

Abstract
Background
The mammalian outflow tract (OFT) and primitive right ventricle arise by accretion of newly differentiated cells to the arterial pole of the heart tube from multi-potent progenitor cells of the second heart field (SHF). While mounting evidence suggests that the genetic pathways regulating SHF development are highly conserved in zebrafish, this topic remains an active area of investigation.
Results
Here, we extend previous observations demonstrating that zebrafish tbx1 (van gogh, vgo) mutants show conotruncal defects consistent with a conserved role in SHF-mediated cardiogenesis. Surprisingly, we reveal through double in situ analyses that tbx1 transcripts are excluded from cardiac progenitor cells or differentiated cardiomyocytes, suggesting a non-autonomous role in SHF development. Further, we find that the diminuitive ventricle in vgo animals results from a 25% decrease in cardiomyocyte numbers that occurs subseqent to heart tube stages. Lastly, we report that although SHF progenitors are specified in the absence of Tbx1, they fail to be maintained due to compromised SHF progenitor cell proliferation.
Conclusion
These studies highlight conservation of the Tbx1 program in zebrafish SHF biology.
Keywords: tbx1, zebrafish, heart, second heart field, cardiac
Introduction
In higher vertebrates cardiac crescent cells of the first heart field (FHF) generate the primitive heart tube, which is elongated at both poles by late differentiation of second heart field (SHF) progenitors in pharyngeal mesoderm (Vincent and Buckingham, 2010). At the arterial pole, SHF precursors give rise to new myocardial segments that become the future right ventricle and outflow tract (OFT). Anomolies in OFT development comprise ~30% of congenital heart defects (Srivastava and Olson, 2000) and often result from hemizygous microdeletions in a 3 Mb region on chromosome 22q11.2 (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001). This genomic disruption results in a variable constellation of congenital malformations and cognitive impairments known collectively as DiGeorge Syndrome (DGS). Among the most life-threatening phenotypes are cardiovascular (CV) anomolies, such as Tetralogy of Fallot and Interrupted Aortic Arch, that appear to arise from reduced expression of TBX1, a gene in the typically deleted region required for cardiac neural crest and SHF development (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001; Yagi et al., 2003). Despite the prevalence and severity of CV malformations in the DGS population, ~20% of genetically affected individuals lack any detectable cardiovascular pathology (Ryan et al., 1997). This observation underscores the profound influence of genetic and/or environmental modifiers over the DGS phenotype and reveals the complexity of the requirement for TBX1 in CV development. As such, a more detailed understanding of the molecular mechanisms by which TBX1 influences OFT development are essential.
Much of our understanding of how TBX1 influences CV development comes from gene expression and inactivation studies in the mouse (Aggarwal and Morrow, 2008; Parisot et al., 2011; Scambler, 2010). Murine Tbx1 is expressed in tissues that form the pharyngeal system - including pharyngeal surface ectoderm, pharyngeal endoderm, and pharyngeal mesoderm, which contains SHF progenitors (Chapman et al., 1996; Garg et al., 2001; Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001; Vitelli et al., 2002a). In regards to the heart, cre/loxP lineage tracing of Tbx1+ cells showed substantial contribution to the inferior myocardial wall and endothelium of the OFT and some contribution to the myocardium at the base outlet of the RV, consistent with Tbx1 expression in a subset of SHF precursors (Huynh et al., 2007; Xu et al., 2004). Loss of function analyses revealed that homozygous Tbx1 neonates die at birth from severe craniofacial and CV malformations, the latter of which include the loss of the pharyngeal apparatus (pharyngeal arches, pouches, and clefts), OFT hypoplasia, and ventricular septal defects (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001). It has been proposed that TBX1 provides a pro-proliferation signal to SHF progenitors (Chen et al., 2009; Liao et al., 2008; Xu et al., 2004; Zhang et al., 2006b) that is likely mediated, at least in part, by FGF8 (Abu-Issa et al., 2002; Brown et al., 2004; Hu et al., 2004; Park et al., 2006; Vitelli et al., 2010; Vitelli et al., 2002b; Zhang et al., 2006b). This idea is supported by the ability of TBX1 to activate an Fgf8 enhancer in cell culture (Hu et al., 2004) and by genetic interaction studies between Tbx1 and Fgf8 for OFT development in vivo (Brown et al., 2004; Vitelli et al., 2010; Vitelli et al., 2002b; Zhang et al., 2006b).
Why only a fraction of patients hemizygous for a deletion in the TBX1 containing region present with DGS while others have no observable abnormalities is not understood. Moreover, the spectrum of defects in affected DGS individuals suggests the existence of genetic or environmental modifiers, most of which are not known. The zebrafish model organism offers distinct strategies for identifying such modifiers, such as forward genetic or small molecule based screening. Despite being comprised of only two cardiac chambers, the zebrafish heart is partially derived from a SHF population (de Pater et al., 2009; Hami et al., 2011; Lazic and Scott, 2011; Zhou et al., 2011) that expresses nkx2.5 (Lazic and Scott, 2011; Zhou et al., 2011), mef2cb (Hinits et al., 2012; Lazic and Scott, 2011), and ltbp3 (Zhou et al., 2011). Cre/loxP lineage tracing demonstrated that approximately half of the single ventricular chamber and entire OFT is derived via late differentiation and accretion of SHF progenitors following heart tube formation (Zhou et al., 2011). Impairment of SHF-mediated cardiogenesis results in loss of ventricular cardiomyocytes that normally comprise the distal portion of the chamber and loss or diminution of Elastin2+ (Eln2+) smooth muscle precursor cells of the OFT.
The genetic programs regulating SHF biology in the zebrafish appear largely conserved with that of higher vertebrates. Using small molecule, morpholino, or genetic means of inhibition, FGF (de Pater et al., 2009; Lazic and Scott, 2011; Marques et al., 2008), BMP (Hami et al., 2011), Hedgehog (Hami et al., 2011), and TGFβ (Zhou et al., 2011) signaling have all been implicated as critical SHF pathways in zebrafish. Islet1 is arguably the best known SHF marker in mice. Despite recent reports suggesting conserved expression of isl1 in zebrafish SHF progenitors (Hami et al., 2011; Witzel et al., 2012), isl1−/− mutants show normal arterial pole development (de Pater et al., 2009). Thus, while evidence of gentic conservation between zebrafish and mammalian SHF-mediated cardiogenesis is mounting, this topic is still an active area of investigation.
In regards to tbx1, an initial study in zebrafish showed that tbx1 null embryos (van gogh, vgo) (Piotrowski et al., 2003) have an undersized ventricle, a small Eln2+ smooth muscle component of the outflow tract, and impaired migration of dye-labeled pharyngeal cells into the heart tube (Hami et al., 2011). Taken together, these observations suggest preliminarily that zebrafish Tbx1 is required for SHF development. However, a more comprehensive characterization of the vgo cardiac phenotype is required to determine the degree to which Tbx1 function is conserved. Thus, we sought to confirm and extend initial observations suggesting that Tbx1 function is required for zebrafish SHF development as in mice and presumably humans.
Here, we characterized tbx1 expression in relation to cardiac progenitors and differentiated cardiomyocytes in zebrafish and analyzed tbx1/vgo null embryos for molecular and morphological evidence of SHF perturbations. Unexpectedly, we found that tbx1 expression appears non-overlapping with cardiac progenitors cell (CPC) markers of the first or second heart fields or differentiated cardiomyocytes that comprise the early zebrafish heart tube. However, vgo mutant ventricles show a clear diminution in size. We discovered that this reduction in ventricular chamber size is due to an approximately 25% decrease in cardiomyocyte numbers that occurrs subsequent to linear heart tube establishment. This finding demonstrates that FHF development occurs normally in the absence of Tbx1 function, but that SHF development is specifically disrupted. Further, we discovered that the SHF defect in vgo mutants is caused by compromised SHF progenitor cell proliferation. Overall, our data suggest that the tbx1 pathway is conserved in zebrafish for SHF-mediated cardiogenesis.
Results
Although the developmental expression pattern of zebrafish tbx1 has been reported previously (Kochilas et al., 2003; Piotrowski et al., 2003; Zhang et al., 2006a), we sought to characterize the location of tbx1 transcripts relative to cardiac markers during the initial phases of zebrafish cardiogenesis. First, we performed double in situ hybridization prior to early myocardial differentiation with tbx1 and the conserved cardiac progenitor marker, nkx2.5, which is expressed in both the first and second heart fields. At the 8 somite-stage [8ss; 13 hours post-fertilization (hpf)], tbx1 transcripts are located medial (Fig. 1A,B) and dorsal (Fig. 1A,D) to nkx2.5+ CPCs. Interestingly, fgf8a transcripts were reported to have a similar dorsomedial relationship with nkx2.5 at similar developmental stages (Reifers et al., 2000). Cross-section analysis provided further evidence that tbx1 expression is non-overlapping with nkx2.5 (Fig. 1C). Next, we examined the localization of tbx1 transcripts relative to a cardiomyocyte marker, cardiac myosin light chain 2 (cmlc2; also called myl7) (Yelon et al., 1999) and a SHF marker latent TGF-beta binding protein-3 (ltbp3). At 16ss (17hpf), cmlc2 transcripts mark FHF-derived myocardial cells in the anterior lateral plate mesoderm (ALPM) (de Pater et al., 2009; Lazic and Scott, 2011). At this stage, tbx1 transcripts were observed lateral to the medially migrating cmlc2+ cardiomyocytes of the heart forming region (Fig. 1E). Similarly at 23ss (20.5hpf), tbx1 transcripts were readily visible in the developing pharyngeal arches (Piotrowski et al., 2003), but absent from cmlc2+ cells that comprise the cardiac cone, a precursor to the linear heart tube in zebrafish (Fig. 1F). At the same developmental stage, ltbp3 expression commenced in SHF progenitors adjacent to the cardiac cone, but failed to overlap spatially with tbx1 (Fig. 1G). Finally, after establishment of the linear heart tube (24hpf), tbx1 transcripts were absent from the heart tube proper (Fig. 1H) and the arterial pole of the heart tube inhabited by SHF progenitor cells (Fig. 1H, inset) (Hami et al., 2011; Lazic and Scott, 2011; Zhou et al., 2011). Taken together, these data suggest that zebrafish tbx1 transcripts are non-overlapping with CPC markers of the first or second heart fields or differentiated cardiomyocytes that comprise the early zebrafish heart tube.
Figure 1. tbx1 transcripts fail to co-localize with cardiac cells during heart field or heart tube stages.

Whole-mount double in situ hybridization. (A–D) tbx1 (red) and nkx2.5 (blue) transcripts are non-overlapping during heart field stages (8 somite stage). (A) Flat mount, dorsal view, anterior up, 10X maginification. (B) 20X magnification of the ALPM (boxed region from A) (C) Transverse cryo-section, 20X magnification of the ALPM (location of section shown by the solid line in A) (D) Dorso-lateral view showing that tbx1-expressing cells lie dorsal to nkx2.5-expressing CPCs at the 12ss. (E,F) tbx1+ cells are lateral to medially migrating cmlc2+ cardiomyocytes at 16ss and 23ss. (G) tbx1+ cells are lateral to ltbp3+ SHF progenitors at 20.5hpf/23ss. Dorsal view, anterior up. (H) At 24hpf, the linear heart tube lies dorsal to the tbx1 expression domain. Dorsal view, anterior up. 10X magnification. (inset) 20X magnification of boxed region from F. tbx1 is not expressed at the end of the linear heart tube where SHF progenitors reside (red arrowhead).
To determine if zebrafish embryos devoid of tbx1 display evidence of defective cardiogenesis, we analyzed embryos homozygous for the tbx1 null alleles, van goghtu285 (vgotu285) and van goghtu208 (vgotu208), which produce indistinguishable embryonic phenotypes (Piotrowski et al., 2003; Piotrowski and Nusslein-Volhard, 2000). To visualize cardiogenesis in absence of tbx1, we crossed the vgotu285 allele with our previously described Tg(nkx2.5:ZsYellow)fb7 line that reports yellow fluorescence in nkx2.5+ cells (henceforth called nkx2.5:ZsY) (Zhou et al., 2011). Consistent with a previous report (Hami et al., 2011), we observed a diminuitive ventricle in vgo mutants compared to controls at 72hpf (Fig. 2A–D), a timepoint following SHF-mediated myocardial accretion (de Pater et al., 2009; Lazic and Scott, 2011; Zhou et al., 2011). This size difference could be due to a decrease in the number of ventricular cardiomyocytes or equivalent numbers of ventricular cells that are smaller and more densely organized. To distinguish between these alternatives, we quantified the number of cardiomyocytes in each chamber of control and vgotu285 mutant embryos at 72hpf using a transgenic strain in which cardiomyocyte nuclei express DsRed2 fluorescent protein (Mably et al., 2003). From this analysis, we learned that vgo embryos display no alterations in the number of atrial cells but exhibit an approximately 25% decrease in the number of ventricular cardiomyocytes compared to controls (Fig. 2E–G). Moreover, OFT smooth muscle precursors that are marked by Elastin2 (Eln2) (Miao et al., 2007) were severely diminished or absent in vgo embryos compared to control siblings (Fig. 2H–J). Together, these data demonstrate that the small ventricle observed in vgo mutants is a result of a ventricular cardiomyocyte deficit and suggest that tbx1 is essential to build the arterial pole of the zebrafish heart.
Figure 2. tbx1 mutants have diminutive ventricles caused by decreased cardiomyocyte numbers and diminished OFT smooth muscle.

(A–D) Fluorescent microscopy images of Tg(nkx2.5:ZsYellow) control and vgo embryos; 10X magnification. At 72hpf, the ventricular chamber and OFT appears small in vgo (C, D) compared to control (A,B). A,C; lateral view, anterior right. B,D; ventral view, anterior up. (E,F) Flattened confocal images of 72hpf cardiomyocyte (CM) nuclei in Tg(cmlc2::DsRednuc) control (E) and vgo (F) hearts. (G) Graph depicting the average number of CMs at 72hpf in control (n=4) and vgo (n=4) embryos. Asterisks indicate statistical significance as determined using unpaired students T-test. Error bars represent +/− s.e.m. Atrial CM numbers remain unchanged (P=0.47), while ventricular and total CM numbers are significantly lower in vgo mutants (P=0.00008 and 0.0002, respectively). (H,I) Flattened confocal images following double immunofluorescence at 72 hpf to visualize OFT smooth muscle precursors (Eln2; green) and chamber cardiomyocytes (MF20; red) in control (n=21; H) and vgo (n=20; I) embryos. (J) Graph depicting the percentage of control or vgo embryos with normal (n), reduced (r), or absent (a) Eln2 staining.
A decrease in the number of ventricular cardiomyocytes could arise from a defect in FHF-mediated cardiogenesis. Therefore, we first compared the spatial and temporal expression of cardiac markers at the cone stage (23ss/20.5hpf), a timepoint prior to SHF-mediated myocardial accretion, between vgo tu285 and sibling embryos. At 23ss, nkx2.5:ZsY reporter fluorescence (Fig. 3A,B) and ventricular myosin heavy chain (vmhc) transcripts (Fig. 3C,D), which highlight differentiated FHF-derived ventricular cardiomyocytes and undifferentiated SHF progenitors (Lazic and Scott, 2011; Zhou et al., 2011), appeared grossly normal in vgo animals. Next, we quantified the number of cardiomyocytes in the linear heart tube of control and vgo embryos that derive from early-differentiating FHF cells. We found that although the heart tube of vgo animals appeared somewhat misshapen compared to their sibling controls, the total number of cardiomyocytes was similar (Fig. 3E–G). These data provide conclusive evidence that FHF-derived cardiogenesis is unaffected in vgo mutants, formally demonstrating that the ventricular phenotype observed is not due to compromised FHF development.
Figure 3. Cardiac progenitors within the heart forming region are specified in tbx1 mutants and differentiate appropriately into ventricular cardiomyocytes.

(A,B) Flattened confocal images of Tg(nkx2.5::ZsYellow) control (n=37; A) and vgo (n=14; B) embryos at 20.5hpf/23somite-stage (ss). (C,D) Whole-mount in situ hybridization of vmhc at 20.5hpf/23ss in both control (n=16; C) and vgo (n=16; D) embryos. (E,F) Flattened confocal images following immunofluorescence at 26hpf to visualize DsRed+ nuclei in control (E) and vgo (F) embryos. Dorsal view, Anterior up in all images. (G) Graph depicting the average number of CMs at 26hpf in control (n=7) and vgo (n=3) embryos.
Recently, we and others have demonstrated that zebrafish cardiogenesis relies on a second (or secondary) heart field (SHF) that resides in pharyngeal mesoderm and accretes myocardium to the arterial pole of the linear heart tube (de Pater et al., 2009; Hami et al., 2011; Lazic and Scott, 2011; Zhou et al., 2011). The molecular signature of the zebrafish SHF to date is nkx2.5, mef2cb, and ltbp3. ltbp3 is the most specific marker of the SHF because it overlaps only partially with differentiated myocardium of the heart tube (Zhou et al., 2011) whereas nkx2.5 (Lazic and Scott, 2011; Zhou et al., 2011) and mef2cb (Hinits et al., 2012; Lazic and Scott, 2011) label the SHF and the heart tube. To determine if the ventricular deficit in vgo tu285 mutants is a result of perturbations in ltbp3 expression, we performed in situ hybridization using an ltbp3 riboprobe at progressive developmental stages. At 23ss (20.5hpf), when ltbp3 transcripts are first visualized by in situ analyses, we learned that ltbp3 expression was properly initiated in vgo embryos (Fig. 4A,B). However, at linear heart tube stages (24hpf), ltbp3 transcripts were no longer detected in SHF progenitors at the arterial pole in vgo mutants (Fig. 4 C,D). We confirmed the absence of SHF precursors at heart tube stages in vgo tu208 embryos using a complementary approach that relies on the fluorescent reporter transgenic strains Tg(nkx2.5:ZsY) and Tg(cmlc2:GFP). To distinguish the two fluorophores by confocal microscopy, double fluorescent immunohistochemistry was performed to highlight cmlc2+ differentiated cardiomyocytes in green and nkx2.5+ SHF progenitors and differentiated ventricular cardiomyocytes in red. Although a distinct nkx2.5+ population of SHF progenitors could be easily visualized at the arterial pole of the heart tube at 24–26 hpf in control embryos, this population was absent in vgo mutants (Fig. 4E,F). Together, these data suggest that Tbx1 is required for SHF progenitor cell maintenance, but is dispensible for their initial specification.
Figure 4. tbx1 is required for SHF progenitor cell maintenance.

(A–B) ltbp3 was observed via whole-mount in situ hybridization at 20.5hpf/23ss in control (A) and vgo (B) embryos (n>12). Dorsal view, anterior down. 20X Magnification. (C, D) Whole-mount double in situ hybridization at 26 hpf shows ltbp3+ (blue) cells at the arterial pole (arrowhead) of the cmlc2+ (red) heart tube in control embryos. ltbp3 expression is drastically reduced (21%) or absent (79%) at the arterial pole of vgo hearts (n=14). Asterisk indicates ltbp3 expression within the notochord. (E, F) Double transgenic Tg(nkx2.5::ZsYellow); Tg(cmlc2::GFP) control (E) and vgo (F) embryos co-immunostained with GFP antibody (anti-GFP, green) and ZsYellow antibody (anti-RCFP, red) at 26 hpf. The future atrial segment of the linear heart tube (LHT, green arrow) expresses cmlc2 alone (green), while the future proximal ventricular myocardium co-expresses cmlc2 and nkx2.5 (yellow). Non-myocardial nkx2.5+ second heart field (SHF) progenitors (red arrow) can be visualized in control animals (n=8), but are lacking in vgo mutants (n=7).
To elucidate the cellular mechanisms underlying the vgo SHF phenotype, we evaluated mutants soon after cardiac cone stages for cellular defects in the extra-cardiac population of nkx2.5+ SHF cells at the arterial pole. First, we used TUNEL (terminal deoxynucleotidyl transferase biotin-dUTP nick end-labeling) labelling to highlight apoptotic cells in control and vgotu285 mutants carrying the nkx2.5:ZsY transgene. Although TUNEL+ apoptotic cells were observed throughout the bodies of both control and vgo embryos, no TUNEL+ cells were detected in the ZsY+ SHF domain (data not shown). Next, we used the thymidine analog EdU to examine the proliferative state of nkx2.5:ZsY+ SHF cells at the arterial pole. Specifically, embryos were pulsed with 5-ethynyl-2′deoxyuriding (EdU) at 20ss (19 hpf) and collected at 26ss (22hpf) for processing using Click-iT Alexa-555 along with antibody staining for ZsYellow. We detected extensive proliferation throughout the bodies of both vgo tu285 and sibling controls (Fig. 5A,B). Interestingly, we observed abundant proliferative ZsY+ cells that predominantly localized to the arterial pole/SHF region of the cardiac cone (Fig. 5C,E). However, while cardiac development appeared similar between vgo and sibling controls (Fig 5A,B; insets), a significant decrease in the number of proliferative EdU+ SHF progenitors was observed in vgo mutants (Fig. 5C–E). These findings support a model in which Tbx1 stimulates proliferation or self-renewal of ltpb3+; nkx2.5+ SHF progenitors to maintain the CPC pool as new myocardium is accreted to the linear heart tube. Based on this model, we speculate that loss of Tbx1 results in a premature depletion of undifferentiated CPCs from the SHF.
Figure 5. SHF progenitor cells fail to proliferate in the absence of Tbx1.
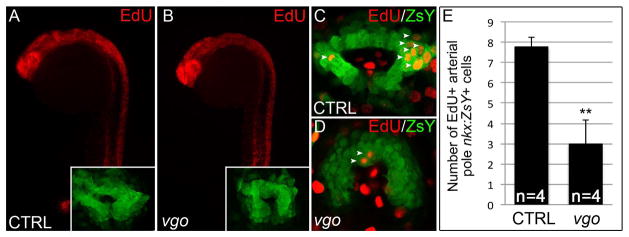
(A–D) Click-iT EdU labeling in Tg(nkx2.5:ZsYellow) vgo and control siblings. (A–B) Flourescent microscopy images of EdU+ cells (red) in control (A) and vgo (B) embryos. 10X maginificaition, anterior up, dorsal right. Insets show flattened confocal images of ZsY+ cells at the arterial pole (C–D) Composite of two confocal sections showing EdU+ cells (red) within the ZsYellow+ (green) SHF (white arrowheads). (E) Graph depicting the total number of EdU+ cells in the entire confocal stack in control (n=4) and vgo (n=4) embryos.
Discussion
Because SHF perturbations are predicted to be common causes of conotruncal malformations in patients, studies designed to decipher the molecular regulation of SHF biology are significant. Specifically, any gene implicated in SHF development becomes a candidate that, when mutated, may cause OFT-related congenital heart defects. Mounting evidence suggests that the genetic pathways regulating SHF development are highly conserved in zebrafish. Here, we extend previous observations showing that zebrafish tbx1 mutants show conotruncal defects consistent with a role in SHF development. These studies serve to further solidify the use of the zebrafish model organism for discovery based approaches that may uncover new and potentially clinically relevant genes important for arterial pole development.
Deciphering the tissue-specific roles of TBX1 in mammalian OFT morphogenesis has been extremely complex due in part to the high degree of interaction between the Tbx1-expressing pharyngeal endoderm and pharyngeal mesoderm, the latter of which contains SHF progenitors. While conditional loss-of-function studies have yielded valuable information, the interpretations have been clouded by cre-mediated recombination in both tissue compartments. For example, although deletion of Tbx1 in the Nkx2.5 expression domain results in OFT hypoplasia, recombination occurs in the pharyngeal endoderm, pharyneal mesoderm, and SHF (Xu et al., 2005; Xu et al., 2004). In another study, Arnold et al. showed that deletion of Tbx1 in the Foxg1+ pharyngeal endoderm results in severe OFT defects that resemble those occurring in Tbx1−/− nulls (Arnold et al., 2006). Although Foxg1-mediated recombination was reported to be limited to pharyngeal endoderm in this study (Arnold et al., 2006), Zhang et al. observed additional Foxg1-induced cre activity in pharyngeal mesoderm when examining recombination on a different genetic background (Zhang et al., 2005). Thus, the specific interplay between the pharyngeal endoderm and mesoderm in regards to TBX1 function remains unclear.
The most compelling argument that TBX1 functions cell non-autonomously for SHF development in the mouse was shown through mosaic analyses (Xu et al., 2004). Specifically, Tbx1−/− cells were mixed with wild-type cells to generate embryos of mixed genetic origin. In these mice, Tbx1−/− cells contributed to the SHF, showed no proliferative disadvantage, and were accreted at the same frequency as wild-type cells to the developing OFT. Interestingly, inactivation of Tbx1 in Mesp1+ mesoderm results in severe OFT defects that can be rescued by mesodermal restoration of Tbx1 (Zhang et al., 2006b). Although these data show a strict requirement for Tbx1 in the mesoderm, Mesp1 is expressed rather broadly leaving open the possibility that Tbx1 is required in a mesodermal population distinct from the SHF. Thus, the outcome of these lineage-specific inactivation/reactivation experiments remains consistent with those from the chimera analyses that demonstrated a non-autonomous role for TBX1 in SHF-mediated OFT formation.
Our studies in zebrafish show that Tbx1 is required for SHF proliferation and, based on its expression pattern, most likely acts cell non-autonomously. Although our in situ analyses are unable to definitively exclude tbx1 expression in medial nkx2.5+ cells in the heart forming region that may represent SHF progenitors, the domains appear separate. Moreover, we failed to observe transcripts in the region occupied by SHF progenitors at cardiac cone or heart tube stages. Our observations coupled with previous reports showing tbx1 transcripts in the pharynx (Piotrowski et al., 2003) makes it tempting to speculate that perhaps the role of Tbx1 in pharyngeal tissues has been conserved in zebrafish for OFT development. However, chimera analyses to determine autonomy in OFT development and cre/loxP-mediated genetic lineage tracing of tbx1-expressing cells in zebrafish will be required to appreciate the complete tbx1 expression profile and tissue derivatives during cardiac development.
Although tbx1 does not appear to be expressed in SHF progenitors in zebrafish, our data and that of Hami et al. show that Tbx1 is required for SHF development. Experimental evidence garnered from ltbp3 cre/loxP lineage tracing (Zhou et al., 2011) and a photoconversion assay that measures the timing of cardiomyocyte differentiation (Lazic and Scott, 2011) suggests that approximately 40–50% of the ventricular chamber is derived from SHF progenitors following heart tube formation. However, our ventricular cardiomyocyte counts in tbx1 mutants revealed only a 25% reduction. This outcome could be explained several ways. It is possible that only 25% of the ventricle is derived from SHF cells and that our previous cre/loxP lineage tracing overestimated contribution as ltbp3 expression partially overlaps with differentiated cardiomyocytes at the end of the heart tube. Alternatively, because ltbp3 expression is initiated in tbx1 null animals, it is possible that there is only a partial myocardial deficit in vgo animals. Another explanation, which is consistent with its role in the mouse (Parisot et al., 2011), is that Tbx1 is required for proper development of a sub-domain within a larger SHF. As new SHF markers and lineage traces emerge in zebrafish, a better understanding of the full complement of cardiovascular derivatives of the SHF will be revealed, ultimately aiding our understanding of mutant phenotypes and gene function.
Experimental Procedures
Zebrafish lines and maintenance
Zebrafish embryos, larvae, and adults were produced, grown and maintained according to standard protocols approved by the Institutional Animal Care and Use Committees of Massachusetts General Hospital. Ethical approval was obtained from the Institutional Animal Care and Use Committees of Massachusetts General Hospital. The previously published (Piotrowski et al., 2003; Piotrowski et al., 1996; Schilling et al., 1996) vgotu285 and vgotu208 mutant tbx1 alleles were used in this study. The vgo285 and vgo208 alleles of tbx1 contain a C -> T and an A-> T transition at nucleotide positions 364 and 879 respectively, each creating premature stop codons. Transgenic strains Tg(cmlc2::DsRed-nuc) (Mably et al., 2003), Tg(nkx2.5::ZsYellow) (Zhou et al., 2011) and Tg(cmlc2::GFP) (Burns et al., 2005) were described previously.
Double in situ hybridization, immunohistochemistry, and cryosectioning
Single and double in situ hybridization was performed as described (Thisse et al., 1993), with digoxygenin-labeled antisense RNA probes to ltbp3 (EcoRI/Sp6), nkx2.5(EcoRI/T7), tbx1 (EcoRI/T7), vmhc (EcoRI/T3), and fluorescein-labeled antisense RNA probes to cmlc2 (NotI/T7) and tbx1. For cryo-sections, stained embryos were washed in PBS and embedded in 1.2% agarose in a 5% sucrose solution. Embedded embryos were placed in 30 % sucrose solution overnight at 4° C. Agarose blocks were covered with Optimal Cutting Temperature compound (OCT) (Tissue-Tek) and 50um sections were taken using Leica CM 3050 S cryostat. Immunohistochemistry was performed as described (Zhou et al., 2011), using primary antibodies anti-GFP mouse mono-clonal (1:50; Santa Cruz Biotechnology, Santa Cruz, CA), anti-RCFP rabbit polyclonal pan (1:50; Clontech, Mountain View, CA), anti-dsRed rabbit polyclonal (1:50; Clontech, Mountain View, CA), muscle-specific MF20 (1:50; University of Iowa, Iowa City, IA), Elastin-2 (1:1000; Fred Keeley) and secondary antibodies Alexa Fluor 555 goat anti-rabbit IgG2b, Alexa Fluor 488 goat anti-mouse IgG2b and Alexa Fluor 546 goat anti-mouse IgG2b (all 1:200; Invitrogen, Carlsbad, CA).
Bright Field, Fluorescence and Confocal microscopy
Confocal imaging was performed as described previously (Zhou et al., 2011) on a LS5 confocal microscope (Zeiss) using a 40× water immersion objective. ImageJ (v1.43U; National Institute of Health, USA) software was used to count cardiomyocyte (CM) nuclei in Z-stack confocal images. Statistical significance for CM counts was determined using unpaired Student’s t-test. Bright field and fluorescent microscopy was performed with a 10x objective on an Eclipse 80i microscope (Nikon, Melville, NY) using a Retiga 2000R camera (Q-imaging, Surrey, BC Canada) and NIS-Elements AR 3.00 imaging software (Nikon).
TUNEL and Click-iT EdU staining
TUNEL (terminal deoxynucleotidyl transferase biotin-dUTP nick end-labeling) was performed as described previously (Zhou et al., 2011). Briefly, Tg(nkx2.5:ZsYellow) vgo285 mutant and control embryos were collected at 23ss and fixed overnight at 4°C in 4% paraformaldehyde before being transferred to 100% methanol and stored at −20°C. Embryos were rehydrated in methonol/PBST series, permeabilized with proteinase K and subjected to a TUNEL assay via the In Situ Cell Death Detection Kit, TMR red (Roche Applied Science) according to the manufacturer’s instructions. Proliferation experiments were performed as previously described (Mahler et al., 2010) using the Click-iT EdU imaging kit (Invitrogen). Briefly, embryos were incubated on ice for 30 minutes in 10mM 5-ethynyl-2′deoxyuriding (EdU), rinsed 3 times in embryo medium, and chased for 3 hours at 28°C. Immunofluorescent staining with anti-rCFP (Alexa-488) and Click-iT-Alexa-555 was performed and embryos were analyzed using confocal microscopy.
Acknowledgments
We thank L. Zon and I. Scott for providing zebrafish strains; J Holzschuh for providing a detailed Click-iT EdU labeling protocol; Fred Keeley for providing Elastin2 anti-serum, and the Developmental Studies Hybridoma Bank under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, for providing the MF20 antibody. This work was supported by awards from the National Institutes of Health to K.N. (5F32HL110627), the American Heart Association to B.G-A. (10POST4170037), the American Heart Association (Grant in Aid no. 10GRNT4270021) and the National Heart Lung and Blood Institute (5R01HL096816) to C.G.B, and the Harvard Stem Cell Institute (Young Investigator Award and Cardiovascular Program Award), National Heart Lung and Blood Institute (5R01HL111179) and the March of Dimes Foundation (1-FY12-467) to C.E.B.
References
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Aggarwal VS, Morrow BE. Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev Disabil Res Rev. 2008;14:19–25. doi: 10.1002/ddrr.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JS, Werling U, Braunstein EM, Liao J, Nowotschin S, Edelmann W, Hebert JM, Morrow BE. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development. 2006;133:977–987. doi: 10.1242/dev.02264. [DOI] [PubMed] [Google Scholar]
- Brown CB, Wenning JM, Lu MM, Epstein DJ, Meyers EN, Epstein JA. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev Biol. 2004;267:190–202. doi: 10.1016/j.ydbio.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Burns CG, Milan DJ, Grande EJ, Rottbauer W, MacRae CA, Fishman MC. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat Chem Biol. 2005;1:263–264. doi: 10.1038/nchembio732. [DOI] [PubMed] [Google Scholar]
- Chapman DL, Garvey N, Hancock S, Alexiou M, Agulnik SI, Gibson-Brown JJ, Cebra-Thomas J, Bollag RJ, Silver LM, Papaioannou VE. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev Dyn. 1996;206:379–390. doi: 10.1002/(SICI)1097-0177(199608)206:4<379::AID-AJA4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Chen L, Fulcoli FG, Tang S, Baldini A. Tbx1 regulates proliferation and differentiation of multipotent heart progenitors. Circ Res. 2009;105:842–851. doi: 10.1161/CIRCRESAHA.109.200295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pater E, Clijsters L, Marques SR, Lin YF, Garavito-Aguilar ZV, Yelon D, Bakkers J. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136:1633–1641. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol. 2001;235:62–73. doi: 10.1006/dbio.2001.0283. [DOI] [PubMed] [Google Scholar]
- Hami D, Grimes AC, Tsai HJ, Kirby ML. Zebrafish cardiac development requires a conserved secondary heart field. Development. 2011;138:2389–2398. doi: 10.1242/dev.061473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinits Y, Pan L, Walker C, Dowd J, Moens CB, Hughes SM. Zebrafish Mef2ca and Mef2cb are essential for both first and second heart field cardiomyocyte differentiation. Dev Biol. 2012 doi: 10.1016/j.ydbio.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131:5491–5502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- Huynh T, Chen L, Terrell P, Baldini A. A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis. 2007;45:470–475. doi: 10.1002/dvg.20317. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Kochilas LK, Potluri V, Gitler A, Balasubramanian K, Chin AJ. Cloning and characterization of zebrafish tbx1. Gene Expr Patterns. 2003;3:645–651. doi: 10.1016/s1567-133x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- Lazic S, Scott IC. Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev Biol. 2011;354:123–133. doi: 10.1016/j.ydbio.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Liao J, Aggarwal VS, Nowotschin S, Bondarev A, Lipner S, Morrow BE. Identification of downstream genetic pathways of Tbx1 in the second heart field. Dev Biol. 2008;316:524–537. doi: 10.1016/j.ydbio.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Mably JD, Mohideen MA, Burns CG, Chen JN, Fishman MC. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13:2138–2147. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- Mahler J, Filippi A, Driever W. DeltaA/DeltaD regulate multiple and temporally distinct phases of notch signaling during dopaminergic neurogenesis in zebrafish. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:16621–16635. doi: 10.1523/JNEUROSCI.4769-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques SR, Lee Y, Poss KD, Yelon D. Reiterative roles for FGF signaling in the establishment of size and proportion of the zebrafish heart. Dev Biol. 2008;321:397–406. doi: 10.1016/j.ydbio.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Miao M, Bruce AE, Bhanji T, Davis EC, Keeley FW. Differential expression of two tropoelastin genes in zebrafish. Matrix Biol. 2007;26:115–124. doi: 10.1016/j.matbio.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Parisot P, Mesbah K, Theveniau-Ruissy M, Kelly RG. Tbx1, subpulmonary myocardium and conotruncal congenital heart defects. Birth Defects Res A Clin Mol Teratol. 2011;91:477–484. doi: 10.1002/bdra.20803. [DOI] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–2433. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski T, Ahn DG, Schilling TF, Nair S, Ruvinsky I, Geisler R, Rauch GJ, Haffter P, Zon LI, Zhou Y, et al. The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development. 2003;130:5043–5052. doi: 10.1242/dev.00704. [DOI] [PubMed] [Google Scholar]
- Piotrowski T, Nusslein-Volhard C. The endoderm plays an important role in patterning the segmented pharyngeal region in zebrafish (Danio rerio) Dev Biol. 2000;225:339–356. doi: 10.1006/dbio.2000.9842. [DOI] [PubMed] [Google Scholar]
- Piotrowski T, Schilling TF, Brand M, Jiang YJ, Heisenberg CP, Beuchle D, Grandel H, van Eeden FJ, Furutani-Seiki M, Granato M, et al. Jaw and branchial arch mutants in zebrafish II: anterior arches and cartilage differentiation. Development. 1996;123:345–356. doi: 10.1242/dev.123.1.345. [DOI] [PubMed] [Google Scholar]
- Reifers F, Walsh EC, Leger S, Stainier DY, Brand M. Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar) Development. 2000;127:225–235. doi: 10.1242/dev.127.2.225. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scambler PJ. 22q11 Deletion Syndrome: A Role for TBX1 in Pharyngeal and Cardiovascular Development. Pediatr Cardiol. 2010 doi: 10.1007/s00246-009-9613-0. [DOI] [PubMed] [Google Scholar]
- Schilling TF, Piotrowski T, Grandel H, Brand M, Heisenberg CP, Jiang YJ, Beuchle D, Hammerschmidt M, Kane DA, Mullins MC, et al. Jaw and branchial arch mutants in zebrafish I: branchial arches. Development. 1996;123:329–344. doi: 10.1242/dev.123.1.329. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- Thisse C, Thisse B, Schilling TF, Postlethwait JH. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development. 1993;119:1203–1215. doi: 10.1242/dev.119.4.1203. [DOI] [PubMed] [Google Scholar]
- Vincent SD, Buckingham ME. How to make a heart: the origin and regulation of cardiac progenitor cells. Curr Top Dev Biol. 2010;90:1–41. doi: 10.1016/S0070-2153(10)90001-X. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Lania G, Huynh T, Baldini A. Partial rescue of the Tbx1 mutant heart phenotype by Fgf8: genetic evidence of impaired tissue response to Fgf8. J Mol Cell Cardiol. 2010;49:836–840. doi: 10.1016/j.yjmcc.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002a;11:915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002b;129:4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- Witzel HR, Jungblut B, Choe CP, Crump JG, Braun T, Dobreva G. The LIM Protein Ajuba Restricts the Second Heart Field Progenitor Pool by Regulating Isl1 Activity. Dev Cell. 2012;23:58–70. doi: 10.1016/j.devcel.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Cerrato F, Baldini A. Timed mutation and cell-fate mapping reveal reiterated roles of Tbx1 during embryogenesis, and a crucial function during segmentation of the pharyngeal system via regulation of endoderm expansion. Development. 2005;132:4387–4395. doi: 10.1242/dev.02018. [DOI] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–3227. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yelon D, Horne SA, Stainier DY. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol. 1999;214:23–37. doi: 10.1006/dbio.1999.9406. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhong T, Wang Y, Jiang Q, Song H, Gui Y. TBX1, a DiGeorge syndrome candidate gene, is inhibited by retinoic acid. Int J Dev Biol. 2006a;50:55–61. doi: 10.1387/ijdb.052036lz. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A, et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132:5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006b;133:3587–3595. doi: 10.1242/dev.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cashman TJ, Nevis KR, Obregon P, Carney SA, Liu Y, Gu A, Mosimann C, Sondalle S, Peterson RE, et al. Latent TGF-beta binding protein 3 identifies a second heart field in zebrafish. Nature. 2011 doi: 10.1038/nature10094. [DOI] [PMC free article] [PubMed] [Google Scholar]