

Abstract
Background
Myocardial contractile depression develops 4–24 hrs after major burn injury. We have reported previously that in a rat burn injury model (≈40% of total body surface area burn), mesenteric lymph duct ligation (LDL) prior to burn prevented myocardial dysfunction. However, the underlying cellular and molecular mechanisms are not well understood.
Materials and Methods
Left ventricular myocytes were isolated from sham burn (control), sham burn with LDL (sham+LDL), burn, and burn with LDL (burn+LDL) rats at 4 and 24 hrs after burn or sham burn. Electrophysiological techniques were used to study myocyte size, contractility and L-type Ca2+ channel current (ICa). Further studies examined changes in the mRNA expression levels of pore-forming subunit of the L-type Ca2+ channel, α1C and its auxiliary subunits, β1, β2, β3 and α2δ1 which modulate the abundance of the ICa in post burn hearts
Results
Depressed myocyte contractility (≈20%) developed during 4–24 hrs post-burn compared with control, sham+LDL or burn+LDL groups, a pattern of changes consistent with whole heart studies. There was no significant alteration in myocyte size. The ICa density was significantly decreased (≈30%) at 24 hrs post-burn whereas the mRNA expression levels of Ca2+ channel gene were not significantly altered at 4 and 24 hrs after burn injury.
Conclusions
These results suggest that the post-burn contractile phenotype in vivo was also present in isolated myocytes in vitro, but cellular remodeling was not a major factor. The results also suggest that changes in ICa regulation, but not from Ca2+ channel gene modification may be a key element involved in post-burn contractile depression and the beneficial effects of LDL.
Keywords: Burn injury, cardiac myocyte, L-type Ca2+ channel, mesenteric lymph ligation
Introduction
Clinical and experimental evidence shows that major burn injury causes significantly impaired left ventricular contractile function despite fluid resuscitation to replace or exceed intravascular fluid loss [1–3]. The time-course of changes of cardiac function in clinical and experimental studies have shown that myocardial contractile depression develops 4–24 hrs post-burn [3, 4]. Although the pathogenesis responsible for burn-induced myocardial dysfunction is not well known, the concept that the gastrointestinal tract (i.e., gut) plays a pivotal role in multiple organ dysfunction following a variety of shock states, including burn injury is now well accepted [5–7]. Furthermore, there is increasing evidence that gut-derived factors transported in mesenteric lymph, but not the portal venous system, contribute to the development of post shock organ dysfunction including lung and cardiovascular systems [8–11]. Consistent with these studies, we have reported previously that lymph diversion by mesenteric lymph duct ligation (LDL) prior to burn injury prevented myocardial contractile dysfunction [12]. However, the mechanisms responsible for burn-induced myocardial contractile dysfunction and preventive effects of LDL have not been well understood.
There is not substantial direct evidence for a causative role of myocyte contractile depression in the initiation of post-burn myocardial dysfunction. For example, alterations within myocardium such as myocardial edema and histological changes could result in cell death. Loss of myocyte through apoptosis leads to negative inotropic effects with compensatory morphometric changes with hypertrophy of the remaining myocytes [13–15]. Although signs of apoptosis have been shortly after severe burn [16], isolated myocyte size associated with altered cardiac function and the effects of LDL have not been investigated.
To improve our understanding of the mechanisms underlying the burn-induced acute cardiac dysfunction, we first examined myocyte contractility at 4 and 24 hrs after burn injury to test whether changes are associated with cardiac dysfunction in vivo. Cardiac contraction is activated by Ca2+ release from the sarcoplasmic reticulum (SR), triggered by a rapid transsarcolemmal Ca2+ influx through the L-type Ca2+ channels (ICa). It is also the major source of Ca2+ loading to the SR [14, 17]. Therefore, an abnormal ICa density or regulation in post-burn myocardium could have profound effects of cardiac contractile function and may explain the beneficial effects of LDL. Accordingly, the specific aims of this study were: (1) whether changes in myocyte size are associated with the development of burn-induced myocardial depression? (2) whether depressed contractility in vivo is associated with cardiac myocyte contractile depression? (3) whether L-type Ca2+ channel current density (Peak ICa amplitude, normalized relative to cell size) is altered during the development of myocardial contractile dysfunction? Morphology of myocytes was examined by measuring cell length and cell capacitance (which is proportional to the cell surface area) using whole-cell patch-clamp technique.
Based on biochemical purification, molecular cloning and functional studies, L-type Ca2+ channels are heteromultimers, composed of the principal pore-forming α1C and auxiliary β (β1–β4) and α2δ (α2δ1–α2δ4) subunits. The α1C subunit forms the pore of the Ca2+ channel and specifies basic voltage-dependent characteristics [18, 19]. The auxiliary subunits modulate the biophysical properties and/or trafficking of the α1C subunit to increase Ca2+ influx.[20] Cardiac tissues express β1, β2, β3 and α2δ1 subunits which have been shown to modulate ICa amplitude and gating [20, 21]. However, until now there have been no report which examined changes in Ca2+ channel gene expression and Ca2+ channel current function in post-burn hearts. Therefore, additional studies were performed to examine whether changes in α1C, β1, β2, β3 or α2δ1 mRNA expression levels were altered at 4 and 24 hrs after burn injury.
Materials and Methods
Changes in myocyte size, function and gene expression were investigated at 4 and 24 hrs after burn injury because the majority of studies on burn injury have reported that acute myocardial contractile dysfunction develops during the 4–24 hrs post-burn period [3, 4]. Furthermore, mesenteric lymph generated 1–4 hrs after the burn injury exhibits the highest level of biological activity [22], suggesting the association between a mesenteric lymph-mediated events and myocardial dysfunction.
Animals
Male Sprague Dawley rats (250–350g) were used in this study. The animals were maintained in accordance with the rules of the New Jersey Medical School Animal Care and Use Committee approved the experiments.
Burn injury model and mesenteric lymph duct ligation (LDL)
The procedures used to induce burn injury were similar to those described by Walker and Mason [23]. Briefly, the rats were anesthetized with pentobarbital sodium (50 mg/kg) and a 40% total body surface area (TBSA) scald burn was induced by immersing the back of the animal through a template into boiling water (100°C) for 10 seconds following which an abdominal burn was induced by immersion for 5 seconds [24]. The sham-burned rats were anesthetized, placed in the plastic template, and immersed in room temperature water.
Mesenteric lymph duct ligation (LDL) was performed on anesthetized rats immediately before sham or burn injury as previously described [12].
Left ventricular myocyte isolation, measurements of contraction and Patch-clamp
Single left ventricular myocytes were isolated with techniques described previously.[24]
Cell contraction (% cell shortening) was measured by video edge detection [24]. Myocytes were perfused with Tyrode solution (mmol/L): NaCl, 120; KCl, 2.6; CaCl2, 1.0; MgCl2, 1.0; glucose, 11; HEPES, 5 (pH 7.3) at 32°C. Myocytes were field stimulated at 1.0 Hz.
Whole-cell patch clamp studies were performed as described previously [24]. Cell capacitance was measured using voltage ramps of 0.8V/s from a holding potential of −50 mV. ICa was recorded in external solution containing (mmol/L) CaCl2, 2; MgCl2, 1; tetraethyl ammonium chloride, 135; 4-aminopyridine, 5; glucose, 10; HEPES, 5 (pH 7.3). The pipette solution contained (mmol/L) Cs-aspartate, 100; CsCl, 20; MgCl2, 1; MgATP, 2; GTP, 0.5; EGTA, 5 and HEPES, 5 (pH 7.3).
RNA measurement
Left ventricular tissue was rapidly dissected from rats and immediately frozen on dry ice. Total RNA was isolated by a one-step extraction with acid-phenol guanidium thiocyanate, followed by a column-based purification (RNeasy, Invitrogen Carlsbad, CA). The concentration of RNA was spectrophotometically determined using A260=40 μg/ml.
First-strand cDNA was synthesized with d(T)20 using thermostable reverse transcriptase at 50°C according to the manufacture’s instruction (Thermoscript RT-PCR system, Invitrogen Carlsbad, CA). Primers for Ca2+ channel subunits were designed to avoid the generation of a band from genomic contamination at the same size. Due to variation in sample-to-sample loading, glyceraldehydes-3-phosphate dehydrogenase (GAPDH) mRNA was used as housekeeping gene for normalization [25]. For detection, PCR was performed under the following conditions: denature at 95°C for 5 seconds, annealing at 58 or 60°C for 5 seconds and extension at 72°C for 1 minute for 16–25 cycles, with the final extension at 72°C for 4 minutes. A SYBR green dye-based method was used to quantify mRNA levels with a commercial master mix (Applied Biosystem, Foster, CA) using an Opticon DNA engine (MJ Research, Waltham, MA). Complementary DNA prepared without reverse transcriptase was used as a negative control, whereas a common RNA preparation prepared from normal adult rat hearts was used as a positive control to account for the deviations in individual experiments. Semi-quantitative measurement of standard PCR bands was also performed using a CCD camera-based system as described previously [26].
Statistics
Data are reported as mean values ± SE. Between groups/conditions analyses were conducted by using a Student’s t-test. Differences among multiple groups were tested with analysis of variance (ANOVA) followed by Bonferroni post hoc testing. A value of P<0.05 considered to be significant.
Results
Myocyte size
The yield of quiescent, regularly striated, viable cells was not different for myocytes isolated from control and post-burn hearts at both 4 and 24 hrs after burn injury. Similarly, LDL did not cause any effects on the cell isolation. Furthermore, there was no obvious difference in apparent cell size under transmitted light among the groups examined. To confirm this observation, we measured cell length and cell capacitance (which is proportional to the cell surface area) using cell image system and patch-clamp, respectively. As summarized in Table 1, cell length and the surface area of the myocytes were not significantly different between control and burn or burn+ LDL hearts at 4 and 24 hrs after burn injury.
Table 1.
Morphology of myocytes isolated from control (sham burn), sham+LDL, burn and burn+LDL rats measured at 4 and 24 hrs after burn injury. Individual cell size were examined by measuring cell length using cell image system and cell capacitance (which is proportional to the cell surface area) using patch-clamp protocols.
| Cell Length (μm) | Cell Capacitance (pF) | N | |
|---|---|---|---|
| Control (4 and 24 hrs) | 125.9 ± 2 (n=154) | 129.8 ± 3 (n=168) | 22 |
| Burn (4 hrs) | 123.3 ± 2.6 (n=64 ) | 130.6 ± 5 (n=59) | 8 |
| Burn (24 hrs) | 123.3 ± 2.6 (n=104) | 130.5 ± 4 (n=115) | 14 |
| Burn+LDL (4 hrs) | 126.3 ± 2 (n=54) | 128.7 ± 5 (n=35) | 6 |
| Burn+LDL (24 hrs) | 125.5 ± 2 (n=64) | 130.0 ± 3 (n=58) | 7 |
N= number of rats. n= number of cells. Values are means ± SE.
Cardiomyocyte Contractility
Figure 1 shows representative cell shortening recorded in myocytes isolated from control, 4 and 24 hrs post-burn hearts, and the effects of LDL. Cell shortening from post-burn hearts at 4 hrs did not significantly differ from other groups, but the amplitude at 24 hrs post-burn was significantly reduced (Fig. 1A). As summarized in Fig. 1B, contraction magnitude was significantly smaller at 24 hrs post-burn and this contractile depression was prevented by LDL.
Figure 1.

Cell shortening in myocytes at 4 and 24 hrs after burn injury and the effects of LDL. (A) Typical example of the contraction tracings recorded from each group during field stimulation at 1.0 Hz. (B) Pooled data for cell shortening (%) obtained from the same group shown in Table 1. Data are mean ± SE. *, P<0.01 vs. Control.
It is well established that there is significant slowing of contraction and relaxation velocity in myocytes isolated from hypertrophied and failing hearts due to changes in Ca2+ handling including slower rates of SR Ca2+ uptake or the sarcolemmal Na+-Ca2+ exchanger function [17, 27]. We thus compared the time for 50% decay (T50%) of cell shortening between control and post-burn hearts. There was no significant difference between the control and burn group (94.3±2.5 vs.90.0± 3.8 ms, respectively).
Properties of ICa
Whole-cell ICa was measured using patch-clamp. Figure 2A shows typical ICa density determined by peak ICa normalized relative to myocyte size (pA/pF) recorded in myocytes isolated from control, burn at 4 and 24 hrs, and burn+LDL at 24 hrs post-burn. ICa density was significantly lower in myocytes from post-burn hearts at 24 hrs versus control, while there was no significant decrease in 4 hrs post burn or burn+LDL compared to controls, as summarized in Table 2. The voltage-dependence of ICa was comparable in control, burn and burn+LDL groups (Fig. 2B); i.e., ICa activated around −30 mV and reached its maximum value near +10 mV. Thus, significantly reduced ICa was not caused by abnormal voltage-dependence of Ca2+ channel characteristics at 24 hrs post-burn.
Figure 2.

ICa in ventricular myocytes. (A) Whole-cell ICa recorded in myocyte from control, burn at 4 and 24 hrs, and burn+LDL at 24 hrs. Currents were elicited from a holding potential of −50 mV to the indicated test potentials at 0.1 Hz. (B) I–V relationships for peak ICa. ICa was normalized to the cell capacitance to give current densities (pA/pF). Data are mean ± SE, from the same cells shown in Table 2.
Table 2.
ICa density measured in myocytes isolated from control (sham burn), burn and burn+LDL rats at 4 and 24 hrs after burn injury.
| ICa density (pA/pF) | N | |
|---|---|---|
| Control (4 and 24 hrs) | −7.9 ± 0.4 (n=68) | 10 |
| Burn (4 hrs) | −7.8 ± 0.6 (n=16) | 4 |
| Burn (24 hrs) | −5.4 ± 0.5* (n=48) | 6 |
| Burn+LDL (24 hrs) | −7.0 ± 0.6 (n=30) | 4 |
N= number of rats. n= number of cells examined. Values are means ± SE.
p<0.05 vs. Control.
Expression of L-type Ca2+ channel mRNA
We examined the expression levels of the α1C and the auxiliary subunits, β1, β2, β3 and α2δ1. PCR detected significant expression of all target channel subunit mRNAs including splicing variants of α1C with and without the 75-bp fragment in the C-terminus (Fig. 3). As summarized in figure 4, real-time PCR analysis indicated that burn injury by itself (Burn) did not cause significant changes in mRNA levels of any channel subunits compared with control. The results are consistent with the observation that the voltage-dependence of ICa was not significantly altered after burn injury.
Figure 3.

Representative results of the reverse transcription-PCR analysis showing that burn injury does not influence the expression of Ca2+ channel subunit mRNAs. PCR analysis was performed with left ventricular RNA obtained from the indicated group of animals. Alpha 1C mRNAs were detected with primers that yield two bands at 378 (major strong signal) and 453 (faint signal) bp. The latter band represents a splicing variant the insertion of 75-bp fragment in the carboxyl cytosolic portion of channel polypeptide.
Figure 4.

Determination of Ca2+ channel transcripts. Expression of the corresponding transcripts was normalized to GAPDH transcript level in the same sample. Columns and error bars indicate the mean and SEM. * P<0.05 and ** P<0.01 compared to control, whereas # P<0.05 and ## P<0.01 compared to the corresponding burn group.
In contrast to the lack of the burn injury-associated change in Ca2+ channel mRNA expression, the tissues from burn+LDL contained significantly higher levels of α1C, β2, β3 and α2δ1 than those in burn alone or control (Fig. 4), suggesting that LDL itself may cause complex changes in Ca 2+ channel mRNA expression. Consistent with this notion, sham+LDL exhibited significant changes in α1C and β3 mRNA levels: LDL for 24 hrs significantly increased the expression of the pore-forming subunit mRNA, whereas a significant transient reduction in β3 mRNA level was seen in LDL at 4 hrs after the operation (Fig. 5). However, these changes in Ca2+ channel mRNA expression appears not account for the prevention of the burn-induced reduction in L-type Ca2+ current by LDL.
Figure 5.
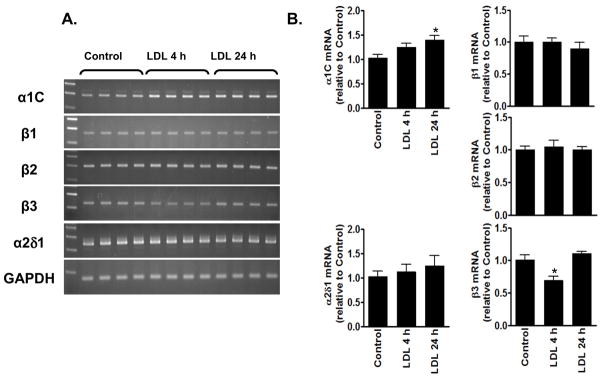
The effects of LDL on the expression of Ca2+ channel subunit mRNAs. (A) PCR results obtained from sham+LDL are shown with the left lanes representing DNA size markers. (B) Real-time PCR data are shown with columns and error bars indicating the mean and SEM. *p<0.05 compared to Control.
DISCUSSION
In the present study, using electrophysiological methods and gene expression analysis, we examined the cellular basis of cardiac dysfunction caused by burn injury as well as the beneficial effect of LDL.
We observed that (1) basal myocyte contraction was significantly reduced (≈20%) during the 4–24 hrs post-burn period compared with sham burn (control), sham+LDL or burn+LDL hearts, (2) myocyte size from post-burn hearts did not differ from other groups, and (3) basal ICa density was significantly decreased (≈30%) at 24 hrs post-burn, with unaltered voltage-dependent properties, however (4) the expression levels of Ca2+ channel mRNAs, α1C, β1, β2, β3 and α2δ1 were not significantly altered after burn injury. Thus the cellular studies suggest that depressed myocyte contractility and reduced ICa are involved in acute post-burn myocardial contractile dysfunction. Our observations also indicate that a potential mechanism for the consistent yet unexplained beneficial effect of LDL is, to a large extent, due to prevention of burn-induced inhibitory modulation of ICa activity.
Characteristic of isolated myocytes: correlation of cellular findings with myocardial contractility
Poor cardiac contractile function does not have to be the result of defects in myocyte contractile properties. Other factors such as tissue injury or edema which causes changes in cardiac structure and/or cell death due to apoptosis also could contribute to the cardiac dysfunction that occur during the early stage of burn injury. For example, in chronic heart failure loss of myocytes through apoptosis, and the presence of dedifferentiated cells, lead to a reduced number of functional myocytes, with hypertrophy of the remaining myocytes [13]. This possibility was validated by pathological studies which suggest that apoptosis contributes to burn-induced acute myocardial contractile dysfunction [16].
Our results show that myocyte contractile function was significantly decreased during the early phase (4–24 hrs) post-burn. These changes in contractility obtained at the cellular level agree with the previously published whole heart measurements [3, 4]. However, a reduction in myocyte function and a beneficial effect of LDL were not associated with changes in myocyte size. These results suggest that depressed myocyte contractility, but not cellular remodeling, was the major factor in burn-induced myocardial contractile dysfunction. Nevertheless, we should consider that in vivo situation, even minor loss of myocytes (apoptosis) or changes in cardiac structure (dilation) may also contribute to local stretch of myocardium and reduced contractility.
Mechanisms underlying the reduced cellular contractility
Numerous studies indicate that the depressed contractile function in failing hearts is associated with altered electrophysiological and excitation-contraction changes due to abnormalities of cellular Ca2+ regulation. Specifically, slowing contraction and relaxation and overall prolongation of contraction duration are common features of cardiac hypertrophy and heart failure [14, 28]. Changes in contraction time course have been implicated in the diminished capacity of the SR to increase its Ca2+ content, secondary to depressed SR Ca2+ uptake function.
In our study, when the time course of relaxation was assessed by the time for 50% decay (T50%) of cell shortening, there was no significant difference between myocytes from sham and post-burn hearts. These findings suggest that the cellular mechanisms underlying burn-induced myocardial contractile dysfunction is not primarily caused by defective SR and Na+-Ca2+ exchanger function. In contrast, our results indicate that alterations in the density and regulation of the L-type Ca2+ channel contribute to the depressed contractility of post-burn hearts. Thus, important features of the acute myocyte dysfunction from post-burn hearts are distinct from cardiac hypertrophy or heart failure which generally develops slowly.
Expression of L-type Ca2+ channel mRNA
The mechanism of the functional down-regulation of L-type Ca2+ channels observed during the early stage after burn injury is unknown, but may involve altered gene expression. We examined the expression levels of the α1C which forms the pore of the Ca2+ channel and the auxiliary subunits, β1, β2, β3 and α2δ1. In the present study, we found that burn itself does not affect either pore-forming α1C or auxiliary subunit mRNAs. The results are consistent with the observation that the voltage-dependence of ICa activation was not significantly different in any of the groups examined.
In the present study, real-time PCR analysis indicated that the burn injury by itself did not cause significant changes in mRNA levels of any channel subunits compared with controls. Lymph duct ligation (LDL) resulted in significant changes in α1C and β3 mRNA levels: Interestingly, sham+LDL exhibited changes in mRNA expression, indicating that LDL results in complex changes in Ca2+ channel mRNA expression, but this was not associated with a significant change in contractile function. It is thus, more likely that LDL prevented a decrease in the amount of Ca2+ influx by blocking mediators which regulate L-type Ca2+ channel function. The underlying cellular mechanisms are presently under investigation.
Possible mediators responsible for burn-induced down-regulation of ICa activity
Although the exact cellular signaling pathways that are responsible for the contractile dysfunction of post-burn hearts are unknown, an increasing body of experimental and clinical evidence now suggest that endogenously produced pro-inflammatory cytokines play an important role in a variety of cardiac pathophysiological conditions. These include traditional inflammatory mediators such as the cytokines TNF-α, IL-1 and IL-6 which can trigger nitric oxide (NO) activity. Changes in NO production can lead to cardiac contractile depression and their inhibition or neutralization has been shown to prevent burn- or trauma-shock-induced myocardial depression [3, 29, 30]. NO can also directly regulate normal contractility by interacting with the proteins involved in normal contractility, such as eNOS and nNOS isoforms, which are compartmentalized within the myocytes [31].
In the classic pathway, cAMP-mediated activation of protein kinase A (PKA) results in the phosphorylation of several intracellular proteins, including the α1C subunit of L-type Ca2+ channels [32]. A blunted response to this adenylyl cyclase step in the β-adrenergic receptor signaling cascade is reported in failing hearts [14]. It has also been reported that nitrosylation of L-type Ca2+ channels increases current amplitude, while phosphorylation of the channel by cGMP/Protein kinase G (PKG) decreases amplitude [33, 34]. Thus, modulation of these signal-transduction pathways may also be responsible for depressed ICa activity observed in post-burn hearts and the beneficial effects of LDL [35].
In summary, our results suggest that depressed cardiomyocyte contractility contributes to myocardial dysfunction that develops during the early stage after burn injury, in the absence of cellular remodeling. Our experiments also suggest that LDL effects results from modification of signaling pathways responsible for the regulation of L-type Ca2+ channel activity.
Acknowledgments
This research was supported by grants from NIH, HL 077480, GM 059841 and T32 GM 069330.
Footnotes
There is no other potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baxter CR, Shires T. Physiological response to crystalloid resuscitation of severe burns. Ann N Y Acad Sci. 1968;150(3):874. doi: 10.1111/j.1749-6632.1968.tb14738.x. [DOI] [PubMed] [Google Scholar]
- 2.Adams HR, Baxter CR, Parker JL. Contractile function of heart muscle from burned guinea pigs. Circ Shock. 1982;9(1):63. [PubMed] [Google Scholar]
- 3.Horton JW. Left ventricular contractile dysfunction as a complication of thermal injury. Shock. 2004;22(6):495. doi: 10.1097/01.shk.0000145205.51682.c3. [DOI] [PubMed] [Google Scholar]
- 4.Maass DL, Naseem RH, Garry M, et al. Echocardiography assessment of myocardial function after burn injury. Shock. 2006;25(4):363. doi: 10.1097/01.shk.0000217814.93. [DOI] [PubMed] [Google Scholar]
- 5.Deitch EA. Multiple organ failure. Pathophysiology and potential future therapy. Ann Surg. 1992;216(2):117. doi: 10.1097/00000658-199208000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassoun HT, Kone BC, Mercer DW, et al. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15(1):1. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 7.Charalambous BM, Stephens RC, Feavers IM, et al. Role of Bacterial Endotoxin in Chronic Heart Failure: the Gut of the Matter. Shock. 2007;28(1):15. doi: 10.1097/shk.0b013e318033ebc5. [DOI] [PubMed] [Google Scholar]
- 8.Magnotti LJ, Upperman JS, Xu DZ, et al. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. 1998;228(4):518. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zallen G, Moore EE, Johnson JL, et al. Posthemorrhagic shock mesenteric lymph primes circulating neutrophils and provokes lung injury. J Surg Res. 1999;83(2):83. doi: 10.1006/jsre.1999.5569. [DOI] [PubMed] [Google Scholar]
- 10.Cox CS, Jr, Fischer UM, Allen SJ, et al. Lymphatic diversion prevents myocardial edema following mesenteric ischemia/reperfusion. Microcirculation. 2004;11(1):1. doi: 10.1080/10739680490266135. [DOI] [PubMed] [Google Scholar]
- 11.Gosain A, Gamelli RL. Role of the gastrointestinal tract in burn sepsis. J Burn Care Rehabil. 2005;26(1):85. doi: 10.1097/01.bcr.0000150212.21651.79. [DOI] [PubMed] [Google Scholar]
- 12.Sambol JT, White J, Horton JW, et al. Burn-induced impairment of cardiac contractile function is due to gut-derived factors transported in mesenteric lymph. Shock. 2002;18(3):272. doi: 10.1097/00024382-200209000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Jacob R, Gulch RW. The functional significance of ventricular geometry for the transition from hypertrophy to cardiac failure. Does a critical degree of structural dilatation exist? Basic Res Cardiol. 1998;93(6):423. doi: 10.1007/s003950050111. [DOI] [PubMed] [Google Scholar]
- 14.Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92(4):350. doi: 10.1161/01.RES.0000060027.40275.A6. [DOI] [PubMed] [Google Scholar]
- 15.Levy RJ, Piel DA, Acton PD, et al. Evidence of myocardial hibernation in the septic heart. Crit Care Med. 2005;33(12):2752. doi: 10.1097/01.ccm.0000189943.60945.77. [DOI] [PubMed] [Google Scholar]
- 16.Zhang JP, Ying X, Liang WY, et al. Apoptosis in cardiac myocytes during the early stage after severe burn. J Trauma. 2008;65(2):401. doi: 10.1097/TA.0b013e31817cf732. [DOI] [PubMed] [Google Scholar]
- 17.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 18.Sather WA, McCleskey EW. Permeation and selectivity in calcium channels. Annu Rev Physiol. 2003;65:133. doi: 10.1146/annurev.physiol.65.092101.142345. [DOI] [PubMed] [Google Scholar]
- 19.Catterall WA, Perez-Reyes E, Snutch TP, et al. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacological reviews. 2005;57(4):411. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 20.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Current opinion in neurobiology. 2003;13(3):298. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 21.Colecraft HM, Alseikhan B, Takahashi SX, et al. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541(Pt 2):435. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deitch EA, Adams C, Lu Q, et al. A time course study of the protective effect of mesenteric lymph duct ligation on hemorrhagic shock-induced pulmonary injury and the toxic effects of lymph from shocked rats on endothelial cell monolayer permeability. Surgery. 2001;129(1):39. doi: 10.1067/msy.2001.109119. [DOI] [PubMed] [Google Scholar]
- 23.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8(6):1049. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Yatani A, Xu DZ, Kim SJ, et al. Mesenteric lymph from rats with thermal injury prolongs the action potential and increases Ca2+ transient in rat ventricular myocytes. Shock. 2003;20(5):458. doi: 10.1097/01.shk.0000090602.26659.5c. [DOI] [PubMed] [Google Scholar]
- 25.Zhang TT, Takimoto K, Stewart AF, et al. Independent regulation of cardiac Kv4. 3 potassium channel expression by angiotensin II and phenylephrine. Circ Res. 2001;88(5):476. doi: 10.1161/01.res.88.5.476. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Takimoto K. Differential expression of Kv4 pore-forming KChIP auxiliary subunits in rat uterus during pregnancy. Am J Physiol Endocrinol Metab. 2005;288(2):E335. doi: 10.1152/ajpendo.00250.2004. [DOI] [PubMed] [Google Scholar]
- 27.Winslow RL, Rice J, Jafri S, et al. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84(5):571. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]
- 28.Bers DM. Calcium cardiac rhythms: physiological pathophysiological. Circ Res. 2002;90(1):14. [PubMed] [Google Scholar]
- 29.Mehra VC, Ramgolam VS, Bender JR. Cytokines cardiovascular disease. Journal of leukocyte biology. 2005;78(4):805. doi: 10.1189/jlb.0405182. [DOI] [PubMed] [Google Scholar]
- 30.Carlson DL, Horton JW. Cardiac molecular signaling after burn trauma. J Burn Care Res. 2006;27(5):669. doi: 10.1097/01.BCR.0000237955.28090.41. [DOI] [PubMed] [Google Scholar]
- 31.Ziolo MT, Bers DM. The real estate of NOS signaling: location location location. Circ Res. 2003;92(12):1279. doi: 10.1161/01.RES.0000080783.34092.AF. [DOI] [PubMed] [Google Scholar]
- 32.Ganesan AN, Maack C, Johns DC, et al. Beta-adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1C but not serine 1928. Circ Res. 2006;98(2):e11. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallo MP, Malan D, Bedendi I, et al. Regulation of cardiac calcium current by NO and cGMP-modulating agents. Pflugers Arch. 2001;441(5):621. doi: 10.1007/s004240000475. [DOI] [PubMed] [Google Scholar]
- 34.Sun J, Picht E, Ginsburg KS, et al. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circulation research. 2006;98(3):403. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 35.Buys ES, Cauwels A, Raher MJ, et al. sGC(alpha)1(beta)1 attenuates cardiac dysfunction and mortality in murine inflammatory shock models. Am J Physiol Heart Circ Physiol. 2009;297(2):H654. doi: 10.1152/ajpheart.00367.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]