

Abstract
A series of compounds containing an α,β-unsaturated carbonyl moiety, such as chalcones and coumarins were designed, synthesized and tested in a variety of assays to assess their potential as anti-Alzheimers’ disease (AD) agents. The investigations included the inhibition of cholinesterases (AChE, BuChE), the inhibition of amyloid beta (Aβ) self-assembly and the disassembly of preformed Aβ oligomers. Several compounds showed excellent inhibition in multiple assays and thus are potential multifunctional compounds for AD. Docking studies for 16 that performed well in all the assays gave a clear interpretation of various interactions in the gorge of AChE. Based on the results, the long-chain coumarin scaffold appears to be a promising structural template for further AD drug development.
Alzheimer’s disease (AD) is associated with a loss of presynaptic markers of the cholinergic system in the areas of the brain related to memory and learning, and is also characterized by the presence of amyloid deposits and neurofibrillary tangles in the brain.1 The deterioration of cholinergic neurotransmission is responsible for the decline in memory and cognition in patients suffering from AD.2 One of the ways to enhance cholinergic function by preserving acetylcholine (ACh) levels is to inhibit acetylcholinesterase (AChE) responsible for the metabolic breakdown of ACh. Preclinical experiments and clinical trials have shown butyrylcholinesterase (BuChE) to be an important contributor for the occurrence, symptoms, progression, and responses to treatment in dementia.3,4 Several anti-cholinergic drugs have been launched including tacrine, rivastigmine, donepezil, and galanthamine.5 It has also been shown that dual AChE/BuChE agents might be superior to the selective AChE inhibitors.2
AChE has a long and narrow gorge (20 Å) with two binding sites; a catalytic site (active site) and a peripheral anionic site (PAS).6 It was reported that the peripheral site of AChE could promote the formation of amyloid beta (Aβ) deposits.7,8 The formation of such deposits is a hallmark of AD pathology.9,10 Detailed studies11 indicated that both fibrillar and oligomeric species of Aβ are neurotoxic.12,13 Thus the design of multifunctional inhibitors, which can control the hydrolytic activity of cholinesterases and interfere with the self-assembly of Aβ and disassemble the preformed aggregates is highly desirable.14, 15, 16
Continuing our earlier work on novel anti-AD compounds,17,18,19,20,21 we describe novel compounds with an α,β-unsaturated carbonyl moiety (chalcones and coumarins) that can act as multifunctional anti-cholinesterase/Aβ agents. Chalcone22 and coumarin23 derivatives have been described as AChE and BuChE inhibitors. While coumarins have also been applied as Aβ aggregation inhibitors,24 chalcones are more commonly used as Aβ imaging agents.25,26 There are no reports that such compounds synthesized or applied as multifunctional agents such as inhibiting both cholinesterases as well as Aβ self-assembly.
We designed α,β-unsaturated carbonyl compounds with an open chain (chalcones) and cyclic (coumarins) structures. In order to have dual binding property in the gorge of AChE the α,β-unsaturated carbonyl unit was linked to different amines. The designed structures were then screened through the 4-point pharmacophore developed in our group27 and molecules that matched 3 of the 4 points of our pharmacophore (Fig. 1) were synthesized.
Figure 1.

Pharmacophore features of two of the designed compounds. Light blue sphere-H-acceptor; red sphere–H-donor; ring – aromatic ring and dark blue sphere – positively charged center
The syntheses of the compounds are summarized in Schemes 1 and 2.28 The synthesis of the target compounds applied readily available starting materials and was completed using well established methods.25
Scheme 1.
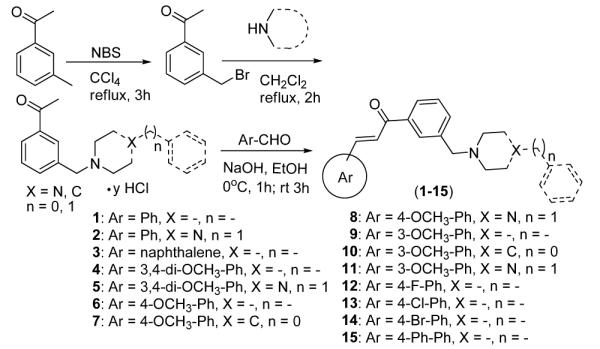
Synthesis and structures of chalcone derivatives used
Scheme 2.
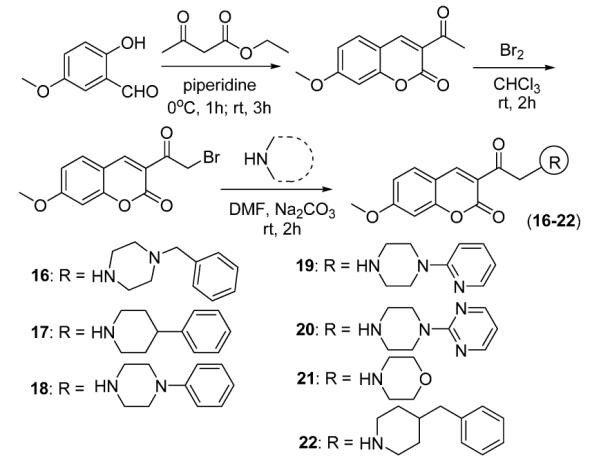
Synthesis and structures of coumarin derivatives used
The compounds were tested by the Ellman colorimetric assay for cholinesterase inhibition.29,30,31 For comparison, galanthamine (GAL), a known cholinesterase inhibitor, was used. The experiments were carried out with 2 μM and 10 μM inhibitor concentration, respectively (IC50 of GAL for AChE and BuChE) (Fig. 2).6,7 GAL was selected due to its low toxicity and ready commercial availability.
Figure 2.

Inhibition of (A) AChE and (B) BuChE hydrolytic activity by chalcones and coumarins. The experiments were carried out at 2 μM (A) and 10 μM (B) inhibitor concentration and 0.02 unit/mL enzyme concentration.29
The data (Fig. 2) indicate that the compounds possess appreciable dual cholinesterase inhibition. Coumarins are potent AChE inhibitors while chalcones show stronger activity in BuChE inhibition. In AChE inhibition most inhibitors showed comparable but somewhat weaker inhibition than that of the reference compound (GAL). Compound 16 exhibited higher activity than GAL and showed excellent (57%) inhibition at 2 μM (IC50 of GAL) with an IC50 of 1.76 μM.
The compounds behaved similarly in BuChE inhibition assays. Chalcones possess activity similar to GAL and a few compounds showed more than 50 % inhibition at 10 μM (IC50 of GAL), however, most of their IC50 values were determined to be slightly higher than that of GAL except 15 with an IC50 of 8.27 μM. While the initial data suggested 11 being a slightly better inhibitor than GAL, the IC50 determination did not confirm this likely due to the deviation of the data.
In order to understand the interactions of 16 with the enzyme, it was docked in the active site of AChE (PDB code: 1E66) (Fig. 3) using SP mode of glide module of Schrödinger.32 For comparison compound 16 was superimposed with galanthamine and another well-known AChE inhibitor, donepezil (Fig. 3). Unlike galanthamine, 16 spanned the active site and peripheral anionic site of the enzyme similarly to donepezil. As shown, 16 is oriented properly in the active site (Fig. 3). The coumarin ring was stabilized by hydrophobic interactions with Trp 333, Phe 290 and Phe 288 residues. The phenyl ring at the amino end was stabilized by hydrophobic and π-π interactions through Trp 84 and Trp 130. The methoxy group of coumarin ring formed a hydrogen bond (Phe 288) and the –NH of the piperazine ring formed a hydrogen bond with Ser 122. These observations explain the potency of 16 in AChE inhibition.
Figure 3.
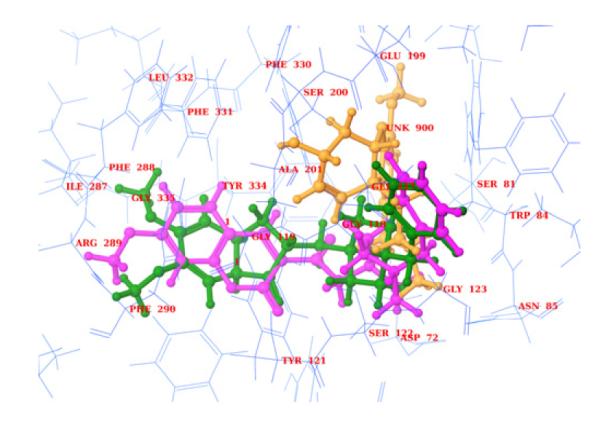
Superimposition of 16 (pink, ball and stick) with donezepil (green, ball and stick) and galanthamine (brown, ball and stick) in the active site of 1E66.
As fibrillar and oligomeric aggregates of Aβ are neurotoxic, the activity of the compounds was determined against the formation of these species and in the disassembly of preformed oligomers. The compounds were tested by standard methods, the biotinyl-Aβ(1-42) single-site streptavidin assay for oligomer formation and disassembly33,34 (Fig. 4) and the thioflavine T-fluorescence assay for fibril formation (Fig. 5).35,36,37
Figure 4.

Effect of the designed compounds on the (A) oligomer assembly of Aβ and (B) disassembly of preformed Aβ oligomers (50μM inhibitor; 0.01μM Aβ-inhibition; 0.0028μM Aβ-dissociation)
Figure 5.
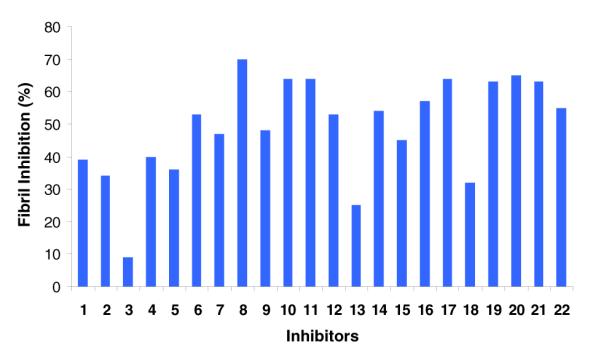
Effect of the designed compounds on the fibrillogenesis of Aβ (100 μM inhibitor, 100 μM Aβ)
In the oligomer assembly assay coumarins (16-22) are excellent inhibitors (> 65%, Fig. 4A) while chalcones showed little or no inhibition (Fig. 4A). The concentration dependence studies revealed the following IC50 values: 16 (36 μM), 17 (14 μM), 18 (21 μM), 20 (1 μM), 21 (29 μM), 22 (36 μM).
The same group of compounds (16-22) effectively (up to 95%) disassembled the preformed oligomers as well (Fig. 4B). The IC50 data for oligomer disassembly are: 17 (29μM), 18 (6.5 μM), 20 (4 μM) and 21 (5 μM). The compounds exhibited similar behavior in the inhibition of Aβ fibril formation (Fig. 5). Both chalcones (1-15) coumarins (16-22) were reasonable fibrillogenesis inhibitors (~ 30-70%). It is worth noting that the compounds did not show significant intrinsic fluorescence thus their presence in the assay did not affect the fluorescence readings.
Atomic force microscopy (AFM) was used to confirm the fibrillogenesis inhibition (Fig. 6). The images revealed the expected dense network of long fibrils in the control sample. While fibrils are still visible in the inhibited samples the density is significantly lower in agreement with the assay results. It also can be observed that shorter assemblies formed in the presence of 16.
Figure 6.
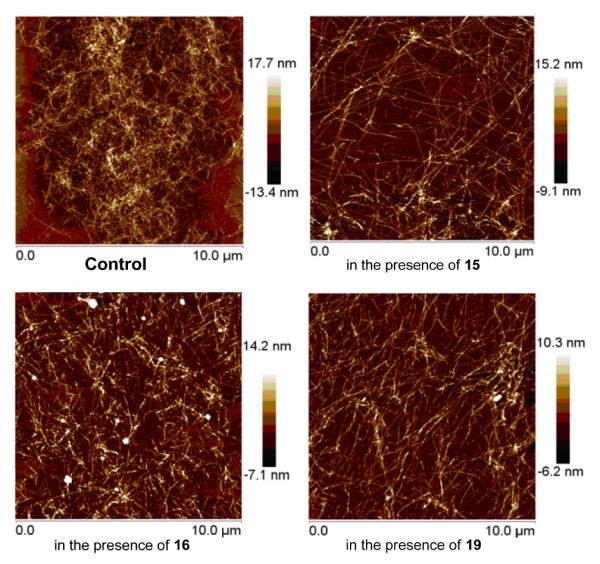
Illustrative images of the control and selected samples in the presence of compounds 15, 16 and 19, respectively.
Chalcones and coumarins have already been investigated to modulate cholinesterase activity or inhibit Aβ self-assembly, respectively.22-26 In an attempt to combine these activities in a single chemical structure and develop multifunctional anti-AD compounds we synthesized 22 compounds with chalcone and coumarin head and varied tail groups. The aim was to build compounds of sufficient length to span the active center of the cholinesterases and at the same time interfere with Aβ self-assembly. The structure-activity relationship reveals important information for future design. It appears that the above results obtained with coumarins support the hypothesis. Chalcones showed strong activity in BuChE and fibrillogenesis inhibition only. They were relatively weak inhibitors of AChE and Aβ oligomer formation. In contrast, coumarins performed more consistently showing significant inhibition in all assays, except against BuChE where they were moderate to weak inhibitors. At this point the coumarin derivatives were limited to one head group and different tail groups possessing aryl-substituted piperidine, piperazine and morpholine units. Compounds with the two nitrogen containing piperazine ring (16, 18-20) performed the best overall. While the observed differences in activity within this subgroup are not decisive, the data suggest that the most beneficial tail groups are phenyl and benzyl piperazines (16, 18). It appears that the presence of basic nitrogens in the middle ring is advantageous; however, too many nitrogens (e.g. pyrimidinyl-piperazine group, 20), result in a slight decrease in activity in all assays.
Comparing the anti-oligomer and anti-fibril assays one may notice that while coumarins almost completely inhibit the formation of oligomers the same compounds are moderate inhibitors (~65%) of the fibril formation. It is most likely due to the fact that there are multiple pathways involved in the amyloid formation and the oligomers are not obligatory precursors to the fibrils.38
In conclusion, a variety of compounds with α,β- unsaturated moiety were synthesized with the aim of preparing multifunctional compounds with anticholinesterase and anti-Aβ assembly activities for AD. It was observed that while the open chain derivatives (chalcones) were inactive in two important assays the cyclic coumarins showed excellent to reasonable effects in several assays, e.g. cholinesterase activity and Aβ self-assembly. Based on the structure-activity relationship, compounds with a coumarin-based head group and an aryl/benzyl-piperazine tail group are promising leads for further inhibitor design. The structures of our lead compounds represent a relatively nonpolar character with an extended network of hetero- and carbocyclic rings. This feature suggests favorable membrane permeability, which is an important factor for drug candidates.
Supplementary Material
Acknowledgements
Financial support provided by the University of Massachusetts Boston, and National Institute of Health (R-15 AG025777-03A1 and R21AG028816-01 to H. L.) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Lancet. 2011;377:1019. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 2.Chen X, Tikhonova IG, Decker M. Bioorg. Med. Chem. 2011;19:1222. doi: 10.1016/j.bmc.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 3.Ballard CG, Greig NH, Guillozet-Bongaarts AL, Enz A, Darvesh S. Curr. Alzheimer Res. 2005;2:307. doi: 10.2174/1567205054367838. [DOI] [PubMed] [Google Scholar]
- 4.Darvesh S, Hopkin DA, Geula C. Nat. Rev. Neurosci. 2003;4:131. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 5.McGleenon BM, Dynan KB, Passmore AP. Br. J. Clin. Pharmacol. 1999;48:471. doi: 10.1046/j.1365-2125.1999.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Science. 1991;253:872. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Wang X, Wang T, Kong L. Bio. Org. Med. Chem. 2008;16:8011. doi: 10.1016/j.bmc.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 8.Sheng R, Xu Y, Hu C, Zhang J, Lin X, Li J, Yang B, He Q, Hu Y. Eur. J. Med. Chem. 2009;44:7. doi: 10.1016/j.ejmech.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Murphy RM. Annu. Rev. Biomed. Eng. 2002;4:155. doi: 10.1146/annurev.bioeng.4.092801.094202. [DOI] [PubMed] [Google Scholar]
- 10.Erion MD, van Poelje PD, Dang Q, Kasibhatla SR, Potter SC, Reddy MR, Reddy KR, Jiang T, Lipscomb WN. Proc. Natl. Acad. Sci. USA. 2005;102:7970. doi: 10.1073/pnas.0502983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erion MD, Dang Q, Reddy MR, Kasibhatla SR, Huang J, Lipscomb WN, van Poelje PD. J. Am. Chem. Soc. 2007;129:15480. doi: 10.1021/ja074869u. [DOI] [PubMed] [Google Scholar]
- 12.Dang Q, Kasibhatla SR, Reddy KR, Jiang T, Reddy MR, Potter SC, Fujitaki JM, van Poelje PD, Huang J, Lipscomb WN, Erion MD. J. Am. Chem. Soc. 2007;129:15491. doi: 10.1021/ja074871l. [DOI] [PubMed] [Google Scholar]
- 13.Re F, Airoldi C, Zona C, Masserini M, Ferla BL, Quattrocchi N, Nicotra F. Curr. Med. Chem. 2010;17:2990. doi: 10.2174/092986710791959729. and ref cited therein. [DOI] [PubMed] [Google Scholar]
- 14.Bolognesi ML, Bartolini M, Tarozzi A, Morroni F, Lizzi F, Milelli A, Minarini A, Rosini M, Hrelia P, Andrisano V, Melchiorre C. Bioorg. Med. Chem. Lett. 2011;21:2655. doi: 10.1016/j.bmcl.2010.12.093. [DOI] [PubMed] [Google Scholar]
- 15.Weinstock M. CNS Drugs. 1999;12:307. [Google Scholar]
- 16.Pisani L, Catto M, Giangreco I, Leonetti F, Nicolotti O, Stefanachi A, Cellamare S, Carotti A. Chem. Med. Chem. 2010;5:1616. doi: 10.1002/cmdc.201000210. [DOI] [PubMed] [Google Scholar]
- 17.Török M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. J. Biol. Chem. 2002;277:40810. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 18.Török M, Abid M, Mhadgut SC, Török B. Biochemistry. 2006;45:5377. doi: 10.1021/bi0601104. [DOI] [PubMed] [Google Scholar]
- 19.Sood A, Abid M, Hailemichael S, Foster M, Török B, Török M. Bioorg. Med. Chem. Lett. 2009;19:6931. doi: 10.1016/j.bmcl.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sood A, Abid M, Sauer C, Hailemichael S, Foster M, Török B, Török M. Bioorg. Med. Chem. Lett. 2011;21:2044. doi: 10.1016/j.bmcl.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Török B, Sood A, Bag S, Kulkarni A, Borkin D, Lawler E, Dasgupta S, Landge SM, Abid M, Zhou W, Foster M, LeVine H, III, Török M. ChemMedChem. 2012;7:910. doi: 10.1002/cmdc.201100569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasan A, Khan M, Sher M, Maharvi GM, Nawaz SA, Choudhary MI, Atta-Ur-Rahman, Supuran CT. J. Enz. Inh. Med. Chem. 2005;20:41. doi: 10.1080/14756360400015231. [DOI] [PubMed] [Google Scholar]
- 23.Anand P, Singh B, Singh N. Bioorg. Med. Chem. 2012;20:1175. doi: 10.1016/j.bmc.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 24.Soto-Ortega DD, Murphy BP, Gonzalez-Velasquez FJ, Wilson KA, Xie F, Wang Q, Moss MA. Bioorg. Med. Chem. 2011;19:2596. doi: 10.1016/j.bmc.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Cui M, Ono M, Kimura H, Liu BL, Saji H. Bioorg. Med. Chem. Lett. 2011;21:980. doi: 10.1016/j.bmcl.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 26.Ono M, Ikeoka R, Watanabe H, Kimura H, Fuchigami T, Haratake M, Saji H, Nakayama M. ACS Chem. Neurosci. 2010;1:598. doi: 10.1021/cn100042d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bag S, Tulsan R, Sood A, Datta S, Török M. Curr. Comp.- Aided Drug Des. 2012 in press. [PubMed] [Google Scholar]
- 28.The synthesis and spectral characterization of the compounds is provided in the Supporting Information.
- 29.The cholinesterase assay description is provided in the Supporting Information.
- 30.Ellman GL, Courtney KD, Andres BJ, Featherstone RM. Biochem. Pharmacol. 1961;7:88. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 31.Greenblatt HM, Kryger G, Lewis T, Silman I, Sussman JL. FEBS Lett. 1999;463:321. doi: 10.1016/s0014-5793(99)01637-3. [DOI] [PubMed] [Google Scholar]
- 32.Glide. Schrödinger, LLC; New York, NY: 2009. version 9.0. [Google Scholar]
- 33.LeVine H., III Anal. Biochem. 2006;356:265. doi: 10.1016/j.ab.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 34.The biotin-streptavidin assay description is provided in the Supporting Information.
- 35.Naiki H, Higuchi K, Hosokawa M, Takeda T. Anal. Biochem. 1989;177:244. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 36.LeVine H., III Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The THT fluorescence assay description is provided in the Supporting Information.
- 38.Necula M, Kayed R, Milton S, Glabe CG. J. Biol. Chem. 2007;282:10311. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.