioRad Mini-Protean II system was used for transfer according to the manufacturers instructions. Western blotting using the following antibodies: donkey anti rabbit IgG F(ab')2-peroxidase (GE Healthcare), goat anti mouse IgG F(ab')2-peroxidase (GE Healthcare), mouse anti beta-actin monoclonal antibody (Sigma), rabbit anti PKC beta 1 polyclonal antibody (Sapphire Biosciences and Cell Signalling), rabbit anti PKC beta 2 phosphorylated at T642 polyclonal antibody (Sapphire Biosciences) and rabbit anti PKC beta 2 polyclonal antibody (Sapphire Biosciences and Cell Signalling). Blots were developed using the Western Lightning Western Blot Chemiluminescence Reagent (Perkin Elmer).
Bioinformatics
The following bioinformatic tools were used: Boxshade 3.2.1 (http://www.ch.embnet.org:80/software/BOX_form.html), ClustalW (http://www.ebi.ac.uk/clustalw/), Cn3D (http://0-www.ncbi.nlm.nih.gov.ilsprod.lib.neu.edu/Structure/CN3D/cn3d.shtml), ENSEMBL Genome Browser (http://www.ensembl.org/index.html), Mouse Genome Informatics (http://www.informatics.jax.org/) and Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi).
Statistical analysis
Data were analysed using the One-way ANOVA and Bonfferonni post-test to compare pairs of columns unless otherwise stated, using the GraphPad prism software. Results were considered significant if P<0.05.
Supplementary Material
{kind=link}
Acknowledgements
We thank the staff of the ANU Bioscience Services, the Scripps Research Institute vivarium, the Australian Phenomics Facility, and the JCSMR Flow Cytometry Facility for expert technical services. This work was supported by grants from the Clive and Vera Ramaciotti Foundation, NIH, Wellcome Trust and the NHMRC.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Scher I. The CBA/N mouse strain: an experimental model illustrating the influence of the X-chromosome on immunity. Adv Immunol. 1982;33:1–71. doi: 10.1016/s0065-2776(08)60834-2. [DOI] [PubMed] [Google Scholar]
- 2.Moresco EM, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21(13):R488–R493. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 3.Brunswick M, Finkelman FD, Highet PF, Inman JK, Dintzis HM, Mond JJ. Picogram quantities of anti-Ig antibodies coupled to dextran induce B cell proliferation. J Immunol. 1988;140(10):3364–3372. [PubMed] [Google Scholar]
- 4.Dintzis RZ, Middleton MH, Dintzis HM. Studies on the immunogenicity and tolerogenicity of T-independent antigens. J Immunol. 1983;131(5):2196–2203. [PubMed] [Google Scholar]
- 5.Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 6.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21(3):379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23(1):7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Mond JJ, Vos Q, Lees A, Snapper CM. T cell independent antigens. Curr Opin Immunol. 1995;7(3):349–354. doi: 10.1016/0952-7915(95)80109-x. [DOI] [PubMed] [Google Scholar]
- 9.Carr TF, Koterba AP, Chandra R, Grammer LC, Conley DB, Harris KE, et al. Characterization of specific antibody deficiency in adults with medically refractory chronic rhinosinusitis. American journal of rhinology & allergy. 2011;25(4):241–244. doi: 10.2500/ajra.2011.25.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faden H. The microbiologic and immunologic basis for recurrent otitis media in children. European journal of pediatrics. 2001;160(7):407–413. doi: 10.1007/s004310100754. [DOI] [PubMed] [Google Scholar]
- 11.Kaur R, Casey JR, Pichichero ME. Serum antibody response to three non-typeable Haemophilus influenzae outer membrane proteins during acute otitis media and nasopharyngeal colonization in otitis prone and non-otitis prone children. Vaccine. 2011;29(5):1023–1028. doi: 10.1016/j.vaccine.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coventry A, Bull-Otterson LM, Liu X, Clark AG, Maxwell TJ, Crosby J, et al. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nature communications. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, et al. Demographic history and rare allele sharing among human populations. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(29):11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keinan A, Clark AG. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science (New York, N.Y. 2012;336(6082):740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, et al. An Abundance of Rare Functional Variants in 202 Drug Target Genes Sequenced in 14,002 People. Science (New York, N.Y. 2012 doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science (New York, N.Y. 2012 doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science (New York, N.Y. 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vinuesa CG, Goodnow CC. Illuminating autoimmune regulators through controlled variation of the mouse genome sequence. Immunity. 2004;20(6):669–679. doi: 10.1016/j.immuni.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Nelms KA, Goodnow CC. Genome-wide ENU mutagenesis to reveal immune regulators. Immunity. 2001;15(3):409–418. doi: 10.1016/s1074-7613(01)00199-6. [DOI] [PubMed] [Google Scholar]
- 20.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, et al. Immunodeficiency in protein kinase cbeta-deficient mice. Science. 1996;273(5276):788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- 21.Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101(8):2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 22.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Curr Biol. 1998;8(25):1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 23.Behn-Krappa A, Newton AC. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr Biol. 1999;9(14):728–737. doi: 10.1016/s0960-9822(99)80332-7. [DOI] [PubMed] [Google Scholar]
- 24.Toellner KM, Luther SA, Sze DM, Choy RK, Taylor DR, MacLennan IC, et al. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. The Journal of experimental medicine. 1998;187(8):1193–1204. doi: 10.1084/jem.187.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3(3):283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 26.Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13(1):25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 27.Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18(6):751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 28.Shinohara H, Maeda S, Watarai H, Kurosaki T. IkappaB kinase beta-induced phosphorylation of CARMA1 contributes to CARMA1 Bcl10 MALT1 complex formation in B cells. The Journal of experimental medicine. 2007;204(13):3285–3293. doi: 10.1084/jem.20070379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity. 2005;23(6):561–574. doi: 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 30.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science (New York, N.Y. 1987;238(4834):1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 31.House C, Robinson PJ, Kemp BE. A synthetic peptide analog of the putative substrate-binding motif activates protein kinase C. FEBS letters. 1989;249(2):243–247. doi: 10.1016/0014-5793(89)80632-5. [DOI] [PubMed] [Google Scholar]
- 32.Orr JW, Newton AC. Intrapeptide regulation of protein kinase C. The Journal of biological chemistry. 1994;269(11):8383–8387. [PubMed] [Google Scholar]
- 33.Hansra G, Garcia-Paramio P, Prevostel C, Whelan RD, Bornancin F, Parker PJ. Multisite dephosphorylation and desensitization of conventional protein kinase C isotypes. The Biochemical journal. 1999;342(Pt 2):337–344. [PMC free article] [PubMed] [Google Scholar]
- 34.Kawakami Y, Kitaura J, Hartman SE, Lowell CA, Siraganian RP, Kawakami T. Regulation of protein kinase CbetaI by two protein-tyrosine kinases, Btk and Syk. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(13):7423–7428. doi: 10.1073/pnas.120175097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patke A, Mecklenbrauker I, Erdjument-Bromage H, Tempst P, Tarakhovsky A. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. The Journal of experimental medicine. 2006;203(11):2551–2562. doi: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, et al. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nature immunology. 2002;3(8):780–786. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 37.Eris JM, Basten A, Brink R, Doherty K, Kehry MR, Hodgkin PD. Anergic self-reactive B cells present self antigen and respond normally to CD40-dependent T-cell signals but are defective in antigen-receptor-mediated functions. Proc Natl Acad Sci U S A. 1994;91(10):4392–4396. doi: 10.1073/pnas.91.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sampson SR, Cooper DR. Specific protein kinase C isoforms as transducers and modulators of insulin signaling. Mol Genet Metab. 2006;89(1–2):32–47. doi: 10.1016/j.ymgme.2006.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold CN, Barnes MJ, Berger M, Blasius AL, Brandl K, Croker B, et al. ENU-induced phenovariance in mice: inferences from 587 mutations. BMC research notes. 2012;5:577. doi: 10.1186/1756-0500-5-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity. 1998;8(4):497–508. doi: 10.1016/s1074-7613(00)80554-3. [DOI] [PubMed] [Google Scholar]
- 41.Miosge LA, Blasioli J, Blery M, Goodnow CC. Analysis of an ethylnitrosourea-generated mouse mutation defines a cell intrinsic role of nuclear factor kappaB2 in regulating circulating B cell numbers. J Exp Med. 2002;196(8):1113–1119. doi: 10.1084/jem.20020959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.