

Abstract
Mitochondria are known to be central to the cell's response to ischemia, because of their role in energy generation, in free radical generation, and in the regulation of apoptosis. Heat shock protein 75 (Hsp75/Grp75/mortalin/TRAP1) is a member of the HSP70 chaperone family, which is targeted to mitochondria. Overexpression of Hsp75 was achieved in rat brain by DNA 7transfection, and expression was observed in both astrocytes and neurons. Rats were subjected to 100 mins middle cerebral artery occlusion followed by assessment of infarct volume, neurological score, mitochondrial function, and levels of oxidative stress at 24 h reperfusion. Overexpression of Hsp75 reduced infarct area from 44.6%±21.1% to 25.7%±12.1% and improved neurological outcome significantly. This was associated with improved mitochondrial function as shown by protection of complex IV activity, marked reduction of free radical generation detected by hydroethidine fluorescence, reduction of lipid peroxidation detected by 4-hydroxy-2-nonenol immunoreactivity, and increased preservation of ATP levels. This suggests that targeting mitochondria for protection may be a useful strategy to reduce ischemic brain injury.
Keywords: Grp75, mitochondria, mortalin, oxidative stress, stroke, TRAP1
Introduction
Ischemic brain injury is caused by the interruption of cerebral blood flow leading to both acute and delayed degeneration of brain cells. Mitochondrial function is involved in the maintenance of cellular homeostasis and in life/death decisions (Bambrick et al, 2004; Fiskum, 2000; Zipfel et al, 2000). Multiple mechanisms contribute to mitochondrial dysfunction during and after ischemia. Mitochondrial electron transport constitutively results in production of reactive oxygen species (ROS), including superoxide radicals and hydrogen peroxide, under normal physiological conditions (Boveris and Chance, 1973). Ischemia and subsequent reperfusion cause ROS overproduction by mitochondria, leading to an increase in oxidative stress (Saito et al, 2005; Siesjo et al, 1989).
Many cell functions including neurotransmitter turnover and ion homeostasis require ATP. Mitochondrial ATP production through oxidative phosphorylation is the major energy source for brain cells (Edmond et al, 1987). Ischemia-induced mitochondrial damage leads to severe ATP depletion, thus compromising ionic balance, neuronal signaling, and other vital processes (Dienel and Hertz, 2005; Hata et al, 2000). Severe ischemia induces loss of mitochondrial membrane potential, which initiates both apoptotic and necrotic mechanisms of cell death (Gottlieb et al, 2003; Honda et al, 2005). Loss of mitochondrial potential has been shown to be a key event in the demise of neurons and astrocytes under ischemic conditions (Juurlink and Hertz, 1993). Therefore, protection of mitochondrial homeostasis and function during ischemic injury represents a promising therapeutic target for the reduction of ischemic brain injury.
Heat shock protein 75 (Hsp75/mtHsp70/Grp75/mortalin/TRAP-1) is the mitochondrial localized member of the heat shock protein 70 (HSP70) family and is an essential mitochondrial chaperone. It binds translocase of the inner membrane to form an ATP-dependent motor that imports mitochondrial proteins into the matrix (Voos et al, 1999). Heat shock protein 75 also associates with other mitochondrial proteins including Hsp60, voltage-dependent anion-selective channel, and nicotinamide adenine dinucleotide (NADH) dehydrogenase, thus making it an important part of the mitochondrial machinery (Bhattacharyya et al, 1995; Schwarzer et al, 2002). Heat shock protein 75 is not heat-inducible, but like other HSP70 members, has been shown to be upregulated by various cellular insults including glucose deprivation, oxidative stress, thyroid hormone treatment, and ultraviolet A radiation (Carette et al, 2002; Hadari et al, 1997; Lee, 2001; Mitsumoto et al, 2002). Heat shock protein 75 induction was also found after focal cerebral ischemia by Massa et al (1995). Increased Hsp75 levels have been shown to be associated with protection against apoptotic death in smooth muscle (Taurin et al, 2002).
So far, several in vitro studies have demonstrated the protective potential of Hsp75 overexpression against ischemia-like injury. In cardiac myocytes exposed to hypoxia/reoxygenation Hsp75 overexpression protected from mitochondrial injury and development of apoptosis (Williamson et al, 2008). Protective effects of Hsp75 overexpression in brain cells have also been reported (Liu et al, 2005; Voloboueva et al, 2007). Increased levels of Hsp75 suppressed rapid ROS accumulation in a neuronal cell line (Liu et al, 2005), and decreased ROS production, preserved mitochondrial function, and increased cell viability in primary astrocytes exposed to ischemia-like injury in vitro (Voloboueva et al, 2007). Despite the evidence of protection by Hsp75 in vitro, studies of the effect of Hsp75 overexpression on brain ischemia/reperfusion injury or mitochondrial function in vivo are lacking. The purpose of this study was to investigate the effects of Hsp75 overexpression in the brains of rats subjected to transient middle cerebral artery occlusion on the size of the infarct area, levels of oxidative stress, and mitochondrial function.
Materials and methods
Overexpression of Hsp75 in Brain
The Hsp75 coding sequences from pBluescript-Hsp75 (a kind gift from R Morimoto, Northwestern University) were PCR amplified using the following primers: GGCCCGTCGGGCCTGCCTCGTACTCCT and GGCCCGATAGGCCGGAAGTCTCTTCACTCCTAAG, to produce a product flanked by two sites for the restriction endonuclease SfiI. This was subcloned into pCR2.1 (Invitrogen, Carlsbad, CA, USA), cut out with SfiI and subcloned into a modification of pL_UGIN carrying two SfiI sites in place of the enhanced green fluorescent protein (eGFP) sequences. This was used as an expression construct with Hsp75 expression driven by the human ubiquitin c promoter. Previous work demonstrated that this is a strong, ubiquitous promoter for expression in mice (Schorpp et al, 1996). Controls were injected with the original pL_UGIN plasmid encoding eGFP, a kind gift from Dr Iain Fraser (California Institute of Technology, USA).
DNA/lipid complex was prepared and injected intracerebroventricularly as described previously (Sun et al, 2006). Briefly, adult male Sprague–Dawley rats (280 to 310 g) were anesthetized by face mask with isoflurane in 70% N2O and 30% O2 and placed in a stereotaxic frame with a rat head holder. DNA (10 µg) of the expression plasmid encoding Hsp75 or plasmid encoding eGFP, referred to as vector in the following text, was mixed with the cationic lipid DOTAP (1:3 µg/µl; Roche Applied Science, Indianapolis, IN, USA). After mixing for 5 secs, and incubating at 37°C for 15 mins the mixture was infused into the right lateral cerebral ventricle at a speed of 2 µl/mins via a burr hole. After that the bone wound was closed with bone wax, anesthesia was discontinued, and rats were returned to their cages. Two days after the injection rats were either killed to assess Hsp75 expression, or subjected to focal ischemia on the contralateral side.
Focal Cerebral Ischemia
Transient ischemia was induced using the suture occlusion technique, as previously described (Sun et al, 2006), with slight modifications. Male, 280 to 310 g Sprague–Dawley rats were anesthetized, and the left external carotid artery (ECA) was exposed and dissected. A 3–0 monofilament nylon suture (from Doccol Co., Redlands, CA, USA) was inserted from the ECA into the internal carotid artery to occlude the left middle cerebral artery (MCA) at its origin. After 100 mins, the suture was removed for reperfusion, the ECA was ligated, and the wound was closed. Sham-operated animals underwent identical procedures, except for the suture insertion. Rectal temperature were maintained at 37°C±0.5°C controlled by a Homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, USA). Temperature, respiratory rate, oxygen saturation, and heart rate were monitored during the surgery, and after reanesthetizing the animals to remove the suture at 100 mins, with a small animal Oximeter (STARR Life Sciences Corp., Allison Park, PA, USA). At 24 h after surgery, rats were tested for neurological score before being killed.
Infarct Volume Measurements
The rats were anesthetized with isoflurane and brains were rapidly removed 24 h after middle cerebral artery occlusion (MCAO). Sections were incubated in 2% 2,3,5-triphenyltetrazolium chloride in saline for 20 mins at 37°C. The infarct area was measured by a blinded observer on 6 sections per brain using digital imaging and image analysis software (ImageJ, version 1.37; Wayne Rasband, available through NIH) corrected for edema as previously described (Sun et al, 2006).
Neurological Deficit Score
The neurological deficit score was assessed 24 h after MCAO as described previously (rating scale: 0=no deficit, 1=failure to extend the right forepaw, 2=circling to the right, 3=falling to the right, 4=unable to walk spontaneously) (Menzies et al, 1992).
Immunohistochemistry, Lipid Peroxidation Assessment of HNE
Ischemic or sham-operated rats were anesthetized and perfused with 0.9% saline, followed by 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4). The brains were kept in 4% paraformaldehyde in PBS (pH 7.4) for 48 h then cut in 50 µm sections. Sections were immunostained using anti-glial fibrillary acidic protein (anti-GFAP, 1:3 dilution; Immunostar, Hudson, WI, USA) to label astrocytes or rabbit anti-microtubule-associated protein 2 (anti- MAP2) (1:2000 dilution; Chemicon, Temecula, CA, USA) to identify neurons, and anti-Hsp75 antibody (1:500; Abcam, Cambridge, MA, USA). Antibody to 4-hydroxy-2-nonenal (HNE) (Oxis, 1:200) was used to determine levels of lipid oxidation. After washing, the slices were incubated with corresponding Alexa Fluor 488- or 594-conjugated secondary antibodies (Invitrogen; 1:200), washed and mounted on glass slides using Vectashield mounting medium with 4′,6- diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). The border between normal appearing cells and infarcted tissue is readily identified after 24 h (Dingman et al, 2006) by the morphological appearance of nuclei. The core was identified as the region in which the majority of 4′,6-diamidino-2-phenylindole stained nuclei were shrunken, whereas the penumbra was defined as the region of generally morphologically normal cells, approximately 500 µm wide, surrounding the core. To quantify levels of lipid oxidation, HNE reactivity was visualized and the average fluorescence intensity was measured in three brain sections (−1.5 to 2.5 mm relative to Bregma). At least six areas of 0.05 mm2, in both cortical and striatal areas, were quantified in the penumbra and in the corresponding non-ischemic contralateral areas of the brain for each of three animals per group.
Measurement of ROS
ROS in brain parenchyma oxidizes HEt to ethidium, the fluorescence of which can be visualized in brain sections. HEt (Molecular Probes, Eugene, OR, USA) 0.5 mg in 200 µl saline was administered by tail vein 23 hours after MCAO; animals were killed one hour later. The brains were fixed in 4% paraformaldehyde in PBS for 48 h, 50 µm sections were prepared, and HEt fluorescence intensity determined and quantified as described above for HNE measurements.
Western Blotting
For western blotting rats were killed 48 h after Hsp75 or vector DNA injection. The brains were removed and immediately homogenized in cold lysis buffer (10 mmol/L 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (pH 7.9), 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1 mmol/L dithiothreitol) plus protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Protein concentrations were determined by the bicinchoninic acid method (Pierce, Rockford, IL, USA). Equal amounts (50 µg) of protein were loaded and separated on a 4% to 15% polyacrylamide gel (Bio-Rad, Hercules, CA, USA), and electrotransferred to Immobilon polyvinylidene fluoride membrane (Millipore Corp., Bedford, MA, USA). Membranes were blocked with 5% nonfat dry milk in PBS with 0.1% Tween 20 for 1 h, incubated overnight with 1:1000 primary anti-Hsp75 antibody (Stressgen, Ann Arbor, MI, USA), washed three times with 0.1% Tween in PBS, and incubated with 1:2,000 anti-mouse antibody, 5% milk, and 0.1% Tween in PBS, for 90 mins. Immunoreactive bands were visualized with the enhanced chemiluminescence detection system (Amersham, Piscataway, NJ, USA) according to the manufacturer's protocol. Equal protein loading was assessed by Ponceau S solution staining.
Measurements of Mitochondrial Complex Activities
Preparation of mitochondria: brain tissue was finely minced in cold isolation medium containing 0.32 mol/L sucrose, 1 mmol/L potassium–ethylenediaminetetraacetic acid and 10 mmol/L Tris- HCl (pH 7.4) and adjusted to a final concentration of 1 g of tissue/10 ml of medium. The minced tissue was homogenized with a Polytron tissue processor. The homogenate was centrifuged at 1,000g (4°C) for 10 mins. The supernatant was transferred to a clean tube and the mitochondrial pellet was obtained by centrifugation at 12,000g (4°C) for 15 mins. The assays for complex activities were modified from the procedure described by Pandey et al (2008). NADH– ubiquinone oxidoreductase (complex I): the reaction was followed spectrophotometrically using a microplate reader at 340 nm for 3 mins at 37°C in a solution containing 40 to 50 µg of the submitochondrial particles, 35 mmol/L potassium phosphate buffer, pH 7.4, 2.65 mmol/L NaN3, 1 mmol/L ethylenediaminetetraacetic acid, 5 mmol/L MgCl2, 200 µmol/L NADH as donor, and 100 µmol/L coenzyme Q0 as acceptor. Succinate dehydrogenase (complex II): activity was monitored spectrophotometrically at 600 nm for 1 min in a reaction mixture containing 50 mmol/L potassium phosphate buffer, pH 7.5, 40 mmol/L sodium succinate, 750 µmol/L NaN3, 290 µmol/L phenazonium methosulphate, and 50 µmol/L DCIP. Ubiquinol–cytochrome c oxidoreductase (complex III): the reduction of cytochrome c was monitored as increase in absorbance at 550 nm for 3 mins in the presence of 5 µmol/L rotenone. The reaction mixture consisted of 50 mmol/L potassium phosphate buffer, pH 7.4, containing 1 mmol/L ethylenediaminetetraacetic acid potassium salt, 20 mmol/L NaN3, 50 µmol/L cytochrome c, and 5 to 10 µg sub-mitochondrial protein, which was incubated at 30°C for 1 mins, before starting the reaction by the addition of 100 µmol/L NADH. Cytochrome c oxidase (complex IV): the activity of complex-IV was measured using a cytochrome c oxidase assay kit (CYTOX-OX1; Sigma, St Louis, MO, USA), taking reduced cytochrome c as the donor. The oxidation of cytochrome c was monitored as the decrease in absorbance at 550 nm using a microplate reader and the initial rate of cytochrome c reduction was used for the calculation of activity. Protein concentration was measured using the Bio-Rad protein assay kit and the mitochondrial complex activities were normalized by dividing them by protein concentration and expressing as a ratio to normal control.
ATP Measurements
At 24 h after MCAO rats (four per group) were anesthesized with isoflurane and decapitated. Brains were removed and immediately dissected on a cold −20°C board. In each hemisphere, the region −1.5 to 2.5 mm Bregma, which our previous studies have indicated show the most pronounced injury, was collected and quick-frozen in liquid nitrogen. The brain specimens were next placed in 0.4 mol/L perchloric acid (10 mL/g), homogenized, and centrifuged at 500g for 5 mins. The supernatant was neutralized with 150 µL of 2 mol/L K2CO3 added to 1 mL of supernatant, and recentrifuged. The resulting supernatants were stored at −80°C until measurement. Cellular ATP concentrations were measured using the CellTiter-Glo luminescent ATP assay kit, based on the luciferase/luciferin reaction (Promega, Madison, WI, USA) according to the manufacturer's instructions. The luminescence measurements of the samples in opaque white 96-well plates were performed using a Veritas luminescence counter (Turner BioSystems, Sunnyvale, CA, USA). ATP standards (Sigma) were used for calibration.
Statistics
Data are presented as the mean±s.d. The differences between groups were determined by T-test for unpaired data and differences between multiple groups for ATP levels and fluorescence were determined using analysis of variance followed by post hoc Tukey's multiple comparison test. Differences were considered significant at P<0.05.
Results
Expression of Hsp75 in Neurons and Astrocytes in Brain
We investigated the anatomic spread and cell type expression of Hsp75 by immunohistochemistry 48 h after unilateral injection of plasmid DNA encoding Hsp75 into the lateral ventricle. As shown in Figure 1, we observe widespread expression of Hsp75 protein throughout the brain, bilaterally and with broad rostral–caudal spread. As shown in Figure 2 by double label immunohistochemistry, Hsp75 overexpression was observed in the bodies and processes of both astrocytes (Figure 2A), and neurons (Figure 2B). We also investigated levels of Hsp75 expression in brain by western blotting of brain protein with Hsp75 antibody. Figures 2C and 2D show that Hsp75-injected brains demonstrated significantly higher levels of Hsp75 expression compared with vector-injected, and control uninjected brain (endogenous Hsp75 levels, were comparable to vector injected, data not shown).
Fig. 1.
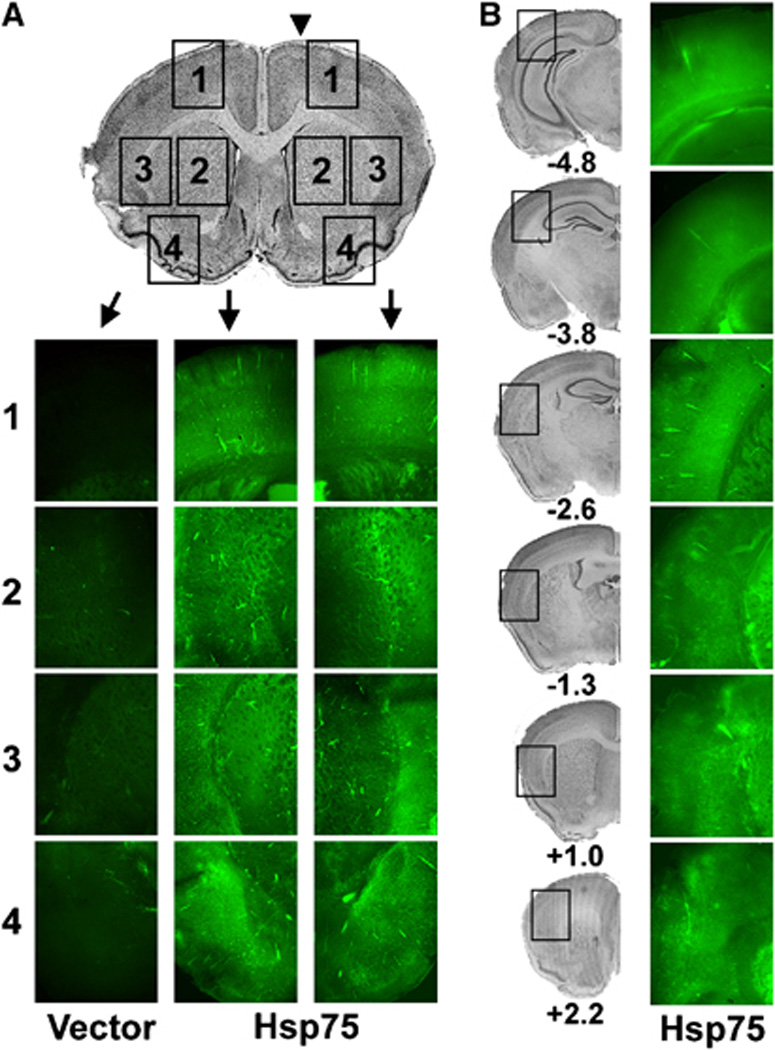
Distribution of Hsp75 protein expression after unilateral intraventricular injection of plasmid (indicated by arrow head in A). (A) Hsp75 immunofluorescence (green) contralateral (left two columns) and ipsilateral to the injected ventricle (right column) from approximate regions indicated on the cresyl violet stained brain section shown at the top. (1) Superior cortex including primary motor cortex (penumbral area), (2) striatum (infarct area), (3) medial cortex plus lateral striatum (infarct and most protected area), and (4) inferolateral cortex (seldom included in infarct area) in both vector- and Hsp75-injected brain slices. Photomicrographs were taken at the same fluorescence exposure for vector- and Hsp75-injected brains. (B) Hsp75 fluorescence from representative coronal sections at different levels relative to bregma of an Hsp75 injected brain. Left column of cresyl violet stained sections indicates the approximate regions from which the images in the right column were taken.
Fig. 2.
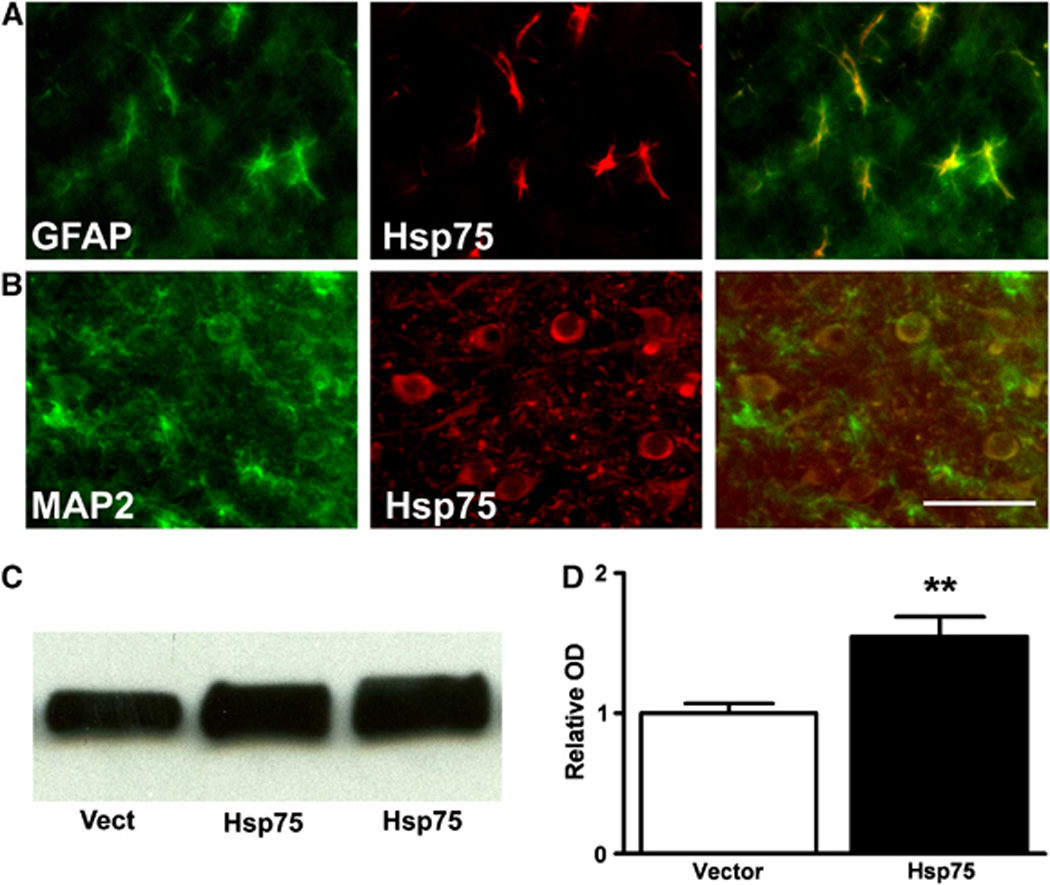
Hsp75 was overexpressed in both astrocytes (A) and neurons (B) 48 h after DNA transfection. Astrocytes and neurons were stained with GFAP and MAP2 antibodies, respectively, and visualized with green Alexa Fluor 488 secondary antibody (left panels). The sections were colabeled with anti-Hsp75 antibody and red Alexa Fluor 594 secondary antibody (middle panels). Right panels show the colocalization of Hsp75 with astrocytes (A) and neurons (B). Scale bar is 50 µm. (C) Western blot of extracts from brains of animals injected with vector DNA (Vect) or Hsp75-encoding DNA, lanes 2 and 3 are from two different animals. (D) Quantification of western blots (**P<0.01; n=2 for vector, n=5 for Hsp75).
Hsp75 Overexpression Decreases Infarct Volume and Improves Neuroscore
Rats were subjected to MCAO 48 h after Hsp75 or vector injection because both immunohistochemistry and western blotting confirmed Hsp75 overexpression at this time point. The physiological variables of temperature, oxygen saturation, and heart rate are given in Table 1. The temperature and heart rate were increased at the end of 100 mins MCAO compared with either before or at the beginning of MCAO (P<0.05), but did not differ between groups at any time point. Figure 3 shows that Hsp75 overexpressing animals (N=10) demonstrated a 43% reduction of infarct volume compared with vector only injected rats (N=10), as evidenced by 2,3,5-triphenyltetrazolium chloride (TTC) staining. The neuroscores were 1.63±0.53 and 2.23±0.75, respectively, for Hsp75 (n=12) and vector-injected (n=11) groups. The difference was significant at P=0.036.
Table 1.
Physiological measurements
| Time | Temperature | Oxygen saturation | Heart rate | |||
|---|---|---|---|---|---|---|
| HSP75 | Control | HSP75 | Control | HSP75 | Control | |
| Before | 37.4±0.4 | 37.5±0.5 | 96.3±0.6 | 96.8±0.4 | 327±16 | 312±38 |
| Start MCAO | 37.3±0.2 | 37.2±0.4 | 96.8±0.3 | 96.6±0.4 | 339±16 | 324±38 |
| End MCAO | 39.0±0.9a | 39.4±0.5a | 96.3±1.4 | 96.9±0.6 | 392±40a | 393±44a |
HSP75, heat shock protein 75.
Significant difference compared with before and at the start of MCAO (P<0.05) for the same condition. There was no difference between groups at the same time point. Control indicates vector control injected animals, Start MCAO is within 5 mins of placing the suture, End MCAO is just before removing the suture.
Fig. 3.
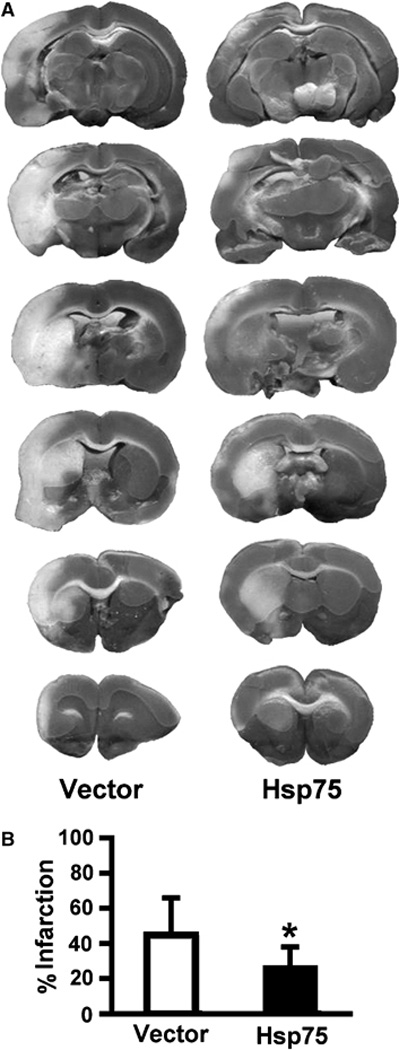
Hsp75 overexpression reduces infarct volume after transient MCAO. (A) Representative TTC-stained coronal sections demonstrate a reduction in infarct size in Hsp75-injected animals compared with vector-injected animals. (B) Quantification of infarct volume shows a significant decrease in infarct volume in Hsp75-injected rats expressed as a percent of the volume of the hemisphere (*P=0.025, n=10 for vector-injected, n=10 for Hsp75-injected groups).
Oxidative Stress is Reduced with Hsp75 Overexpression
Oxidative stress and damage were evaluated using HEt fluorescence and HNE immunocytochemistry. Figure 4A shows that Hsp75 overexpression resulted in decreased levels of HEt fluorescence in the penumbra of the ischemic brains. Vector-injected animals demonstrated a 1.9-fold rise in HEt fluorescence compared with HEt staining of contralateral brain areas, whereas there was only a 1.26-fold rise in HEt fluorescence in HSP75-injected animals (Figure 4B), significantly less. To investigate whether decreased ROS levels were associated with decreased oxidative cell damage, we used HNE antibody to identify oxidized lipids. Figure 4C demonstrates that the penumbra of vector-injected animals demonstrated significantly higher levels of HNE staining compared with the penumbra of HSP75-injected animals. Only a diffuse background HNE staining was observed in the contralateral brain areas of all animals. HNE staining was observed in both core and penumbra, with a 1.9-fold rise on the injured side compared with contralateral of vector-injected animals, whereas the Hsp75 injected animals had only a 1.26-fold rise (Figure 4D).
Fig. 4.
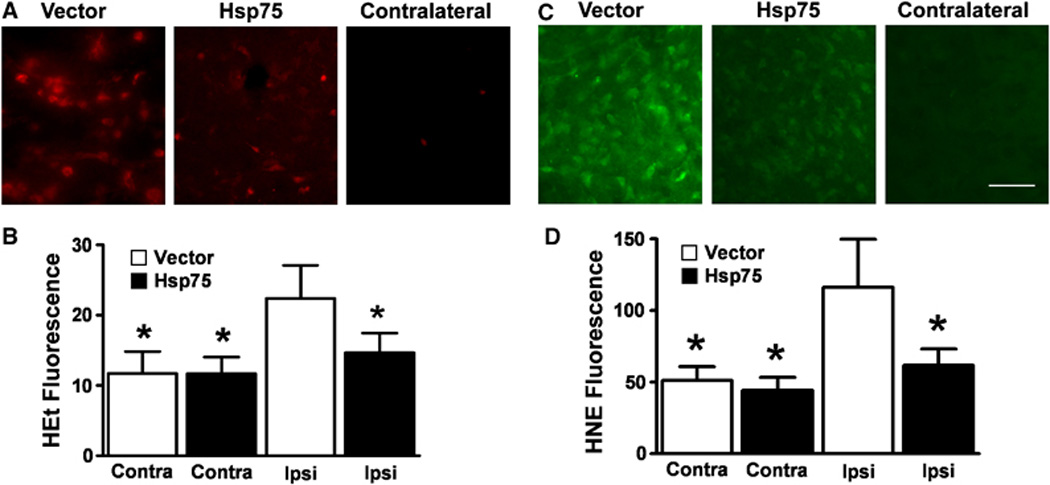
Hsp75 overexpression reduces ROS and oxidized lipid levels after transient MCAO. ROS levels were evaluated by HEt fluorescence (A and B). Representative micrographs show MCAO induced a larger increase in HEt fluorescence in the ipsilateral hemispheres of vector-injected animals compared with Hsp75-injected animals (A, left and middle panels). Contralateral hemispheres in both vector- and Hsp75-injected animals demonstrated comparable low levels of oxidized HEt fluorescence (A, right panel). Quantification of oxidized HEt fluorescence showed a significantly decreased level of HEt signal in Hsp75-injected animals compared with vector-injected animals (B). Asterisk (*) indicates P<0.05 compared with ipsilateral vector-injected hemisphere. Levels of oxidized lipids were evaluated by HNE staining (C and D). Representative micrographs show MCAO injury resulted in higher levels of lipid oxidation in vector-injected animals compared with Hsp75-injected animals ipsilateral to the occlusion (C, left and middle panels). Contralateral hemispheres of both vector- and Hsp75-injected animals demonstrated comparable low levels of HNE staining (C, right panel). Quantification of HNE staining showed a significantly decreased level of oxidized lipid in Hsp75-injected animals compared with vector-injected animals (D). Scale bar is 50 µm. Asterisk (*) indicates P<0.001 compared with ipsilateral vector-injected hemisphere.
Effect of Hsp75 Overexpression on Mitochondrial Function
We measured the activities of individual mitochondrial complexes: NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), ubiquinone dehydrogenase (complex III), and cytochrome c oxidase (complex IV) in Hsp75- and vector-injected animals 24 h after 100 mins MCAO. Figure 5 shows that although no differences were observed in complexes I to III activity between Hsp75 and vector-injected ischemic brains, the activity of complex IV, which has been shown to be particularly sensitive to free-radical damage (Heales et al, 1994), was significantly higher in Hsp75-overexpressing brains, consistent with better preservation.
Fig. 5.
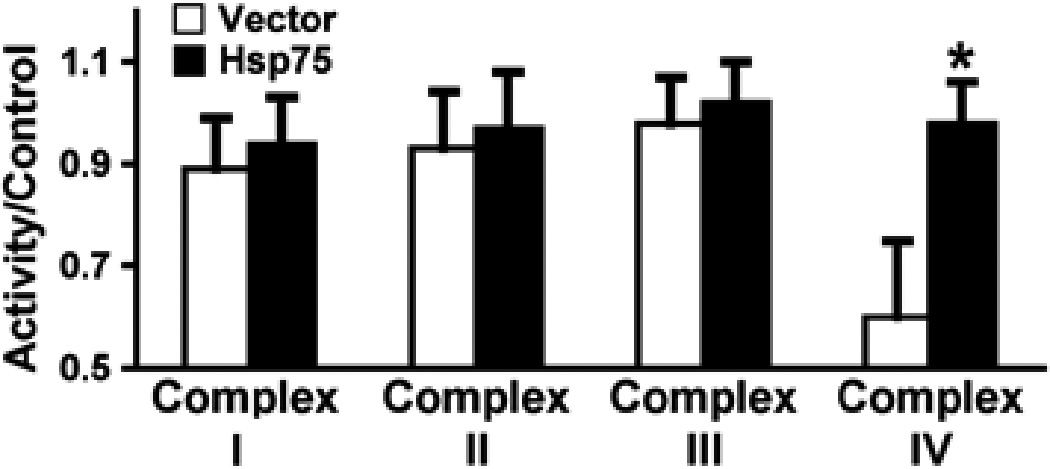
Effect of Hsp75 overexpression on mitochondrial electron transport chain complex activity after transient MCAO. Activities of complexes were measured in brain harvested 24 h after 100 mins MCAO and expressed as mean±s.d. *P=0.02 compared with vector control.
Hsp75 Protects Against Ischemic Depletion of Brain ATP Levels
ATP content was measured in both MCAO and sham-operated animals. As shown in Figure 6 first two bars, there was no significant difference in ATP levels between sham-operated Hsp75 or vector-injected animals. Also, the hemispheres contralateral to MCAO treatment demonstrated ATP levels comparable to those of sham-operated animals. ATP content was significantly decreased in ipsilateral MCAO hemispheres of both Hsp75 and vector-injected animals compared with the contralateral. However, Hsp75-overexpressing animals demonstrated a decrease of only 35%, whereas vector-injected animals demonstrated a decrease of 66%, compared with the corresponding contralateral hemispheres.
Fig. 6.
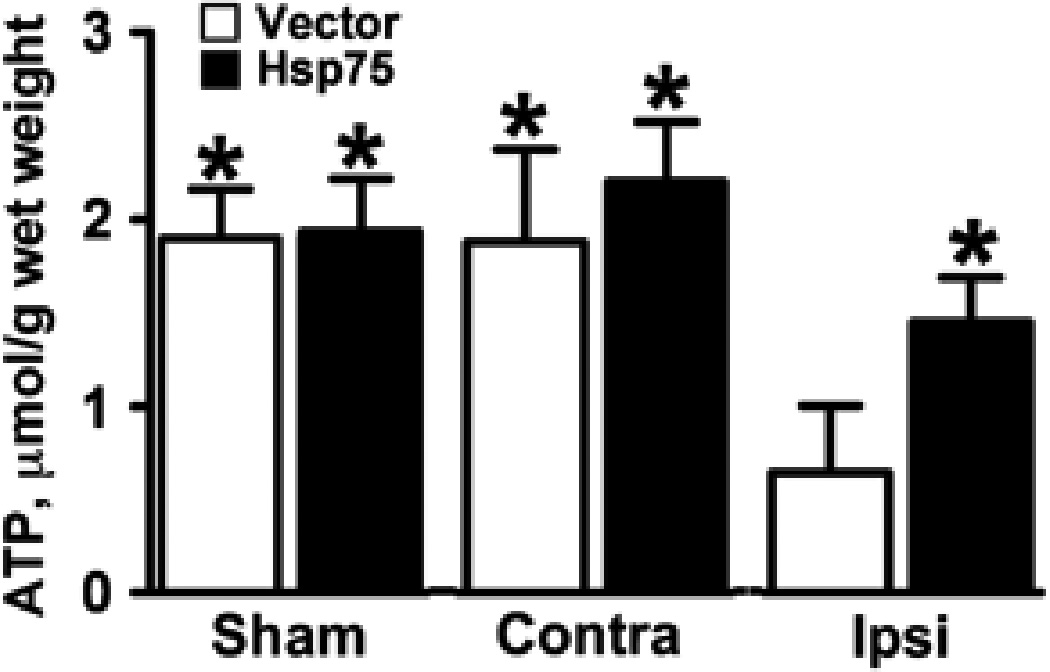
Hsp75 overexpression ameliorates the reduction of ATP levels ipsilateral to MCAO. Brain was harvested 24 h after 100 mins MCAO and separated into ipsilateral to the infarct and contralateral hemispheres. In addition, brain was harvested from sham animals not subjected to MCAO (sham). Asterisk (*) indicates P<0.05 compared with ipsilateral vector-injected hemisphere. Contra, contralateral hemisphere; Ipsi, ipsilateral hemisphere relative to the occlusion. n=4 animals per group.
Discussion
Heat shock protein 75 overexpression was previously shown to be protective against mitochondrial dysfunction, oxidative stress, and cell death induced by various forms of injury in vitro (Liu et al, 2005; Voloboueva et al, 2007; Williamson et al, 2008). Brain ischemia promotes damage to multiple subcellular sites, with mitochondria being considered one of the primary targets (Soane et al, 2007). In this study we targeted mitochondria for protection by testing whether the mitochondrially localized chaperone Hsp75 when overexpressed in brain could protect brain cell function and increase overall brain viability after transient focal ischemic injury. Using DNA transfection, we obtained Hsp75 overexpression in both neurons and astrocytes, and observed a 43% decrease of infarct volume and improved neurological function after MCAO. Further studies are needed to assess whether this protection would still be detected after extended survival times and be reflected in greater functional recovery.
Because Hsp75 overexpression was postulated to protect mitochondria and reduce oxidative stress, we evaluated ROS levels and lipid oxidation. Reactive oxygen species formation decreased 43% in the ischemic penumbra of Hsp75-overexpressing animals compared with animals transfected with control DNA. Lipid peroxidation was reduced 50% with Hsp75 overexpression, demonstrating that oxidative damage to cell structures was indeed reduced with Hsp75 overexpression. Many studies have shown that cerebral ischemia/reperfusion promotes numerous enzymatic oxidation reactions that contribute to infarction (Chan, 2001; Siesjo et al, 1989). Although there is increasing evidence that NADPH oxidase is important in ROS production (Sun et al, 2007), mitochondria are also considered an important source of oxidative stress in cerebral ischemia reperfusion (Christophe and Nicolas, 2006; Fiskum et al, 2004). Several mechanisms have been shown to stimulate mitochondrial ROS production during ischemia reperfusion, including nitric oxide inhibition of mitochondrial electron transport complexes (Brown and Borutaite, 2001) and apoptotic cytochrome c release (Starkov et al, 2002). Also, the phenomenon of mitochondrial ROS-induced ROS release in which exposure to oxidative stress stimulates increased mitochondrial ROS production has been recently described (Zorov et al, 2006).
Consistent with the importance of oxidative damage, earlier work has shown that overexpression of antioxidant enzymes, including cytosolic superoxide dismutase 1 and mitochondrial superoxide dismutase 2, provide protection against ischemic brain injury (Chan, 2001). Heat shock protein 75 is a central component of the mitochondrial protein import machinery. Mitochondrial protein import is important in preservation of mitochondrial function during ischemic injury through delivery and replacement of essential and protective mitochondrial proteins. A previous study demonstrated that Hsp75 overexpression resulted in increased levels of mitochondrial superoxide dismutase 2 in cardiac myocytes (Williamson et al, 2008); thus, indicating one potential mechanism of the observed Hsp75 ischemic protection. Consistent with our work, others have also found reduced ROS generation with Hsp75 (Hua et al, 2007; Liu et al, 2005).
To evaluate the effect of Hsp75 overexpression on mitochondrial function, we measured the activity of the mitochondrial complexes and found significantly higher levels of complex IV activity in Hsp75-overexpressing animals compared with vector-injected ones 24 h after focal ischemia. Complex IV function directly depends on the availability of the mitochondrial phospholipid cardiolipin (Robinson, 1993). Cardiolipin is particularly susceptible to free-radical peroxidation, because of its high content of unsaturated fatty acids (Paradies et al, 2000). Thus, the reduction of ROS levels and the associated reduction of cardiolipin peroxidation might be responsible for the Hsp75 protection of complex IV activity. Complex IV appears to be one of the most susceptible components of mitochondrial electron transport chain during brain ischemia (Heales et al, 1994). Interestingly, complex IV activity in the heart is relatively insensitive to free-radical exposure (Cassina and Radi, 1996). One possible explanation of the differential sensitivity may be that the cardiolipin concentration is several times lower in brain compared with heart (Ushmorov et al, 1999), thus rendering complex IV activity in brain more sensitive to cardiolipin depletion.
The reduction of blood flow during MCAO strongly impairs energy generation, because of both oxygen starvation and glucose deprivation (Hertz, 2008). Secondary postischemic energy failure after transient focal ischemia has been reported in several studies (Folbergrova et al, 1995; Hata et al, 2000; Lust et al, 2002). This secondary failure may in part reflect persistent impairment of blood flow after ischemic injury (Takagi et al, 1995; van Dorsten et al, 1999). Ischemia-induced impairment of mitochondrial oxidative phosphorylation would also contribute to delayed energy failure. Postischemic DNA repair causes additional depletion of intracellular ATP. Free radicals formed during ischemia/reperfusion induce DNA strand breaks that lead to activation of poly (ADP-ribose) polymerase (Kauppinen and Swanson, 2007; Koh et al, 2005). poly(ADP-ribose) polymerase activity consumes NAD+ thus slowing down glycolytic ATP production (Ying et al, 2005). It has been shown that poly(ADP-ribose) polymerase hyperactivation rapidly impairs mitochondrial ATP production (Cipriani et al, 2005). Reduced energy stores play a central role in neuronal loss (Zhu et al, 2004), and several studies have demonstrated that astrocytes have greater protective effects if they do not have severe metabolic impairment (Dienel and Hertz, 2005; Swanson et al, 1997; Voloboueva et al, 2007).
In our experiments, Hsp75 overexpression resulted in only 35% ATP depletion after focal ischemia compared with the 66% decrease measured in vector-injected animals. Thus, the Hsp75-associated decrease in ROS and lipid peroxidation as well as increased preservation of mitochondrial complex IV activity might contribute to the observed greater ATP preservation. These results are consistent with previous in vivo studies which demonstrated bioenergetic failure and mitochondrial dysfunction after 2 h of MCAO (Kuroda et al, 1996). This was not because of permanent, structural damage, rather, transient reduction in complex IV activity was observed (Canevari et al, 1997). Other studies indicate that electron transport chain activity may not be metabolically limiting in mitochondrial ATP production during reperfusion (Fiskum et al, 2004). Decreased production of NADH by pyruvate dehydrogenase and tricarboxylic acid cycle dehydrogenases (Fiskum et al, 2004), impaired flow of reducing equivalents between the components of the respiratory chain because of damage to mitochondrial membranes, and ROS-induced mitochondrial permeability transition activation (Kowaltowski et al, 2000) may all contribute to reduced mitochondrial ATP production without reduction in mitochondrial complex activities as measured under standard conditions in vitro.
Heat shock protein 75 has also been postulated to participate in regulation of apoptotic death and reduce release of cytochrome c from mitochondria after oxidative stress (Pridgeon et al, 2007; Taurin et al, 2002). Thus, Hsp75 by protecting mitochondria likely directly contributes to reducing oxidative stress and reducing the induction of apoptosis, both relevant to the protection against cerebral ischemia observed here.
In conclusion, the results of this study demonstrate that in vivo overexpression of Hsp75 provides protection against ischemic brain injury. The reduced infarction and improved neuroscore was associated with decreased ROS levels, reduced tissue oxidative damage, better preserved mitochondrial function, and attenuated ATP loss. This study demonstrates that the protective mechanisms of Hsp75 reported in vitro also apply in focal ischemia in vivo. The data also provide evidence that protecting mitochondrial function, such as by Hsp75 overexpression, is a valid target for protection from cerebral ischemia.
References
- Bambrick L, Kristian T, Fiskum G. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res. 2004;29:601–608. doi: 10.1023/b:nere.0000014830.06376.e6. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya T, Karnezis AN, Murphy SP, Hoang T, Freeman BC, Phillips B, Morimoto RI. Cloning and subcellular localization of human mitochondrial hsp70. J Biol Chem. 1995;270:1705–1710. doi: 10.1074/jbc.270.4.1705. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Borutaite V. Nitric oxide, mitochondria, and cell death. IUBMB Life. 2001;52:189–195. doi: 10.1080/15216540152845993. [DOI] [PubMed] [Google Scholar]
- Canevari L, Kuroda S, Bates TE, Clark JB, Siesjo BK. Activity of mitochondrial respiratory chain enzymes after transient focal ischemia in the rat. J Cereb Blood Flow Metab. 1997;17:1166–1169. doi: 10.1097/00004647-199711000-00005. [DOI] [PubMed] [Google Scholar]
- Carette J, Lehnert S, Chow TY. Implication of PBP74/mortalin/GRP75 in the radioadaptive response. Int J Radiat Biol. 2002;78:183–190. doi: 10.1080/09553000110097208. [DOI] [PubMed] [Google Scholar]
- Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Christophe M, Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr Pharm Des. 2006;12:739–757. doi: 10.2174/138161206775474242. [DOI] [PubMed] [Google Scholar]
- Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, Chiarugi A. Nuclear poly(ADPribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J Biol Chem. 2005;280:17227–17234. doi: 10.1074/jbc.M414526200. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50:362–388. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- Dingman A, Lee SY, Derugin N, Wendland MF, Vexler ZS. Aminoguanidine inhibits caspase-3 and calpain activation without affecting microglial activation following neonatal transient cerebral ischemia. J Neurochem. 2006;96:1467–1479. doi: 10.1111/j.1471-4159.2006.03672.x. [DOI] [PubMed] [Google Scholar]
- Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res. 1987;18:551–561. doi: 10.1002/jnr.490180407. [DOI] [PubMed] [Google Scholar]
- Fiskum G. Mitochondrial participation in ischemic and traumatic neural cell death. J Neurotrauma. 2000;17:843–855. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, Kowaltowski A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J Bioenerg Biomembr. 2004;36:347–352. doi: 10.1023/B:JOBB.0000041766.71376.81. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Zhao Q, Katsura K, Siesjo BK. N-tert-butyl-alpha-phenylnitrone improves recovery of brain energy state in rats following transient focal ischemia. Proc Natl Acad Sci USA. 1995;92:5057–5061. doi: 10.1073/pnas.92.11.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–717. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- Hadari YR, Haring HU, Zick Y. p75, a member of the heat shock protein family, undergoes tyrosine phosphorylation in response to oxidative stress. J Biol Chem. 1997;272:657–662. doi: 10.1074/jbc.272.1.657. [DOI] [PubMed] [Google Scholar]
- Hata R, Maeda K, Hermann D, Mies G, Hossmann KA. Evolution of brain infarction after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2000;20:937–946. doi: 10.1097/00004647-200006000-00006. [DOI] [PubMed] [Google Scholar]
- Heales SJ, Bolanos JP, Land JM, Clark JB. Trolox protects mitochondrial complex IV from nitric oxide-mediated damage in astrocytes. Brain Res. 1994;668:243–245. doi: 10.1016/0006-8993(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Hertz L. Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology. 2008;55:289–309. doi: 10.1016/j.neuropharm.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–258. doi: 10.1196/annals.1341.022. [DOI] [PubMed] [Google Scholar]
- Hua G, Zhang Q, Fan Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J Biol Chem. 2007;282:20553–20560. doi: 10.1074/jbc.M703196200. [DOI] [PubMed] [Google Scholar]
- Juurlink BHJ, Hertz L. Ischemia-induced death of astrocytes and neurons in primary culture: pitfalls in quantifying neuronal cell death. Brain Res Dev Brain Res. 1993;71:239–246. doi: 10.1016/0165-3806(93)90175-a. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience. 2007;145:1267–1272. doi: 10.1016/j.neuroscience.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Vercesi AE, Fiskum G. Bcl-2 prevents mitochondrial permeability transition and cytochrome c release via maintenance of reduced pyridine nucleotides. Cell Death Differ. 2000;7:903–910. doi: 10.1038/sj.cdd.4400722. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Katsura KI, Tsuchidate R, Siesjo BK. Secondary bioenergetic failure after transient focal ischaemia is due to mitochondrial injury. Acta Physiol Scand. 1996;156:149–150. doi: 10.1046/j.1365-201X.1996.449170000.x. [DOI] [PubMed] [Google Scholar]
- Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu W, Song XD, Zuo J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol Cell Biochem. 2005;268:45–51. doi: 10.1007/s11010-005-2996-1. [DOI] [PubMed] [Google Scholar]
- Lust WD, Taylor C, Pundik S, Selman WR, Ratcheson RA. Ischemic cell death: dynamics of delayed secondary energy failure during reperfusion following focal ischemia. Metab Brain Dis. 2002;17:113–121. doi: 10.1023/a:1015420222334. [DOI] [PubMed] [Google Scholar]
- Massa SM, Longo FM, Zuo J, Wang S, Chen J, Sharp FR. Cloning of rat grp75, an hsp70-family member, and its expression in normal and ischemic brain. J Neurosci Res. 1995;40:807–819. doi: 10.1002/jnr.490400612. [DOI] [PubMed] [Google Scholar]
- Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery. 1992;31:100–106. doi: 10.1227/00006123-199207000-00014. discussion 6–7. [DOI] [PubMed] [Google Scholar]
- Mitsumoto A, Takeuchi A, Okawa K, Nakagawa Y. A subset of newly synthesized polypeptides in mitochondria from human endothelial cells exposed to hydroperoxide stress. Free Radic Biol Med. 2002;32:22–37. doi: 10.1016/s0891-5849(01)00755-9. [DOI] [PubMed] [Google Scholar]
- Pandey M, Varghese M, Sindhu KM, Sreetama S, Navneet AK, Mohanakumar KP, Usha R. Mitochondrial NAD+-linked State 3 respiration and complex-I activity are compromised in the cerebral cortex of 3-nitropropionic acid-induced rat model of Huntington's disease. J Neurochem. 2008;104:420–434. doi: 10.1111/j.1471-4159.2007.04996.x. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000;466:323–326. doi: 10.1016/s0014-5793(00)01082-6. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000;466:323–326. doi: 10.1016/s0014-5793(00)01082-6. [DOI] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NC. Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr. 1993;25:153–163. doi: 10.1007/BF00762857. [DOI] [PubMed] [Google Scholar]
- Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, Yu F, Liu J, Lee YS, Nito C, Kamada H, Dodd RL, Hsieh LB, Hassid B, Kim EE, Gonzalez M, Chan PH. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol. 2005;31:105–116. doi: 10.1385/MN:31:1-3:105. [DOI] [PubMed] [Google Scholar]
- Schorpp M, Jager R, Schellander K, Schenkel J, Wagner EF, Weiher H, Angel P. The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res. 1996;24:1787–1788. doi: 10.1093/nar/24.9.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Barnikol-Watanabe S, Thinnes FP, Hilschmann N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int J Biochem Cell Biol. 2002;34:1059–1070. doi: 10.1016/s1357-2725(02)00026-2. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Agardh CD, Bengtsson F. Free radicals and brain damage. Cerebrovasc Brain Metab Rev. 1989;1:165–211. [PubMed] [Google Scholar]
- Soane L, Kahraman S, Kristian T, Fiskum G. Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. J Neurosci Res. 2007;85:3407–3415. doi: 10.1002/jnr.21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Polster BM, Fiskum G. Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem. 2002;83:220–228. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem. 2007;103:1–16. doi: 10.1111/j.1471-4159.2007.04670.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ouyang YB, Xu L, Chow AM, Anderson R, Hecker JG, Giffard RG. The carboxylterminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab. 2006;26:937–950. doi: 10.1038/sj.jcbfm.9600246. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Farrell K, Stein BA. Astrocyte energetics, function, and death under conditions of incomplete ischemia: a mechanism of glial death in the penumbra. Glia. 1997;21:142–153. doi: 10.1002/(sici)1098-1136(199709)21:1<142::aid-glia16>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Takagi K, Zhao W, Busto R, Ginsberg MD. Local hemodynamic changes during transient middle cerebral artery occlusion and recirculation in the rat: a [14C]iodoantipyrine autoradiographic study. Brain Res. 1995;691:160–168. doi: 10.1016/0006-8993(95)00657-c. [DOI] [PubMed] [Google Scholar]
- Taurin S, Seyrantepe V, Orlov SN, Tremblay TL, Thibault P, Bennett MR, Hamet P, Pshezhetsky AV. Proteome analysis and functional expression identify mortalin as an antiapoptotic gene induced by elevation of [Na+]i/[K+]i ratio in cultured vascular smooth muscle cells. Circ Res. 2002;91:915–922. doi: 10.1161/01.res.0000043020.45534.3e. [DOI] [PubMed] [Google Scholar]
- Ushmorov A, Ratter F, Lehmann V, Droge W, Schirrmacher V, Umansky V. Nitric-oxideinduced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome c release. Blood. 1999;93:2342–2352. [PubMed] [Google Scholar]
- van Dorsten FA, Hata R, Maeda K, Franke C, Eis M, Hossmann KA, Hoehn M. Diffusion- and perfusion-weighted MR imaging of transient focal cerebral ischaemia in mice. NMR Biomed. 1999;12:525–534. doi: 10.1002/(sici)1099-1492(199912)12:8<525::aid-nbm597>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voos W, Martin H, Krimmer T, Pfanner N. Mechanisms of protein translocation into mitochondria. Biochim Biophys Acta. 1999;1422:235–254. doi: 10.1016/s0304-4157(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Williamson CL, Dabkowski ER, Dillmann WH, Hollander JM. Mitochondria protection from hypoxia/reoxygenation injury with mitochondria heat shock protein 70 overexpression. Am J Physiol Heart Circ Physiol. 2008;294:H249–H256. doi: 10.1152/ajpheart.00775.2007. [DOI] [PubMed] [Google Scholar]
- Ying W, Alano CC, Garnier P, Swanson RA. NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res. 2005;79:216–223. doi: 10.1002/jnr.20289. [DOI] [PubMed] [Google Scholar]
- Zhu S, Li M, Figueroa BE, Liu A, Stavrovskaya IG, Pasinelli P, Beal MF, Brown RH, Jr, Kristal BS, Ferrante RJ, Friedlander RM. Prophylactic creatine administration mediates neuroprotection in cerebral ischemia in mice. J Neurosci. 2004;24:5909–5912. doi: 10.1523/JNEUROSCI.1278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma. 2000;17:857–869. doi: 10.1089/neu.2000.17.857. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]