

Abstract
Age-related macular degeneration (AMD) is the leading cause of blindness in developed countries; with the aging population, the negative health impacts and costs of the disease will increase dramatically over the next decade. Although the exact cause of AMD is unknown, genetic studies have implicated the complement system as well as other immune responses in disease pathogenesis and severity. Furthermore, histologic studies have shown the presence of macrophages, lymphocytes, and mast cells, as well as fibroblasts, in both atrophic lesions and with retinal neovascularization. This review summarizes discussions from the fifth annual conference of the Arnold and Mabel Beckman Initiative for Macular Research by the Inflammation and Immune Response Task Force. These deliberations focused on the role of inflammatory immune responses, including complement, inflammasomes, adaptive immune responses, and para-inflammation, unanswered questions and studies to address these questions, and potential immune-related therapeutic targets for AMD.
1. Introduction
Age-related macular degeneration (AMD) is the leading cause of central vision loss in developed countries. The most recent data suggest that more than 3 million people in the United States will be affected by the disease by 2020 [1]. The disease affects the choriocapillaris, Bruch's membrane and the retinal pigment epithelium, with dysfunction and death of overlying photoreceptors. In addition to age, risk factors for the disease include both environmental and epidemiologic factors. Specific disease associations include smoking, light exposure, obesity, and race [2]. Recent genetic studies have implicated roles for the immune system, particularly abnormalities in the complement system, in disease pathogenesis, and severity. Although patients with AMD do not have signs of overt ocular inflammation, histologic studies have shown the presence of macrophages, lymphocytes, and mast cells, as well as fibroblasts, associated with both atrophic lesions and with neovascularization of the retina [3].
Importantly, the retina is a highly metabolically active tissue, with requirements to mediate photoreceptor turnover. As the retina ages, it may be less able to handle these metabolic requirements. Immunologically active deposits called drusen that contain lipids, complement, and other potentially immune activating substances may act as additional triggers for immune responses in the eye. Other inflammatory initiators include oxidative stress and secondary mediators of inflammation such as cytokines. On the other hand, the retina performs well until late in life despite constant stress, suggesting that at least some of the inflammatory responses observed may be beneficial. Equally intriguing, although perhaps less well understood, is a renewed appreciation for the role of the adaptive immune response in the pathogenesis of AMD. Collectively, as a result of previous studies showing inflammatory cells associated with AMD and newer genetic studies implicating the innate immune system in developing the disease, there is heightened interest in studying the role of the immune response in AMD and in determining whether modulating the immune response could help treat the disease.
The extent to which innate and adaptive immune responses play roles in the pathogenesis of AMD, and the ability to target these pathways to effectively treat the disease, remains debatable. This may in part be due to the complexity of the immune response, the number of different inflammatory cell types and cytokines involved, and the kinetics of the inflammatory response. Further, it is as yet difficult to know whether immune responses are driven and controlled locally in the retina, or operate systemically, further complicating interpretations and the development of useful therapeutic approaches.
One key question, however, is whether this immune activation is always pathologic in AMD, or whether it can actually help preserve function and moderate damage at certain stages of the disease. The data support the idea that activated states confer protection. Resident CD200R myeloid cells in the retina are under tonic control by cognate interaction with CD200 [4, 5]. The tissue consequence of microglial activation is context dependent [6, 7]. For example, in photoreceptor neurodegenerative models, microglia do not contribute to the progression of disease despite being activated [8]. In more inflammatory scenarios, a recognized consequence of activated response is contributing toward immune regulation in an attempt to contain further retinal damage [9]. A chronic inflammatory state has also been identified in a number of nonocular diseases, including type 2 diabetes and cardiovascular disease. Could a low-grade immune response be helpful in some circumstances? The intriguing concept has been distilled and developed to infer that tissue stress or malfunction can induce an advantageous response, and has been referred to as para-inflammation [10]. Medzhitov hypothesized that a well-controlled “para-inflammatory” response could be beneficial by either protecting against infection or preserving function in diseased tissues. The experimental evidence and now the concept of para-inflammation have been further articulated and illuminated experimentally by Xu et al., who discuss the potential role of para-inflammation in the aging retina elsewhere [11]. Briefly, and discussed in more detail below, immune activation and recruitment of macrophages may be required to help process photoreceptor and RPE byproducts, thus controlling overt inflammation, tissue dysfunction, and cell death.
In January 2013, the fifth annual conference of the Arnold and Mabel Beckman Initiative for Macular Research was particularly focused on a common form of AMD, namely, atrophic macular degeneration. Meeting participants were divided into task groups devoted to discussing and brainstorming particular aspects of AMD, including one responsible for considering the role of inflammation and immune responses. This review arose in part from the discussions of that task group. Here, therefore, the role of immune responses in regulating or promoting tissue damage, including complement, inflammasomes, and para-inflammation, will be discussed, followed by a summary of the group's thinking on potential research approaches and therapeutic targets.
2. The Complement System and AMD
The complement system is the most widely accepted pathogenic pathway of the immune system implicated in AMD. The genetic evidence from genome wide association studies (GWAS) and rare variant analyses indicate an overactive alternative pathway (AP). Multiple outstanding reports have detailed and reviewed this evidence at the genetic, RNA and protein levels [13, 14, 16–25]. Therefore, these data will primarily be summarized here—the underlying thesis being that excessive engagement of the alternative pathway is a key component in AMD pathogenesis.
In 2005, four GWAS demonstrated that approximately 50% of the inheritance in AMD could be accounted for by a single nucleotide polymorphism (SNP) in an exon encoding the regulator complement factor H (CFH) [26–29]. Moreover, this SNP in CFH at amino acid position 402—a tyrosine (Y) (major allele) or a histidine (H) (minor allele)—has a functional consequence. At sites of tissue injury, the risk variant 402H does not dampen the alternative pathway (AP) of complement activation as efficiently as 402Y [30–34]. While the complement system had been previously implicated in AMD [35–38], it was the GWAS-derived genetic data that cemented the relationship [26–29].
In Caucasian populations of European ancestry, the risk allele (402H) has a gene frequency of 0.3 to 0.4, and the more common allele (Y402) 0.6 to 0.7. The 402H allele is likely replacing the major one because in early life it provides a survival advantage against streptococcal infections [13, 39, 40]. Multiple bacteria and several groups of viruses impair the complement system by hijacking the host's regulators (reviewed in [40]); for example, microbes bind CFH to their surface to inhibit complement activation. The CFH binding protein of group A beta hemolytic streptococcus has a lower affinity for 402H than for Y402. Consequently, the host's complement system has greater activity against the pathogen if the host expresses 402H, thereby reducing the microbes' ability to counteract the AP. CFH adheres to damaged eukaryotic cells and tissue debris via the same anionic (heparin) binding sites that microorganisms employ to attach it to their surface. Two and possibly as many as four such cellular and tissue binding sites are positioned along the linear CFH protein (Figure 1). An unintended consequence later in life of carrying 402H is that it does not bind as well as Y402 to debris in the retina. Differential binding of 402H versus Y402 to multiple constituents of a damaged retina [30–34, 41–44] has been demonstrated for DNA, RNA, lipids, C-reactive protein (CRP), necrotic and apoptotic cells, heparin and other glycosaminoglycans, lipofuscin, bisretinoids, photooxidation byproducts, and amyloid beta. The common finding is that the 402H protein binds with a lower affinity than Y402. Therefore, in the retina of an individual carrying this risk variant, there is a greater degree of AP activation as retinal debris accumulates in AMD patients.
Figure 1.
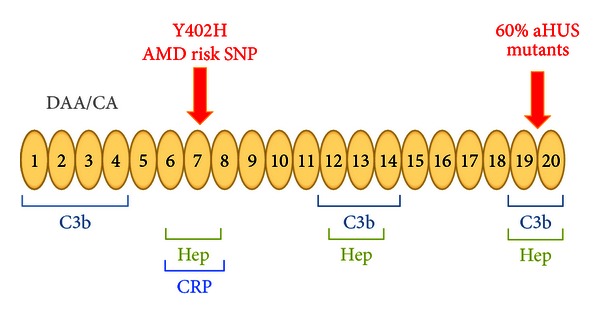
Schematic diagram of complement factor H (CFH). The protein consists entirely of 20 repeating homologous units (complement control repeats or CCPs), each ~60 amino acids in length (like beads on a string). The N-terminal portion houses the regulatory domains (repeats 1–4). The surface-binding recognition motifs are located in repeats 6–8, 12–14 and 19 and 20. They are also known as anionic- or heparin-binding sites. Both Y402H in repeat 7, and a rare variant in repeat 20 [12], are associated with AMD; these regions mediate the binding of factor H to cellular debris such as drusen or damaged retinal cells/tissues. Atypical hemolytic uremic syndrome (aHUS) has been compared to AMD because multiple variants leading to haploinsufficiency of factor H allow for excessive complement activation in this thrombomicroangiopathy [13–15]. Specifically, about 60% of the mutations in aHUS occur in repeats 19 and 20, which decrease factor H's ability to bind to damaged endothelium. DAA, decay accelerating activity; CA, cofactor activity; Hep, heparin binding; CRP, C-reactive protein. Modified from Richards et al. [13].
Thus, the complement hypothesis for the etiopathogenesis of AMD centers on the concept of an “overreaction” to injury and debris in the retina by individuals carrying a “complement hyperinflammatory phenotype” [13, 14, 45]. The AP becomes engaged on a target site if there is a relative lack of inhibitors. To regulate the AP that is continuously turning over, CFH must first transfer from plasma to the foreign material or altered self. To maintain homeostasis and to prevent excessive AP activation, it binds to the target using both C3b and anionic material as ligands for its multiple binding sites (Figure 1). It then serves as a cofactor for the serine protease (Factor I) to cleave C3b. This results in a C3 fragment that does not support amplification via the AP's feedback loop. A host carrying the 402H allele or other risk factors may deposit undesirable quantities of C3b and release C3a in the retina, as well as the downstream effectors C5a and C5b-C9. This scenario for AMD pathogenesis does not preclude triggering of the classical or lectin complement pathways by autoantibodies or lectins, which could then be followed by excessive amplification through the AP. Also, environmental (e.g., smoking) and endogenous (e.g., increased body mass) factors further tip the balance in favor of more inflammation [46–52].
Multiple other CFH variants, both common and rare, influence risk of developing AMD [18, 20, 21, 53–55]. For example, the CFH 62I variant is protective, as is a 84 kb deletion of two CFH-related genes, FHR-1 and FHR-3. The simplest and most likely interpretation of these data is that these genetic changes enhance regulation of the AP by CFH. In contrast, a rare and defective CFH variant confers substantial risk (with high penetrance) for AMD [12]. This recent discovery of a rare variant in CFH with a large effect is probably just the beginning in terms of identification by targeted deep sequencing of highly penetrant mutations in regulators and components of the AP in AMD. Further, haploinsufficiency of C9 conferred a nearly 5-fold reduction in neovascular AMD in the Japanese population, where a nonsense mutation in the C9 gene is frequently found [56]. The interpretation here is that membrane attack complex is less active and thus is protective against retinal damage.
In addition to risk and protective variants of CFH and CFH-related genes, polymorphisms in AP components C3 [57, 58] and factor B [30, 58] are also associated with AMD. A consistent observation is that the protective variants result in less AP activity, whereas risk variants result in more AP activity. Genetic variants in Factor I, the protease employed by CFH to inactivate C3b, have also been associated with AMD by GWAS [59]. Taken together, these findings provide powerful evidence implicating overactivation of the AP predisposing to AMD; thus, common and rare variants in multiple members of a proinflammatory pathway of innate immunity—the AP—are associated with the same disease. Those that decrease function of the pathway are protective, and those that increase function create risk. Moreover, the variants have both independent and additive effects on the risk of developing AMD [25, 47, 48, 52, 58].
Other inhibitors of the AP include membrane cofactor protein (MCP; CD46), decay accelerating factor (DAF; CD55), and complement receptor one (CR1; CD35, the C3b/C4b or immune adherence receptor). MCP and DAF are widely expressed, whereas CR1 has a more limited distribution. DAF and MCP are expressed on the cell surface, where they protect healthy cells from complement attack. MCP is expressed at a high level by RPE cells (particularly at the basal surface) and endothelial cells [24]. A decrease in MCP expression at this RPE location was observed in early AMD. CR1 also has potent regulatory activity for AP C3 and C5 convertases. A surprising recent observation is CR1 expression on the apical surface of RPE cells [24]. These observations concerning the expression and function of DAF, MCP, and CR1 in the retina require further investigation. Although GWAS have not implicated DAF, MCP, or CR1 in susceptibility to AMD, results of targeted next generation deep sequencing of these genes have not been reported.
A role for complement is further evident specifically in “wet” AMD. This severe condition is associated with choroidal neovascularization (CNV) [53], a process characterized by newly formed and leaky vessels invading the sub-retinal space. CNV is associated with fluid accumulation and retinal detachment with loss of the underlying photoreceptors. One animal model of wet AMD is the laser-induced CNV model in rodents. The model is initiated by argon laser photocoagulation, which ruptures Bruch's membrane and triggers complement activation [60]. In mice, the key role of the complement system in the development of CNV is well established. Using knockout and specific inhibitor approaches, it appears that the alternative pathway of complement is the key driver of CNV, in that the removing of the classical or lectin pathway has no protective effect [61, 62]. However, the alternative pathway alone is not sufficient to drive CNV, confirming its importance in the amplification loop [61]. With regard to effector functions, the anaphylatoxins C3a and C5a [60] are important in developing injury. In addition, the membrane attack complex (MAC) contributes to the development of CNV, as CD59 −/− mice lacking the MAC regulator CD59 develop CNV at a higher level than control mice [63], and treatment with recombinant soluble CD59a-IgG2a fusion-protein [64] or gene therapy expressing soluble CD59 [65] both reduce CNV. The CNV model has also been successfully treated with the targeted murine CR2-factor H (muCR2-fH) protein, which consists of a domain which directs the regulatory domain of CFH to sites of complement activation [66], as demonstrated by systemic administration and evaluation of local CNV development [67]. Importantly, in each model evaluated, complement activation amplifies the generation of vascular endothelial growth factor (VEGF), which is strongly implicated in fueling the development of CNV and AMD [68].
3. Inflammasome Activation in AMD
The maintenance of the delicate balance between self and nonself regulates cellular homeostasis. However, during the aging process this system may be more vulnerable to a variety of noxious challenges that may activate host defense systems. The inflammasome is responsible for activation of many inflammatory processes. The inflammasome is a multiprotein complex, comprising of a sensor protein, the adaptor protein ASC (apoptosis-associated speck-like domain containing a caspase recruitment domain), and the inflammatory protease caspase-1. The assembly of the inflammasome signaling platform occurs due to conformational changes in the sensor protein, which in turn recruits caspase-1 to the complex and subsequently promotes the activation of caspase-1. Once activated, caspase-1 cleaves the inactive precursors of two proinflammatory cytokines, interleukin 1β (IL-1β) and IL-18, thereby generating mature forms which are then secreted from cells [69]. The inflammasome forming sensors are different receptor molecules, such as nucleotide-binding domain and leucine-rich repeat containing family pyrin (NLRP), which belong to the Nod-like receptor family of proteins. These include NLRP1, NLRP3, and NLRC4; or Absent In Melanoma (AIM 2), a receptor of the HIN (IFN-inducible nuclear proteins) family of proteins [70]. A growing body of evidence suggests that the NLRP3 inflammasome is clearly involved in host defense and autoinflammatory conditions, and is an integrator of cell damage and stress signals [71].
Activation of IL-1β by an inflammasome is required to efficiently control viral, bacterial, and fungal pathogen infections. However, excess IL-1β activity contributes to a variety of diseases [72]. The NLRP3 inflammasome has been shown to play a central role in the pathogenesis of autoinflammatory disorders; its activity has also been implicated in diseases such as Alzheimer's disease, cancer, type II diabetes, and most recently AMD [71, 73, 74]. The classic pathology of AMD is multiple small or intermediate drusen in the macular area. In a recent study, drusen isolated from donor AMD eyes were shown to activate NLRP3 inflammasome, causing secretion of IL-1β and IL-18 [73]. The authors postulated that NLRP3 may be a sensor for drusen-induced inflammasomes, as NLRP3 has been shown previously to act as a receptor for “danger” signals such as amyloid-like structures. Because laser-induced CNV was considerably greater in NLRP3 knockout mice, but not IL-1R knockout mice, NLRP3 and IL-18 may have a protective role in the progression of AMD [73]. Further, CEP (carboxyethylpyrrole), a biomarker of AMD, was thought to prime the inflammasome. Interestingly, while C1q, another complement component known to contribute to the inflammation and the pathophysiology of AMD [61], can also act as a danger signal that is, sensed by the NLRP3 inflammasome [75], C1q knockout mice develop CNV of similar size to control mice [61]. In addition, a recent study reported that lysosomal destabilization can activate the NLRP3 inflammasome in RPE cells [76].
Regulation of the NLRP3 inflammasome is poorly understood but probably involves the integration of signals from a number of stimuli, such as cellular damage and stress. It is now appreciated that inflammasome-dependent biological effects may be mediated not only by IL-1β and IL-18, but also by the multifaceted activities of caspase-1. Therefore, it is important to determine the mechanisms by which inflammasomes in RPE cells directly or indirectly modulate IL-1β activity that may lead to AMD. In chronically stressed states, where autophagy is increased, there may be secondary effects of protecting against inflammasome activation [77, 78]. Further understanding in context of drusen and RPE behavior may provide pathways to interrogate to maintain RPE function and health and attenuate inflammatory activation. Future studies to better understand how inflammasomes may be activated in AMD, and the molecular mechanisms involved in the assembly of the inflammasome signaling platform, may therefore lead to the development of novel therapeutic approaches for AMD.
4. Para-Inflammation in AMD
Inflammation, both acute and chronic, functions to control danger signals or to respond to pathogens to safeguard a host and maintain tissue health. Disturbances of homeostasis (e.g., infection, tissue injury, foreign bodies, but may also include stresses from aging) trigger inflammatory responses, the purpose of which are to remove or sequester the source of the disturbance and to allow the host to adapt to the abnormal conditions and return to a state of homeostasis. However, the spectrum of inflammation is broad. When appropriate, inflammation can be both adaptive and protective. Conversely, the immune response also has significant pathological potential and can promote tissue damage and facilitate disease progression.
Medzhitov first introduced the idea of para-inflammation as a tissue adaptive response to noxious stress or malfunction that has characteristics intermediate between basal and inflammatory states [10]. Briefly, in the basal state, tissue-resident macrophages (principally retinal microglia and retinal perivascular macrophages or choroidal macrophages) may play a role to promote an adaptive change with short-term benefits, promoting tissue homeostasis. However, if the abnormal conditions are sustained, or if the tissue receives a “danger signal,” this can result in immune cell infiltration, which in turn can become maladaptive. Para-inflammation has characteristics that are intermediate between basal and inflammatory states. The purpose of normal para-inflammation is presumably to maintain tissues homeostasis and to restore tissue function. Nonetheless, if a tissue is exposed to prolonged stress or malfunction, para-inflammation can become chronic and promote disease progression. Dysregulated para-inflammation has been proposed to play an important role in the progression of diabetes, atherosclerosis, and obesity.
Similarly, dysregulated para-inflammation, which is especially relevant in aging tissues dependent on nonproliferative cells and characterized by very high metabolism and other oxidative stress (e.g., the macula), has also been postulated to contribute to the development of AMD [11]. In the aging retina, oxidized lipoproteins and free radicals are major causes of tissue stress and serve as local triggers for retinal para-inflammation. Para-inflammatory responses in the neuroretina may be reflected in microglial activation and subretinal migration, and (potentially) breakdown of blood-retinal barrier. At the retinal/choroidal interface, para-inflammation manifests as complement activation in Bruch's membrane and RPE cells, and accumulation of microglia (and myeloid cells that have recently immigrated) in the subretinal space. In the choroid, para-inflammation may be characterized by increased thickness of choroid, increased macrophages, morphological abnormalities in choroidal melanocytes, mast cell activation and fibrosis.
Recent evidence, derived from the cybrid models of mitochondrial haplotypes into a mitochondrial DNA-null RPE cell line (ARPE19), showed that mitochondrial dysfunction may promote the progression and AMD [79]. The observed distinct polarization of energy cellular energy source and production suggest an approach with promise in further interrogating the influence on immune responses, including para-inflammation. The notion is that switching energy sources, which may be dependent on haplotype, influences the signaling pathways and thus phenotype of any subsequent immune activation of the cell. Further studies may increase our understanding of potential switch of energy sourcing, and the influence on immune activation of RPE that in turn will direct immune responses in cells (i.e., macrophages and choroidal mast cells) to deliver a trigger for progression of disease.
5. Adaptive Immunity in AMD
The role of adaptive immunity in AMD has received increasing attention. Whether adaptive immune responses relay pathogenic or regulatory functions, or are simply bystander effects, remains elusive. In support, there have been numerous reports suggesting involvement due to finding of autoantibodies in AMD patients, not least with the detection of anti-retinal autoantibodies [80, 81]. Whether they have a role as potential pathogenic mediators, or occur as bystanders, it remains to be determined if autoantibodies can act as a prognosticator or biomarker in AMD patients [82]. The search has been driven further with utilization of serum antigen arrays and 2-D gel electrophoresis. Specific targets such as RPB-3, aldolase C and pyruvate kinase IgG have been derived, and altered IgG/IgM ratios of anti-phophsatidylserine are associated with patients with AMD [83, 84]. Autoantibodies have been observed even when investigating responses to complement regulators, such as CFH [85]. The latter finding is enticing in that autoantibodies to CFH were unexpectedly lower in AMD patients, inferring a protective effect. Nevertheless, together there is increasing evidence of the presence of autoantibodies in AMD. The spectrum suggests secondary effects, and indeed also infers the potential of adaptive immune engagement. Consequently further searches for autantibodies, albeit possibly in only a small subset of patients, may be justified to determine whether there is a prevalent autoantibody signature.
More compelling data arises from mouse work. The data from Hollyfield et al. [43, 86] demonstrated that carboxyethylpyrrole (CEP) is present in AMD eye tissue, and mice immunized with this adducted oxidated product generated antibodies and exhibited pathology with some similarities with human AMD. Moreover, in experiments in RAG-deficient animals which lack B and T cells, no anti-CEP antibody was detected. Given the cell infiltrate noted around lesions, both T cell engagement and complement fixation were thought to contribute in this model to the loss of RPE and photoreceptors, and thus progression of AMD.
Most recently, novel observations of cytokine and T cell signatures from AMD patients have been published. First, an intriguing increase in IL-22 and IL-17 levels in serum from AMD patients was shown, supported by the further finding that C5a stimulated IL-22 and IL-17 from T cells [87]. Second, studies of twins and siblings found that the IL-17RC promoter is hypomethylated in AMD patients [88], further suggesting the involvement of adaptive immunity and TH17 cells, as well as potential effect on macrophages. Consequently, a testable hypothesis is that autoantibodies are present early in subsets of AMD patients, and are pathogenic. The notion that autoantibodies may create further complement-mediated damage, or activate myeloid cells to switch from protective para-inflammatory to pathogenic responses, may also be tested. Generation of autoantibodies (i.e., engagement of adaptive immune responses that are pathogenic) may tip the balance from para-inflammatory control, and create an environment that induces further loss of cells, angiogenesis, and an unremitting walk to late stage AMD.
6. Summary of BIMR Conference Discussions
Recent data suggest that dysregulation of immune response could contribute to the pathogenesis of AMD. However, there are a number of questions that remain unanswered. First, if para-inflammation is involved in the pathogenesis of AMD, when and how does a dysregulation of the immune response change from a protective role to a harmful process? Second, although genetic studies point to a role for the complement system and innate immunity in AMD, what role if any does adaptive immunity play in the disease? The group discussed a number of experimental approaches that could help address these questions.
6.1. Human Tissue Studies
Existing tissue banks may be used to interrogate immune response in AMD, with a particular focus on early events. Diseased and fellow eye tissue might first be graded using an established system [89], and then comprehensively characterized in terms of inflammatory cell contents and patterns, presence of complement and autoantibodies, and gene and protein expression profiles. It may be especially useful to compare tissue from different areas of an individual retina (e.g., in the fovea, adjacent to drusen, and “normal” tissue away from drusen).
6.2. Retrospective Clinical Studies
Existing medical records from large databases, such as those available from Medicare in the USA or anti-TNF-α treatment registries in the UK, could be mined to gain insight into selected questions. For example, do patients on immunosuppressive therapies for rheumatologic diseases have a lower prevalence of AMD?
6.3. Prospective Clinical Trials
There are a number of randomized clinical trials examining immunomodulation as a therapy for retinal diseases including AMD. Results from these trials will help guide our knowledge about the role of the immune system in the disease. Examples of such informative trials include prior trials studying treatment with immunomodulators in other diseases, and trials in AMD using immunomodulators such as those that target C5 and other complement components, mTOR, or TNF-α.
6.4. Biomarkers
Identification of direct and/or surrogate biomarkers that are predictive or prognostic for disease susceptibility, disease progression, or treatment response will be beneficial to the study of AMD. Serum, plasma, PBLs, platelets, and aqueous humor could be obtained from patients enrolled in natural history studies, and at the same time, patients encouraged to consent to eventually donate eyes. Samples could be used to assess complement components, cytokines, carboxyethylpyrrole (CEP), and autoantibodies. Collectively, such studies may yield clinical/pathological correlations and genotype/phenotype relationships in individual patients.
6.5. Imaging Modalities
The development of new imaging modalities can detect the trafficking and function of immune cells in the retina and choroid (which may be transient). These tools, if designed to provide quantitative analyses of immune system functions such as the presence in the human eye of ongoing complement activation or specific cellular infiltration could then be applied to both direct imaging studies, particularly immunomodulatory treatments and bioenergetic evaluations, and to studies using transfer of ex vivo labeled cells.
6.6. Animal Models
Although there is no perfect animal model for AMD, preclinical models that reproduce specific aspects of early AMD (e.g., drusen, low grade chronic inflammation, and GA) need to be developed to ask specific questions about the role of inflammation, and to probe specific disease mechanisms. Although eyes in mice do not have macular structures and do have a distinct RPE morphology, an example of a model for para-inflammation may include deliberately inducing low grade inflammation in ob/ob or senescent mice, followed by addition of a systemic insult (e.g., light toxicity) to reproduce the hypothesized dysregulation of para-inflammation.
6.7. In Vitro Cell Biology Studies
Finally, there is interest in studies designed question whether changes in aging RPE and photoreceptors make them more susceptible to damage by dysregulated para-inflammation in AMD. For example, in vitro cultures of photoreceptors could be used to evaluate early changes in rods in AMD, or resistance to injury by cones in AMD. Cultures of RPE could be used to examine metabolic dysfunctions (e.g., mitochondrial dysfunction, haplotypes), and the responses of retinal and choroidal cells to cytokines released by such metabolic change. Of particular interest are epigenetic changes in such cells that may promote or protect cells in aging and AMD.
7. Conclusions
The prevalence of AMD will continue to increase as the population ages. Although we do not know the exact etiology of the disease, recent genetic studies have implicated the complement system in disease pathogenesis and severity. Other studies further support the hypothesis that the immune system is involved in the disease, in concert with or in addition to other factors such as environmental conditions and products of photooxidation (Figure 2). Importantly, understanding how immune responses initiate or exacerbate AMD will allow us to identify novel therapeutic approaches to the disease.
Figure 2.
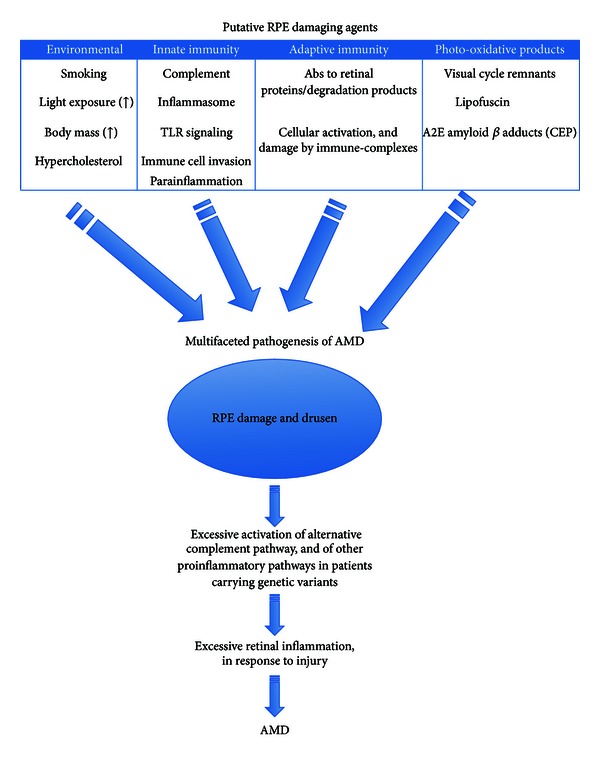
An integrated model of immune regulation of AMD.
Conflict of Interests
Scott M. Whitcup is an employee of Allergen, Inc. John P. Atkinson is funded by Alexion Pharmaceuticals, and Bärbel Rohrer has received royalty payments as well as a sponsored research Grant from Alexion Pharmaceuticals. Michael Holers has received royalty payments from Taligen Therapeutics and Alexion Pharmaceuticals and has received sponsored research grants and served as a consultant to both companies.
Acknowledgments
The authors extend deep thanks to the other members of the 2013 Inflammation and Immune Response task group of the fifth annual conference of the Arnold and Mabel Beckman Initiative for Macular Research: Anthony Adamis, M.D., Genentech, Inc.; Cynthia Grosskreutz, M.D., Ph.D., Novartis Institutes for Biomedical Research; Baruch Kuppermann, M.D., Ph.D., University of California—Irvine; Anthony Nesburn, M.D., University of California—Irvine; and Jerry Niederkorn, Ph.D., University of Texas Southwestern Medical Center. John Atkinson is funded by the following NIH Grants: R01 AI041592-15, R01 GM99111-18, P30 AR48335-12, and U54 HL112303-01; Edward N. and Della L. Thome Memorial Foundation; and Alexion Pharmaceuticals. Debasish Sinha is funded by NIH Grants R01 EY018636 and R01 EY019037 and is a recipient of the Sybil B. Harrington Special Scholar award for Macular Degeneration from Research to Prevent Blindness. Bärbel Rohrer is supported in part by the National Institutes of Health (R01EY019320), Department of Veterans Affairs (I01 RX000444), Foundation Fighting Blindness, and Beckman Initiative for Macular Research. Andrew D. Dick is in part supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Karen L. Elkins provided additional scientific writing and general editorial input during the preparation of the paper.
References
- 1.Friedman DS, O’Colmain BJ, Muñoz B, et al. Prevalence of age-related macular degeneration in the United States. Archives of Ophthalmology. 2004;122(4):564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 2.Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. The Lancet. 2012;379(9827):1728–1738. doi: 10.1016/S0140-6736(12)60282-7. [DOI] [PubMed] [Google Scholar]
- 3.Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Progress in Retinal and Eye Research. 2001;20(3):385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 4.Carter DA, Dick AD. CD200 maintains microglial potential to migrate in adult human retinal explant model. Current Eye Research. 2004;28(6):427–436. doi: 10.1080/02713680490503778. [DOI] [PubMed] [Google Scholar]
- 5.Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. The American Journal of Pathology. 2002;161(5):1669–1677. doi: 10.1016/S0002-9440(10)64444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature Reviews Immunology. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 7.Broderick C, Duncan L, Taylor N, Dick AD. IFN-γ and LPS-mediated IL-10-dependent suppression of retinal microglial activation. Investigative Ophthalmology and Visual Science. 2000;41(9):2613–2622. [PubMed] [Google Scholar]
- 8.Hughes EH, Schlichtenbrede FC, Murphy CC, et al. Minocycline delays photoreceptor death in the rds mouse through a microglia-independent mechanism. Experimental Eye Research. 2004;78(6):1077–1084. doi: 10.1016/j.exer.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Dick AD, Carter D, Robertson M, et al. Control of myeloid activity during retinal inflammation. Journal of Leukocyte Biology. 2003;74(2):161–166. doi: 10.1189/jlb.1102535. [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 11.Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Progress in Retinal and Eye Research. 2009;28(5):348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Raychaudhuri S, Iartchouk O, Chin K, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nature Genetics. 2011;43(12):1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards A, Kavanagh D, Atkinson JP. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory statesthe examples of vascular damage in atypical hemolytic uremic syndrome and debris accumulation in age-related macular degeneration. Advances in Immunology. 2007;96:141–177. doi: 10.1016/S0065-2776(07)96004-6. [DOI] [PubMed] [Google Scholar]
- 14.Goodship THJ. Factor H genotype-phenotype correlations: lessons from aHUS, MPGN II, and AMD. Kidney International. 2006;70(1):12–13. doi: 10.1038/sj.ki.5001612. [DOI] [PubMed] [Google Scholar]
- 15.Lachmann PJ, Smith RAG. Taking complement to the clinic—has the time finally come? Scandinavian Journal of Immunology. 2009;69(6):471–478. doi: 10.1111/j.1365-3083.2009.02258.x. [DOI] [PubMed] [Google Scholar]
- 16.Tuo J, Grob S, Zhang K, Chan CC. Genetics of immunological and inflammatory components in age-related macular degeneration. Ocular Immunology and Inflammation. 2012;20(1):27–36. doi: 10.3109/09273948.2011.628432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sparrow JR, Ueda K, Zhou J. Complement dysregulation in AMD: RPE-Bruch's membrane-choroid. Molecular Aspects of Medicine. 2012;33(4):436–445. doi: 10.1016/j.mam.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zipfel PF, Lauer N, Skerka C. The role of complement in AMD. Advances in Experimental Medicine and Biology. 2010;703:9–24. doi: 10.1007/978-1-4419-5635-4_2. [DOI] [PubMed] [Google Scholar]
- 19.Gehrs KM, Jackson JR, Brown EN, Allikmets R, Hageman GS. Complement, age-related macular degeneration and a vision of the future. Archives of Ophthalmology. 2010;128(3):349–358. doi: 10.1001/archophthalmol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson DH, Radeke MJ, Gallo NB, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Progress in Retinal and Eye Research. 2010;29(2):95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khandhadia S, Cipriani V, Yates JR, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012;217(2):127–146. doi: 10.1016/j.imbio.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 22.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Molecular Aspects of Medicine. 2012;33(4):295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Despriet DDG, Klaver CCW, Witteman JCM, et al. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. Journal of the American Medical Association. 2006;296(3):301–309. doi: 10.1001/jama.296.3.301. [DOI] [PubMed] [Google Scholar]
- 24.Fett AL, Hermann MM, Muether PS, Kirchhof B, Fauser S. Immunohistochemical localization of complement regulatory proteins in the human retina. Histol Histopathol. 2012;27(3):357–364. doi: 10.14670/HH-27.357. [DOI] [PubMed] [Google Scholar]
- 25.Sofat R, Casas JP, Webster AR, et al. Complement factor H genetic variant and age-related macular degeneration: effect size, modifiers and relationship to disease subtype. International Journal of Epidemiology. 2012;41(1):250–262. doi: 10.1093/ije/dyr204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 27.Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 28.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(20):7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montes T, Tortajada A, Morgan BP, De Córdoba SR, Harris CL. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4366–4371. doi: 10.1073/pnas.0812584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sjöberg AP, Trouw LA, Clark SJ, et al. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. Journal of Biological Chemistry. 2007;282(15):10894–10900. doi: 10.1074/jbc.M610256200. [DOI] [PubMed] [Google Scholar]
- 32.Ormsby RJ, Ranganathan S, Tong JC, et al. Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Investigative Ophthalmology and Visual Science. 2008;49(5):1763–1770. doi: 10.1167/iovs.07-1297. [DOI] [PubMed] [Google Scholar]
- 33.Clark SJ, Perveen R, Hakobyan S, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. The Journal of Biological Chemistry. 2010;285(39):30192–30202. doi: 10.1074/jbc.M110.103986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laine M, Jarva H, Seitsonen S, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. The Journal of Immunology. 2007;178(6):3831–3836. doi: 10.4049/jimmunol.178.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Progress in Retinal and Eye Research. 2001;20(6):705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 36.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. American Journal of Ophthalmology. 2002;134(3):411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 37.Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye. 2001;15(3):390–395. doi: 10.1038/eye.2001.142. [DOI] [PubMed] [Google Scholar]
- 38.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Experimental Eye Research. 2001;73(6):887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 39.Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(5):1657–1661. doi: 10.1073/pnas.85.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nature Reviews Microbiology. 2008;6(2):132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lauer N, Mihlan M, Hartmann A, et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. The Journal of Immunology. 2011;187(8):4374–4383. doi: 10.4049/jimmunol.1002488. [DOI] [PubMed] [Google Scholar]
- 42.Clark SJ, Higman VA, Mulloy B, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. The Journal of Biological Chemistry. 2006;281(34):24713–24720. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- 43.Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nature Medicine. 2008;14(2):194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(46):17456–17461. doi: 10.1073/pnas.0606234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lachmann PJ. The amplification loop of the complement pathways. Advances in Immunology. 2009;104:115–149. doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 46.Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Investigative Ophthalmology and Visual Science. 2009;50(5):2044–2053. doi: 10.1167/iovs.08-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Investigative Ophthalmology and Visual Science. 2009;50(12):5818–5827. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scholl HPN, Issa PC, Walier M, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. 2008;3(7) doi: 10.1371/journal.pone.0002593.e2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Ohno-Matsui K, Yoshida T, et al. Amyloid-β up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: another mechanism of complement activation in age-related macular degeneration. Journal of Cellular Physiology. 2009;220(1):119–128. doi: 10.1002/jcp.21742. [DOI] [PubMed] [Google Scholar]
- 50.Droz I, Mantel I, Ambresin A, Faouzi M, Schorderet DF, Munier FL. Genotype-phenotype correlation of age-related macular degeneration: influence of complement factor H polymorphism. British Journal of Ophthalmology. 2008;92(4):513–517. doi: 10.1136/bjo.2007.127811. [DOI] [PubMed] [Google Scholar]
- 51.Sepp T, Khan JC, Thurlby DA, et al. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Investigative Ophthalmology and Visual Science. 2006;47(2):536–540. doi: 10.1167/iovs.05-1143. [DOI] [PubMed] [Google Scholar]
- 52.Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Archives of Ophthalmology. 2005;123(6):774–782. doi: 10.1001/archopht.123.6.774. [DOI] [PubMed] [Google Scholar]
- 53.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration—emerging pathogenetic and therapeutic concepts. Annals of Medicine. 2006;38(7):450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hageman GS, Hancox LS, Taiber AJ, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Annals of Medicine. 2006;38(8):592–604. [PMC free article] [PubMed] [Google Scholar]
- 55.Maller J, George S, Purcell S, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nature Genetics. 2006;38(9):1055–1059. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 56.Nishiguchi KM, Yasuma TR, Tomida D, et al. C9-R95X polymorphism in patients with neovascular age-related macular degeneration. Investigative Ophthalmology & Visual Science. 2012;53(1):508–512. doi: 10.1167/iovs.11-8425. [DOI] [PubMed] [Google Scholar]
- 57.Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nature Genetics. 2007;39(10):1200–1201. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 58.Heurich M, Martínez-Barricarte R, Francis NJ, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(21):8761–8766. doi: 10.1073/pnas.1019338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. European Journal of Human Genetics. 2009;17(1):100–104. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rohrer B, Coughlin B, Kunchithapautham K, et al. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Molecular Immunology. 2011;48(6-7):e1–e8. doi: 10.1016/j.molimm.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bora NS, Kaliappan S, Jha P, et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. The Journal of Immunology. 2006;177(3):1872–1878. doi: 10.4049/jimmunol.177.3.1872. [DOI] [PubMed] [Google Scholar]
- 63.Bora NS, Kaliappan S, Jha P, et al. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. The Journal of Immunology. 2007;178(3):1783–1790. doi: 10.4049/jimmunol.178.3.1783. [DOI] [PubMed] [Google Scholar]
- 64.Bora NS, Jha P, Lyzogubov VV, et al. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. The Journal of Biological Chemistry. 2010;285(44):33826–33833. doi: 10.1074/jbc.M110.153130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cashman SM, Ramo K, Kumar-Singh R. A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0019078.e19078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. The Journal of Immunology. 2008;181(11):8068–8076. doi: 10.4049/jimmunol.181.11.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rohrer B, Long Q, Coughlin B, et al. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Investigative Ophthalmology and Visual Science. 2009;50(7):3056–3064. doi: 10.1167/iovs.08-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bressler SB. Introduction: understanding the role of angiogenesis and antiangiogenic agents in age-related macular degeneration. Ophthalmology. 2009;116(10):S1–S7. doi: 10.1016/j.ophtha.2009.06.045. [DOI] [PubMed] [Google Scholar]
- 69.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunological Reviews. 2011;243(1):136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 70.Dowling JK, O'Neill LA. Biochemical regulation of the inflammasome. Critical Reviews in Biochemistry and Molecular Biology. 2012;47(5):424–443. doi: 10.3109/10409238.2012.694844. [DOI] [PubMed] [Google Scholar]
- 71.Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clinical & Experimental Immunology. 2011;166(1):1–15. doi: 10.1111/j.1365-2249.2011.04440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mankan AK, Kubarenko A, Hornung V. Immunology in clinic review series, focus on autoinflammatory diseases: inflammasomes: mechanisms of activation. Clinical & Experimental Immunology. 2012;167(3):369–381. doi: 10.1111/j.1365-2249.2011.04534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doyle SL, Campbell M, Ozaki E, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nature Medicine. 2012;18(5):791–798. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tarallo V, Hirano Y, Gelfand BD, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149(4):847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. The Journal of Immunology. 2012;188(11):5682–5693. doi: 10.4049/jimmunol.1103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tseng WA, Thein T, Kinnunen K, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Investigative Ophthalmology & Visual Science. 2013;54(1):110–120. doi: 10.1167/iovs.12-10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kenney MC, Chwa M, Atilano SR, et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0054339.e54339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel N, Ohbayashi M, Nugent AK, et al. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology. 2005;115(3):422–430. doi: 10.1111/j.1365-2567.2005.02173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cherepanoff S, Mitchell P, Wang JJ, Gillies MC. Retinal autoantibody profile in early age-related macular degeneration: preliminary findings from the blue mountains eye study. Clinical and Experimental Ophthalmology. 2006;34(6):590–595. doi: 10.1111/j.1442-9071.2006.01281.x. [DOI] [PubMed] [Google Scholar]
- 82.Morohoshi K, Goodwin AM, Ohbayashi M, Ono SJ. Autoimmunity in retinal degeneration: autoimmune retinopathy and age-related macular degeneration. Journal of Autoimmunity. 2009;33(3-4):247–254. doi: 10.1016/j.jaut.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 83.Morohoshi K, Ohbayashi M, Patel N, Chong V, Bird AC, Ono SJ. Identification of anti-retinal antibodies in patients with age-related macular degeneration. Experimental and Molecular Pathology. 2012;93(2):193–199. doi: 10.1016/j.yexmp.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 84.Morohoshi K, Patel N, Ohbayashi M, et al. Serum autoantibody biomarkers for age-related macular degeneration and possible regulators of neovascularization. Experimental and Molecular Pathology. 2012;92(1):64–73. doi: 10.1016/j.yexmp.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 85.Dhillon B, Wright AF, Tufail A, et al. Complement factor H autoantibodies and age-related macular degeneration. Investigative Ophthalmology and Visual Science. 2010;51(11):5858–5863. doi: 10.1167/iovs.09-5124. [DOI] [PubMed] [Google Scholar]
- 86.Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Molecular Neurobiology. 2010;41(2-3):290–298. doi: 10.1007/s12035-010-8110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu B, Wei L, Meyerle C, et al. Complement component C5a promotes expression of IL-22 and IL-17 from human T cells and its implication in age-related macular degeneration. Journal of Translational Medicine. 2011;9:p. 111. doi: 10.1186/1479-5876-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei L, Liu B, Tuo J, et al. Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Reports. 2012;2(5):1151–1158. doi: 10.1016/j.celrep.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olsen TW, Feng X. The minnesota grading system of eye bank eyes for age-related macular degeneration. Investigative Ophthalmology and Visual Science. 2004;45(12):4484–4490. doi: 10.1167/iovs.04-0342. [DOI] [PubMed] [Google Scholar]