

Abstract
Hemostasis and thrombosis represent two sides of the same coin. Hemostasis maintains blood fluidity in the vascular system while allowing for rapid thrombus formation to prevent excessive hemorrhage after blood vessel injury. Thrombosis is a pathologic extension of the normal hemostatic mechanism, occurring when unwanted clot formation develops in certain pathological situations. The molecular mechanisms underlying both phenomena are fundamentally identical. One of the key players in both processes is the plasma glycoprotein von Willebrand factor, which perfectly illustrates this duality between hemostatic and thrombotic mechanisms. The purpose of this review is to discuss novel findings on the role of von Willebrand factor at this interface, and how some of these findings may help develop new therapeutic strategies.
Keywords: Animals, Hemorrhage, blood, genetics, metabolism, Humans, Mutation, Protein Conformation, Protein Multimerization, Thrombosis, blood, genetics, metabolism, von Willebrand Factor, chemistry, genetics, metabolism
Keywords: von Willebrand factor, Hemostasis, Thrombosis, von Willebrand disease
1. Introduction
Von Willebrand factor (VWF) is a multimeric glycoprotein that circulates in plasma at a concentration of 5–10 μg/ml. VWF is also located in platelet α-granules, in Weibel-Palade bodies of endothelial cells, and in the subendothelium. During vessel injury, VWF acts as a molecular bridge between platelets and exposed components of the subendothelium, allowing the formation of the platelet thrombus [1]. The role of VWF is particularly important at high shear rates, as VWF is the only molecule able to promote platelet adhesion and platelet aggregation in a flow-resistant manner in these conditions [2]. Another function of VWF is to serve as a carrier protein for coagulation factor VIII, protecting it from premature clearance [3]. VWF has a highly repetitive structure and is composed of homologous domains arranged in the following order: D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK in which D1-D2 represent the VWF propetide and D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK, the mature VWF subunit [4]. An important feature of VWF relates to its organization in multimers ranging from dimer to very large concatamers reaching up to 15,000 kDa [5]. The biological activity of VWF is regulated by the length of these multimers, with the longest being the most active in binding platelets [6]. Regulation of VWF multimeric size is under the control of a metalloprotease, ADAMTS13 (A Disintegrin And Metalloproteinase with ThromboSpondin type 1 motif, member 13) which cleaves at a specific site in the A2 domain of VWF between residues tyrosine 1605 and methionine 1606 [7].
VWF is a rather unique molecule in its ability to change shape according to type of flow. Elegant work from Dr. Springer has recently shed light on our understanding about how unfolding of VWF by elongational flow can regulate both its hemostatic potential and its cleavage by ADAMTS13 [8].
The aim of the present review is to provide an overview of recent findings on how each of the various aspects mentioned in this introduction contributes to the role of VWF in hemorrhagic and thrombotic conditions (Figure 1).
Figure 1. VWF: A Janus protein in bleeding and thrombosis.
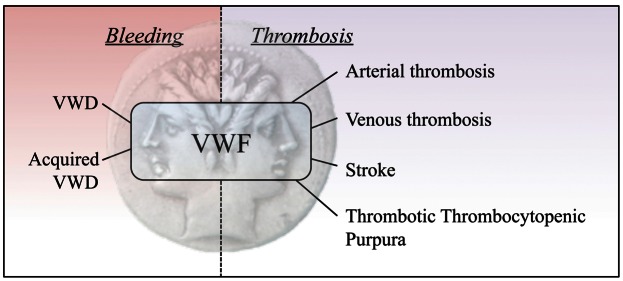
The hemostatic system is designed to react rapidly upon vascular damage in order to minimize blood loss, and needs tight regulation to prevent thrombotic occlusion of the vessels. VWF is a key player of the hemostatic system, illustrated by its liaison with both hemorrhagic and thrombotic conditions. In the present review, novel findings on the role of VWF at this interface between bleeding and thrombosis are being discussed.
2. VWF in Bleeding
2.1. Hereditary von Willebrand disease
The discovery of VWF is intricately linked to the history of bleeding disorders. Von Willebrand disease (VWD), identified in 1926 by Dr. Erik von Willebrand, is caused by inherited quantitative or qualitative deficiencies in VWF. The classification of VWD is based on pathophysiologic mechanisms [9, 10]. Three main types have been recognized, type 1 includes partial quantitative deficiency, type 2 includes qualitative defects, and type 3 corresponds to a complete deficiency of VWF. Type 2 VWD is further divided in several subtypes, including types 2A, 2B, 2M and 2N, each of which is associated with a specific functional or structural defect (for recent review see Schneppenheim and Budde [11]). Briefly, VWD-type 2A is associated with a lack of high multimers, which results in inefficient platelet binding. The lack of high molecular weight multimers originates from mutations impairing intracellular multimerisation (so-called type 2A group 1 mutations, which are mostly located in the D3 domain) or from mutations allowing excessive proteolysis by ADAMTS13 (type 2A group 2 mutations, mostly located in the A2 domain). VWD-type 2B consists of gain-of-function mutations in the A1 domain, increasing not only the affinity for the platelet receptor glycoprotein (Gp) Ibα, but also the susceptibility for proteolysis by ADAMTS13 [12]. Increased platelet affinity may provoke thrombocytopenia, which appears to be the major risk factor for bleeding in VWD-type 2B patients [13]. The absence of high molecular weight multimers seen in these patients apears to be the result of ADAMTS-mediated proteolysis rather than adsorption to platelets [14]. VWD-type 2M mutations also occur in the A1 domain, but contrastingly result in a lack or diminished affinity for GpIbα [11]. Type 2N is characterized by mutations in the N-terminal region (D′-D3 domains) of the mature VWF subunit and is associated with impaired binding of FVIII [15]. As a consequence, FVIII levels are reduced, and the half-life of intravenously administered FVIII is considerably shortened.
Interestingly, recent studies have also revealed series of defects that are not captured by the classic nomenclature for VWD. For instance, several mutations within the A3 domain have been identified that are associated with impaired binding to collagen I and/or III [16–18]. Patients with such mutations are characterized by a mild bleeding tendency or no bleeding tendency at all. Noteworthy, VWF mutants with impaired binding to collagen I and III have recently been examined in a mouse model for VWD. These mutations were associated with a normal tail-bleeding time, confirming the lack of association of these mutations with a severe bleeding tendency (Figure 2) [19]. In contrast, mice expressing these collagen mutants seem to be protected against thrombosis, as illustrated by delayed thrombus growth and significantly increased vessel occlusion times in a ferric chloride-induced thrombosis model (Figure 2) [19]. Another example relates to mutations that are associated with a decreased circulatory half-life of VWF. A first report appeared in 2002, describing mutation Arg1205 to His (also known as the Vicenza mutation) to be associated with a reduced survival of VWF following desmopressin treatment [20]. Additional studies using a purified recombinant VWF mutant confirmed that this mutation results in an increased clearance rate of the molecule [21]. Moreover, the phenotype of this mutation has been reproduced in a mouse model for VWD [22]. Since then, a series of other mutations have been identified that are associated with a reduced circulatory survival [23–25], which may explain the unusually low VWF levels in patients with these mutations.
Figure 2. Effect of deficient collagen binding on VWF function in hemostasis and thrombosis in a murine model.
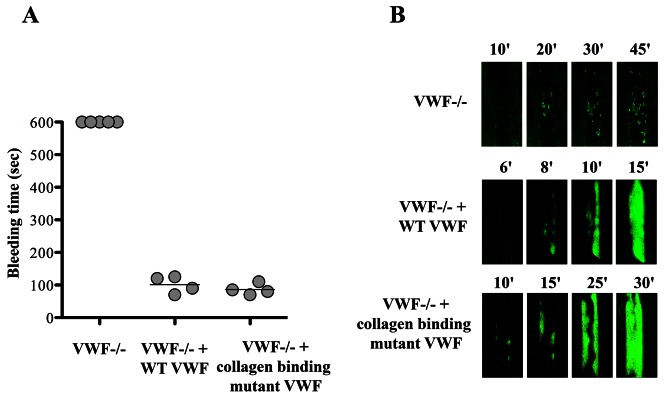
VWF-deficient mice were left untreated or injected with murine Vwf cDNA encoding for wild-type or a collagen binding mutant VWF. Four days after injection, when plasma expression of VWF was stable, mice were challenged in a tail-bleeding time assay (A) or in a ferric-chloride induced thrombosis model. Mice expressing the VWF molecule unable to bind to collagens were able to correct their bleeding time but had significantly prolonged occlusion time in arterioles. Platelet accumulation in vessels is visualized in green fluorescence. This research was originally published in Blood. Marx I et al. Blood. 2008;112:603–609. © the American Society of Hematology.
For both the collagen and clearance mutants, no separate VWD type has (yet) been designated. The specific defects associated with these mutations could be in favor of doing so (for example Type 2C and Type 1C for the collagen and clearance mutations, respectively). On the other hand, this issue may be of poor relevance from a clinical point of view. For instance, desmopressin is often used in type 1 VWD patients, whereas treatment of type 2 VWD patients, except type 2N, merely involves replacement therapy [26]. As for the clearance mutants, one would expect that the short half-life of the endogenous VWF molecules would limit the use of desmopressin in these patients. However, a recent study by Castaman and colleagues involving 90 patients with the Vicenza Arg1205His mutation and 23 with the Cys1130Phe mutation (both associated with increased clearance) revealed that all dental extractions, minor surgeries and deliveries were successfully managed with desmopressin [27]. Apparently, despite the short circulatory half-life of their VWF, the use of desmopressin remains an important therapeutic option in these patients.
2.2. Acquired von Willebrand disease
Deficiency or dysfunction of VWF may also occur secondary to another clinical condition, and as such it is qualified as acquired VWD (AVWD). Recently, a number of excellent reviews of this phenomenon have been published [28, 29]. The diagnosis of AVWD is complicated since its multifactorial etiology requires a series of tests to confirm or exclude AVWD. In addition, AVWD is not always recognized in view of its secondary character and the wide range of underlying disorders that are associated with it. The most common disorders associated with AVWD include lymphoproliferative disorders (such as monoclonal gammopathy), myeloproliferative disorders (such as chronic myeloid leukemia), hypothyroidism, drug use (such as valproic acid), and cardiac disorders. The non-uniformity of these disorders provides an explanation for the different abnormalities found in AVWD. Like in acquired hemophilia, the presence of anti-VWF antibodies may provoke antibody-mediated clearance of the protein resulting in reduced plasma levels and/or reduced recovery of exogenous VWF. Moreover, such antibodies may interfere with VWF function [29]. In addition, AVWD may be characterized by a lack of high molecular weight multimers due to their adsorption onto plasma cells or platelets [30, 31]. Alternatively, the loss of high molecular weight multimers may be related to a shear stress-dependent process, an issue that will be discussed in more detail below (paragraph 2.3).
2.3. Cardiac disorders and loss of high molecular weight VWF multimers
The first report on the association between cardiac disorders and the loss of high molecular weight VWF multimers appeared already in 1986 [32]. In the early 1990s, a link between this loss of high molecular weight multimers and the occurrence of gastro-intestinal bleedings in patients with aortic valve stenosis was proposed [33]. In 2003, a hallmark study was published in which the prevalence and the determinants of hemostatic abnormalities in patients with aortic stenosis was evaluated [34]. From this study it was concluded that bleeding complications occurred in about 20% of the patients with aortic stenosis. A loss of high molecular weight multimers (as assessed directly by multimer analysis or by using collagen-binding as a surrogate marker) was found in the vast majority of patients [34]. Apparently, the loss of VWF multimers is a common phenomenon in aortic stenosis patients, and frequently associated with bleeding symptoms. Interestingly, the loss of VWF multimers associated with increased gastrointestinal hemorrhage has also been observed in other cardiac disorders, such as hypertrophic obstructive cardiomyopathy [35, 36] and patients with left ventricular assist devices (for review see [37]). The frequency at which the bleeding problems associated with the loss of VWF multimers occur, in combination with the large patient groups concerned (aortic stenosis alone affects 3–5% of the population aged over 60 years), suggests that this clinical complication has a wider impact than previously thought. Paradoxically, the disease state of many of the patients with aortic stenosis has been considered to be associated with a prothrombotic risk, and anticoagulant therapy is therefore regularly applied to these patients. However, the reduced hemostatic potential of VWF in these patients may perhaps serve as a “natural” antidote for this thrombotic risk in these patients, and re-assessment of the extent of anticoagulant therapy in these patients may be subject to reconsideration.
Despite the progress on the recognition of the link between cardiac disorders, bleeding risks and VWF abnormalities, not much is known about the molecular basis. An elegant study by Pareti et al. in 2003 suggested that the passage of blood through the stenosed aortic valve may result in shear forces that could trigger the adsorption of higher VWF multimers by platelets, providing an explanation for the reduced hemostatic potential of VWF [38]. In addition, they revealed that the loss of VWF multimers coincided with an increase of VWF degradation products, suggesting that VWF is more susceptible to proteolysis in this clinical condition [38]. Indeed, we now know that proteolysis of VWF by its protease ADAMTS13 proceeds most efficiently upon the unfolding of VWF when exposed to high shear forces [39]. However, additional studies are needed to decipher the underlying cause of the loss of VWF function and its specific relationship with gastrointestinal bleedings. In terms of therapy, it appears that replacement of the stenosed heart valve or removal of the assist device seems sufficient to reverse the loss of VWF multimers and the risk of bleeding [34, 37].
3. VWF in thrombosis
We have seen that during the hemostatic process, VWF plays a crucial role in thrombus formation to prevent excessive bleeding. That same capacity to promote platelet adhesion at high shear stress, conditions that are encountered in stenosed arteries, makes it a crucial player in atherothrombosis. A number of population based studies have suggested that elevated levels of VWF are a risk factor for cardiovascular diseases (CVD) [40] but this issue is still debated for healthy patients for whom it seems difficult to predict occurrence of CVD solely based on this parameter [41]. In contrast, such a link is well established for patients with previous CVD or in older patients where high levels of VWF are clearly associated with higher risk of recurrence and mortality [41–43]. What remains unclear, however, is whether high VWF levels represent the actual cause of CVD or are a marker of endothelial dysfunction, which could be at the origin of CVD. Nevertheless, search for genetic determinants of VWF levels in relation to CVD have led to the identification of single nucleotide polymorphisms (SNPs) on the VWF gene associated with coronary heart disease in both diabetic patients [44, 45] and patients with advanced atherosclerosis [46]. The rs1063856 SNP located in exon 18 seems to be of particular relevance, and has been identified in several studies as associated with VWF antigen levels [45], incident venous thrombosis [47] and other CVDs [40]. In addition to VWF gene SNPs, genome wide association studies have recently identified new genetic determinants of VWF levels [48, 49]. Although at an early stage of analysis, these studies have already led to the recognition of an association between the risk of arterial thrombosis and genetic variations in the SNARE protein gene, STX2 potentially through its capacity to regulate VWF plasma levels [50]. Besides these ongoing epidemiological studies, the role of VWF in a number of thrombotic processes has also been investigated through experimental models on which this review will focus in the next paragraphs.
3.1. VWF and venous thrombosis
Given its prominent role in the recruitment of platelets under conditions of high shear stress, VWF has historically been linked to atherothrombotic complications. In contrast, an eventual link with venous thrombosis was thought to be indirect, via its function as a carrier-protein for coagulation factor VIII [51]. However, a more recent epidemiological study identified VWF as an independent risk factor for venous thrombosis [52]. Indeed, immunohistochemical studies revealed the presence of VWF in ilio-femoral thrombi and in pulmonary thromboemboli of patients who died of venous thromboembolism [53]. Recently, a series of studies using dedicated mouse models has been reported in which the link between VWF and venous thrombosis has been explored in more detail. First, it was found that in the absence of VWF (but with normalized factor VIII levels), no thrombotic occlusion could be provoked in mesenteric venules in a ferric chloride-induced thrombosis model [54]. An unexpected finding in this study was that platelet recruitment by VWF in this venous thrombosis model was not fully mediated by classic VWF-GpIbα interactions, but also involved interactions between VWF and so far unidentified other platelet receptors. The critical role of VWF in platelet recruitment in venous thrombosis was confirmed in a more specific model for deep vein thrombosis [55]. Histological analysis of the thrombi formed in this mouse model resembled to a large extent the thrombi that are formed in humans, consisting of a red part that is rich in fibrin and red blood cells, with few platelets and a white region that is rich in platelets and fibrin. Although both studies are in support of the contribution of VWF to the development of venous thrombi, the underlying mechanism remained to be elucidated. Some intriguing observations have recently been made relating to this point. In 2010, it was reported that extracellular DNA traps (so called Neutrophil Extracellular Traps, NETs) promote thrombosis in vitro and in vivo [56]. NETs are a meshwork of DNA fibers comprising citrullinated histones and antimicrobial proteins. In particular the histone components of these NETs contribute to the prothrombotic property by their capacity to induce platelet aggregation [56, 57]. In venous thrombi formed in a baboon-model for venous thrombosis, a co-localization between the DNA meshwork and VWF was found [56]. The triad NETs-venous thrombosis-VWF was further investigated in a study by Brill and colleagues using their mouse model for deep venous thrombosis [58]. They showed that the presence of histones in the circulation triggers the release of VWF from endothelial cells. Moreover, the citrullinated histones are found throughout the red parts of the thrombi, and often a co-localization with VWF was observed [58]. Taken together, these recent findings have revealed a novel aspect of VWF biology, recognizing VWF as a critical component for venous thrombosis. They also identify VWF and NETs as potential targets for the development of novel therapeutics for the treatment of venous thrombosis.
3.2. VWF and Thrombotic Thrombocytopenic Purpura
The past decade has brought remarkable progress in our understanding of the molecular basis of thrombotic thrombocytopenic purpura (TTP). The link between the presence of ultra-large VWF molecules, the absence of a specific VWF-cleaving protease and this disorder was already recognized in 1982 [59]. The molecular entity of this VWF-cleaving protease, now designated as ADAMTS13, remained unidentified until 2002, when the protein was purified and its cDNA was cloned (for review see [60]). This breakthrough triggered many studies on the genetic and molecular basis of TTP and consequently our knowledge on the structure-function relationships between VWF and ADAMTS13 has been expanding rapidly since then (for review see [61]). As mentioned in the introduction, the ADAMTS13 protease cleaves VWF multimers, leading to shorter, less active VWF molecules. As is always the case for VWF, defects in ADAMTS13 proteolysis have pathological consequences that can be either pro-hemorrhagic or prothrombotic. VWD-type 2A illustrates the pro-hemorrhagic aspect resulting from excessive proteolysis while TTP associated with deficient ADAMTS13 activity illustrates the thrombotic complications that are associated with the presence of ultra-large and supra active VWF multimers.
In the present review, we will focus on recent insights in the interactions between VWF and ADAMTS13 and how these interactions are regulated. Indeed, although merely of experimental nature, the important progress that has been made in that regard, helps us to better understand the pathophysiology of TTP. The first encounter between VWF and ADAMTS13 occurs upon the secretion of VWF from the endothelial cells. Agonist-stimulated secretion of VWF results in the formation of VWF strings that consist of multiple VWF concatamers [62]. Under the influence of hydrodynamic shear forces, these strings elongate along the vascular wall. This elongated conformation not only exposes the binding site for platelets in the VWF A1 domain, but also stretches the VWF A2 domain so that the cleavage site for ADAMTS13 becomes available (Figure 3) [8]. Recently, the elongation process that enables ADAMTS13 cleavage has been visualized in detail, revealing that VWF strings elongate and shrink in a time- and space-dependent manner and that cleavage selectively occurs at sites where VWF is elongated [63]. Following ADAMTS cleavage, VWF is released into the circulation. Due to a drop in shear forces, VWF changes from a stretched into a globular form, in which the ADAMTS13 cleavage site in the A2 domain is inaccessible [64]. Importantly, this does not mean that VWF and ADAMTS13 can no longer interact. Two studies independently showed that ADAMTS13 interact with VWF in the circulation [61]. As expected, this shear force-independent interaction proceeds independently of the A2 domain, but rather involves the C-terminal portion of the VWF subunits. The complementary interactive sites are located away from the catalytic domain of ADAMTS13, and reside in the distal C-terminal region of this enzyme [61]. These C-terminal regions have previously been found to be of functional relevance as well, as ADAMTS13 variants lacking the C-terminal portion appeared less efficient in VWF proteolysis both in vitro and in vivo [65]. However, it remained unclear from these studies whether this reduced efficiency was related to decreased proteolytic activity of truncated ADAMTS13 or a defect independent of VWF cleaving activity.
Figure 3. Events associated with shear stress-induced conformational changes in VWF.
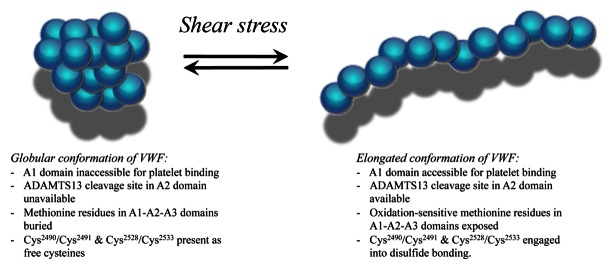
VWF circulates in a globular conformation with its platelet-binding site being inaccessible. In addition, the ADAMTS13-cleavage site in the A2 domain in unavailable. Both parameters protect VWF from premature platelet interactions and ADAMTS13-mediated degradation. Exposure to increased shear stress unfolds the protein, allowing platelet binding and ADAMTS13 cleavage. In addition, buried methionine residues become exposed, which are sensitive to oxidation by reactive oxygen species. Oxidized VWF displays resistance against ADAMTS13 proteolysis and enhanced platelet binding. Platelet binding is further enhanced by the formation of novel disulfide-bridges in the VWF C-domains. Shear stress-induced disulfide bridging is downregulated by thiol reductase activity that resides in the C-terminal region of ADAMTS13.
Insight into this question emerged from a somewhat unexpected angle. It has previously been shown that exposure of VWF to shear stress results not only in unfolding of the molecule, but also in a rearrangement of the disulphide bridges that couple the C-terminal ends of VWF subunits [66, 67]. A study by Yeh et al. revealed that this shear stress-induced reshuffling of thiol residues is inhibited in the presence of ADAMTS13 [68]. In fact, the C-terminal region of ADAMTS13 displays thiol reductase activity towards VWF, representing a second mechanism by which ADAMTS13 regulates VWF multimer size.
As mentioned above, both proteolytic activity and thiol reductase activity of ADAMTS13 towards VWF are only functional when VWF is in its elongated form due to hydrodynamic forces. Interestingly, when elongated, VWF has also recently been found to be susceptible to methionine oxidation in the A-domains in the presence of reactive oxygen species [69]. In fact, also methionine 1606 is susceptible to oxidation [70, 71], a process that renders the VWF resistant against ADAMTS13 mediated proteolysis. This type of posttranslational modification of VWF is just starting to be recognized, and the biological and pathological consequences remain to be discovered. Nevertheless, its physiological significance is perhaps best illustrated by the recent finding that the presence of VWF with its methionine 1606 residue being oxidized (and thereby being resistant against ADAMTS13 proteolysis) appears to represent a risk factor for thrombotic and septic complications in chronic renal failure [72].
3.3. Active VWF
The conformation in which VWF exists is determined by the shear stress to which VWF is exposed [64], resulting in a shear force-dependent equilibrium between a globular latent and an elongated active conformation. This equilibrium allows a shear force-mediated regulation of platelet binding, as this site is hidden and exposed in the respective conformations. Under normal conditions, this equilibrium is dominated by the closed conformation, in order to prevent the premature formation of VWF-platelet aggregates. Several years ago, an assay was developed that was designed to specifically detect the presence of VWF in its active, platelet-binding conformation [73]. Using this assay, it was confirmed that active VWF is present only in limited quantities in normal plasma. However, several pathological conditions appeared to be associated with increased levels of active VWF, in particular VWD-type 2B and TTP [73]. Since then, the presence of active VWF has been examined in various diseases, and it seems that the equilibrium between latent and active VWF is disturbed more often than previously anticipated (for review see [62]). Situations of increased levels of active VWF include infectious diseases (malaria, meningococcal disease), sickle cell disease, HELLP, myocardial infarction and the anti-phospholipid syndrome. Indeed, several of these conditions are associated with thrombotic complications, suggesting that the presence of active VWF may play a role in this regard. Does this also mean that the change of VWF from a latent into an active conformation immediately results in the binding of platelets? Although it is perhaps tempting to assume this in the first instance, it in fact seems to be untrue. In our studies, we have noted that active VWF can interact with a number of proteins in the circulation, with particular reference to β2-glycoprotein I [74]. This protein is able to bind to the A1 domain of active but not latent VWF, and can interfere with platelet binding. As such, β2-glycoprotein I functions as a first line barrier to neutralize initial amounts of active VWF. It should be noted that β2-glycoprotein I is but a weak inhibitor, allowing normal thrombus formation in case of a robust formation of active VWF. The physiological relevance of this defense mechanism is illustrated by the inverse relationship between β2-glycoprotein I plasma levels and myocardial infarction in older men [75]. More specifically, an increase in the ratio β2-glycoprotein I/VWF was accompanied by a 2–3 fold reduced risk of myocardial infarction in aging men [75].
3.4. VWF and stroke
Epidemiological studies of the relationship between VWF and ischemic stoke have yielded inconsistent results (for review see [76]). Recent prospective studies seem to indicate that high VWF levels can indeed increase the risk of first ischemic stroke. However, in most cases, this effect was modest and of unknown clinical utility. An experimental stroke model using transient middle cerebral artery occlusion was used to assess the role of VWF in stroke in a murine model. In the absence of VWF, the infarct volumes were reduced by 40% and neurological function was significantly improved [77]. Importantly, no increased intracerebral bleeding was observed in the VWF-deficient mice. In search for the molecular mechanisms, the authors showed that reconstitution of plasma VWF by gene transfer restored the susceptibility of the VWF-deficient mice to cerebral ischemia, demonstrating that plasma VWF rather than endothelial or platelet VWF is the main player in this process [77]. The use of VWF mutants to reconstitute the plasma compartment pinpointed the crucial role of the VWF-GpIbα and VWF-collagens interactions in mediating stroke development [78], emphasizing the potential interest of blocking these axes as a therapeutic option in ischemic stroke.
4. Conclusion
In recent years, considerable progress has been made in our understanding of the role of VWF in hemostasis and thrombosis, revealing unexpected aspects of VWF biology. It is not surprising therefore, that VWF is now considered as an attractive drug target. Indeed, anti-VWF directed drugs such as nanobody ALX-0081 and aptamer ARC-1779 are under clinical development [41]. In first instance, these agents are being developed for the treatment of arterial thrombotic complications and TTP, but it is well possible that these agents may find applications in other areas, such as venous thrombosis, VWD-type 2B and stroke. VWF may also be targeted using less specific drugs. One example hereof is the use of N-acetyl cysteine, which is currently being used as a mucolytic agent in the treatment of cystic fibrosis and in the management of paracetamol overdose. N-acetyl cysteine has the capacity to break disulphide bonds. In a preclinical setting it has been shown that this agent is also capable of reducing the size of ultra-large VWF multimers, making it a potential drug for TTP treatment [79]. These developments illustrate that continuous research on the molecular basis of how VWF participates in hemostatic and thrombotic processes helps to search for new therapeutic options.
Footnotes
Conflict of interest:
The authors declare that they have no conflict of interest.
References
- 1.Reininger AJ. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia. 2008;14 (Suppl 5):11–26. doi: 10.1111/j.1365-2516.2008.01848.x. [DOI] [PubMed] [Google Scholar]
- 2.Ruggeri ZM. Platelet adhesion under flow. Microcirculation. 2009;16:58–83. doi: 10.1080/10739680802651477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lenting PJ, Van Schooten CJ, Denis CV. Clearance mechanisms of von Willebrand factor and factor VIII. J Thromb Haemost. 2007;5:1353–60. doi: 10.1111/j.1538-7836.2007.02572.x. [DOI] [PubMed] [Google Scholar]
- 4.Denis CV. Molecular and cellular biology of von Willebrand factor. Int J Hematol. 2002;75:3–8. doi: 10.1007/BF02981972. [DOI] [PubMed] [Google Scholar]
- 5.Ruggeri ZM, Zimmerman TS. The complex multimeric composition of factor VIII/von Willebrand factor. Blood. 1981;57:1140–3. [PubMed] [Google Scholar]
- 6.Furlan M. Von Willebrand factor: molecular size and functional activity. Ann Hematol. 1996;72:341–8. doi: 10.1007/s002770050184. [DOI] [PubMed] [Google Scholar]
- 7.Barr J, Motto D. Modulation of von Willebrand factor by ADAMTS13. In: Federici AB, Lee CA, Berntorp EE, Lillicrap D, Montgomery RR, editors. Von Willebrand disease: Basic and Clinical Aspects. Chichester: Wiley-Blackwell; 2011. pp. 49–62. [Google Scholar]
- 8.Springer TA. Biology and physics of von Willebrand factor concatamers. J Thromb Haemost. 2011;9 (Suppl 1):130–43. doi: 10.1111/j.1538-7836.2011.04320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 10.Schneppenheim R, Budde U. Phenotypic and genotypic diagnosis of von Willebrand disease: a 2004 update. Semin Hematol. 2005;42:15–28. doi: 10.1053/j.seminhematol.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Schneppenheim R, Budde U. von Willebrand factor: the complex molecular genetics of a multidomain and multifunctional protein. J Thromb Haemost. 2011;9 (Suppl 1):209–15. doi: 10.1111/j.1538-7836.2011.04324.x. [DOI] [PubMed] [Google Scholar]
- 12.Rayes J, Hommais A, Legendre P, Tout H, Veyradier A, Obert B, et al. Effect of von Willebrand disease type 2B and type 2M mutations on the susceptibility of von Willebrand factor to ADAMTS-13. J Thromb Haemost. 2007;5:321–8. doi: 10.1111/j.1538-7836.2007.02296.x. [DOI] [PubMed] [Google Scholar]
- 13.Federici AB, Mannucci PM, Castaman G, Baronciani L, Bucciarelli P, Canciani MT, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood. 2009;113:526–34. doi: 10.1182/blood-2008-04-152280. [DOI] [PubMed] [Google Scholar]
- 14.Rayes J, Hollestelle MJ, Legendre P, Marx I, de Groot PG, Christophe OD, et al. Mutation and ADAMTS13-dependent modulation of disease severity in a mouse model for von Willebrand disease type 2B. Blood. 2010;115:4870–7. doi: 10.1182/blood-2009-11-254193. [DOI] [PubMed] [Google Scholar]
- 15.Nishino M, Girma JP, Rothschild C, Fressinaud E, Meyer D. New variant of von Willebrand disease with defective binding to factor VIII. Blood. 1989;74:1591–9. [PubMed] [Google Scholar]
- 16.Flood VH, Lederman CA, Wren JS, Christopherson PA, Friedman KD, Hoffmann RG, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost. 2010;8:1431–3. doi: 10.1111/j.1538-7836.2010.03869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ribba AS, Loisel I, Lavergne JM, Juhan-Vague I, Obert B, Cherel G, et al. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost. 2001;86:848–54. [PubMed] [Google Scholar]
- 18.Riddell AF, Gomez K, Millar CM, Mellars G, Gill S, Brown SA, et al. Characterization of W1745C and S1783A: 2 novel mutations causing defective collagen binding in the A3 domain of von Willebrand factor. Blood. 2009;114:3489–96. doi: 10.1182/blood-2008-10-184317. [DOI] [PubMed] [Google Scholar]
- 19.Marx I, Christophe OD, Lenting PJ, Rupin A, Vallez MO, Verbeuren TJ, et al. Altered thrombus formation in von Willebrand factor-deficient mice expressing von Willebrand factor variants with defective binding to collagen or GPIIbIIIa. Blood. 2008;112:603–9. doi: 10.1182/blood-2008-02-142943. [DOI] [PubMed] [Google Scholar]
- 20.Casonato A, Pontara E, Sartorello F, Cattini MG, Sartori MT, Padrini R, et al. Reduced von Willebrand factor survival in type Vicenza von Willebrand disease. Blood. 2002;99:180–4. doi: 10.1182/blood.v99.1.180. [DOI] [PubMed] [Google Scholar]
- 21.Lenting PJ, Westein E, Terraube V, Ribba AS, Huizinga EG, Meyer D, et al. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J Biol Chem. 2004;279:12102–9. doi: 10.1074/jbc.M310436200. [DOI] [PubMed] [Google Scholar]
- 22.Pruss CM, Golder M, Bryant A, Hegadorn CA, Burnett E, Laverty K, et al. Pathologic mechanisms of type 1 VWD mutations R1205H and Y1584C through in vitro and in vivo mouse models. Blood. 2011;117:4358–66. doi: 10.1182/blood-2010-08-303727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denis CV, Christophe OD, Oortwijn BD, Lenting PJ. Clearance of von Willebrand factor. Thromb Haemost. 2008;99:271–8. doi: 10.1160/TH07-10-0629. [DOI] [PubMed] [Google Scholar]
- 24.Haberichter SL, Castaman G, Budde U, Peake I, Goodeve A, Rodeghiero F, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD) Blood. 2008;111:4979–85. doi: 10.1182/blood-2007-09-110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Schooten CJ, Tjernberg P, Westein E, Terraube V, Castaman G, Mourik JA, et al. Cysteine-mutations in von Willebrand factor associated with increased clearance. J Thromb Haemost. 2005;3:2228–37. doi: 10.1111/j.1538-7836.2005.01571.x. [DOI] [PubMed] [Google Scholar]
- 26.Federici AB, Bucciarelli P, Castaman G, Baronciani L, Canciani MT, Mazzucconi MG, et al. Management of inherited von Willebrand disease in Italy: results from the retrospective study on 1234 patients. Semin Thromb Hemost. 2011;37:511–21. doi: 10.1055/s-0031-1281037. [DOI] [PubMed] [Google Scholar]
- 27.Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost. 2011;105:647–54. doi: 10.1160/TH10-11-0697. [DOI] [PubMed] [Google Scholar]
- 28.Shetty S, Kasatkar P, Ghosh K. Pathophysiology of acquired von Willebrand disease: a concise review. Eur J Haematol. 2011;87:99–106. doi: 10.1111/j.1600-0609.2011.01636.x. [DOI] [PubMed] [Google Scholar]
- 29.Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood. 2011;117:6777–85. doi: 10.1182/blood-2010-11-297580. [DOI] [PubMed] [Google Scholar]
- 30.Budde U, Scharf RE, Franke P, Hartmann-Budde K, Dent J, Ruggeri ZM. Elevated platelet count as a cause of abnormal von Willebrand factor multimer distribution in plasma. Blood. 1993;82:1749–57. [PubMed] [Google Scholar]
- 31.Richard C, Cuadrado MA, Prieto M, Batlle J, Lopez Fernandez MF, Rodriguez Salazar ML, et al. Acquired von Willebrand disease in multiple myeloma secondary to absorption of von Willebrand factor by plasma cells. Am J Hematol. 1990;35:114–7. doi: 10.1002/ajh.2830350210. [DOI] [PubMed] [Google Scholar]
- 32.Gill JC, Wilson AD, Endres-Brooks J, Montgomery RR. Loss of the largest von Willebrand factor multimers from the plasma of patients with congenital cardiac defects. Blood. 1986;67:758–61. [PubMed] [Google Scholar]
- 33.Warkentin TE, Moore JC, Morgan DG. Aortic stenosis and bleeding gastrointestinal angiodysplasia: is acquired von Willebrand’s disease the link? Lancet. 1992;340:35–7. doi: 10.1016/0140-6736(92)92434-h. [DOI] [PubMed] [Google Scholar]
- 34.Vincentelli A, Susen S, Le Tourneau T, Six I, Fabre O, Juthier F, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349:343–9. doi: 10.1056/NEJMoa022831. [DOI] [PubMed] [Google Scholar]
- 35.Banerjee AK. Angiodysplasia associated with hypertrophic obstructive cardiomyopathy (HOCM) Br J Clin Pract. 1990;44:326–7. [PubMed] [Google Scholar]
- 36.Le Tourneau T, Susen S, Caron C, Millaire A, Marechaux S, Polge AS, et al. Functional impairment of von Willebrand factor in hypertrophic cardiomyopathy: relation to rest and exercise obstruction. Circulation. 2008;118:1550–7. doi: 10.1161/CIRCULATIONAHA.108.786681. [DOI] [PubMed] [Google Scholar]
- 37.Slaughter MS. Hematologic effects of continuous flow left ventricular assist devices. J Cardiovasc Transl Res. 2010;3:618–24. doi: 10.1007/s12265-010-9222-6. [DOI] [PubMed] [Google Scholar]
- 38.Pareti FI, Lattuada A, Bressi C, Zanobini M, Sala A, Steffan A, et al. Proteolysis of von Willebrand factor and shear stress-induced platelet aggregation in patients with aortic valve stenosis. Circulation. 2000;102:1290–5. doi: 10.1161/01.cir.102.11.1290. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330–4. doi: 10.1126/science.1170905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Schie MC, van Loon JE, de Maat MP, Leebeek FW. Genetic determinants of von Willebrand factor levels and activity in relation to the risk of cardiovascular disease: a review. J Thromb Haemost. 2011;9:899–908. doi: 10.1111/j.1538-7836.2011.04243.x. [DOI] [PubMed] [Google Scholar]
- 41.De Meyer SF, De Maeyer B, Deckmyn H, Vanhoorelbeke K. Von Willebrand factor: drug and drug target. Cardiovasc Hematol Disord Drug Targets. 2009;9:9–20. doi: 10.2174/187152909787581327. [DOI] [PubMed] [Google Scholar]
- 42.Jager A, van Hinsbergh VW, Kostense PJ, Emeis JJ, Yudkin JS, Nijpels G, et al. von Willebrand factor, C-reactive protein, and 5-year mortality in diabetic and nondiabetic subjects: the Hoorn Study. Arterioscler Thromb Vasc Biol. 1999;19:3071–8. doi: 10.1161/01.atv.19.12.3071. [DOI] [PubMed] [Google Scholar]
- 43.Wiman B, Andersson T, Hallqvist J, Reuterwall C, Ahlbom A, deFaire U. Plasma levels of tissue plasminogen activator/plasminogen activator inhibitor-1 complex and von Willebrand factor are significant risk markers for recurrent myocardial infarction in the Stockholm Heart Epidemiology Program (SHEEP) study. Arterioscler Thromb Vasc Biol. 2000;20:2019–23. doi: 10.1161/01.atv.20.8.2019. [DOI] [PubMed] [Google Scholar]
- 44.Klemm T, Mehnert AK, Siegemund A, Wiesner TD, Gelbrich G, Bluher M, et al. Impact of the Thr789Ala variant of the von Willebrand factor levels, on ristocetin co-factor and collagen binding capacity and its association with coronary heart disease in patients with diabetes mellitus type 2. Exp Clin Endocrinol Diabetes. 2005;113:568–72. doi: 10.1055/s-2005-872896. [DOI] [PubMed] [Google Scholar]
- 45.Lacquemant C, Gaucher C, Delorme C, Chatellier G, Gallois Y, Rodier M, et al. Association between high von willebrand factor levels and the Thr789Ala vWF gene polymorphism but not with nephropathy in type I diabetes. The GENEDIAB Study Group and the DESIR Study Group. Kidney Int. 2000;57:1437–43. doi: 10.1046/j.1523-1755.2000.00988.x. [DOI] [PubMed] [Google Scholar]
- 46.van der Meer IM, Brouwers GJ, Bulk S, Leebeek FW, van der Kuip DA, Hofman A, et al. Genetic variability of von Willebrand factor and risk of coronary heart disease: the Rotterdam Study. Br J Haematol. 2004;124:343–7. doi: 10.1046/j.1365-2141.2003.04776.x. [DOI] [PubMed] [Google Scholar]
- 47.Smith NL, Rice KM, Bovill EG, Cushman M, Bis JC, McKnight B, et al. Genetic variation associated with plasma von Willebrand factor levels and the risk of incident venous thrombosis. Blood. 2011;117:6007–11. doi: 10.1182/blood-2010-10-315473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoni G, Oudot-Mellakh T, Dimitromanolakis A, Germain M, Cohen W, Wells P, et al. Combined analysis of three genome-wide association studies on vWF and FVIII plasma levels. BMC Med Genet. 2011;12:102. doi: 10.1186/1471-2350-12-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121:1382–92. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Loon JE, Leebeek FW, Deckers JW, Dippel DW, Poldermans D, Strachan DP, et al. Effect of genetic variations in syntaxin-binding protein-5 and syntaxin-2 on von Willebrand factor concentration and cardiovascular risk. Circ Cardiovasc Genet. 2010;3:507–12. doi: 10.1161/CIRCGENETICS.110.957407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koster T, Blann AD, Briet E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet. 1995;345:152–5. doi: 10.1016/s0140-6736(95)90166-3. [DOI] [PubMed] [Google Scholar]
- 52.Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Tracy RP, Aleksic N, et al. Coagulation factors, inflammation markers, and venous thromboembolism: the longitudinal investigation of thromboembolism etiology (LITE) Am J Med. 2002;113:636–42. doi: 10.1016/s0002-9343(02)01345-1. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi M, Yamashita A, Moriguchi-Goto S, Marutsuka K, Sato Y, Yamamoto H, et al. Critical role of von Willebrand factor and platelet interaction in venous thromboembolism. Histol Histopathol. 2009;24:1391–8. doi: 10.14670/HH-24.1391. [DOI] [PubMed] [Google Scholar]
- 54.Chauhan AK, Kisucka J, Lamb CB, Bergmeier W, Wagner DD. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109:2424–9. doi: 10.1182/blood-2006-06-028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–7. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708–14. doi: 10.1182/blood-2011-01-332676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brill A, Fuchs TA, Savchenko A, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil Extracellular Traps Promote Deep Vein Thrombosis in Mice. J Thromb Haemost. 2011 Nov 1; doi: 10.1111/j.1538-7836.2011.04544.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307:1432–5. doi: 10.1056/NEJM198212023072306. [DOI] [PubMed] [Google Scholar]
- 60.Levy GG, Motto DG, Ginsburg D. ADAMTS13 turns 3. Blood. 2005;106:11–7. doi: 10.1182/blood-2004-10-4097. [DOI] [PubMed] [Google Scholar]
- 61.Crawley JT, de Groot R, Xiang Y, Luken BM, Lane DA. Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood. 2011;118:3212–21. doi: 10.1182/blood-2011-02-306597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lenting PJ, Pegon JN, Groot E, de Groot PG. Regulation of von Willebrand factor-platelet interactions. Thromb Haemost. 2010;104:449–55. doi: 10.1160/TH09-11-0777. [DOI] [PubMed] [Google Scholar]
- 63.De Ceunynck K, Rocha S, Feys HB, De Meyer SF, Uji-i H, Deckmyn H, et al. Local elongation of endothelial cell-anchored von Willebrand factor strings precedes ADAMTS13 protein-mediated proteolysis. J Biol Chem. 2011;286:36361–7. doi: 10.1074/jbc.M111.271890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneider SW, Nuschele S, Wixforth A, Gorzelanny C, Alexander-Katz A, Netz RR, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci U S A. 2007;104:7899–903. doi: 10.1073/pnas.0608422104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lenting PJ, Rastegarlari G. ADAMTS-13: double trouble for von Willebrand factor. J Thromb Haemost. 2010;8:2775–7. doi: 10.1111/j.1538-7836.2010.04124.x. [DOI] [PubMed] [Google Scholar]
- 66.Ganderton T, Berndt MC, Chesterman CN, Hogg PJ. Hypothesis for control of von Willebrand factor multimer size by intra-molecular thiol-disulphide exchange. J Thromb Haemost. 2007;5:204–6. doi: 10.1111/j.1538-7836.2006.02292.x. [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Choi H, Zhou Z, Nolasco L, Pownall HJ, Voorberg J, et al. Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. J Thromb Haemost. 2008;6:1135–43. doi: 10.1111/j.1538-7836.2008.02991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yeh HC, Zhou Z, Choi H, Tekeoglu S, May W, 3rd, Wang C, et al. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. J Thromb Haemost. 2010;8:2778–88. doi: 10.1111/j.1538-7836.2010.04094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu X, Chen J, Gallagher R, Zheng Y, Chung DW, Lopez JA. Shear stress-induced unfolding of VWF accelerates oxidation of key methionine residues in the A1A2A3 region. Blood. 2011;118:5283–91. doi: 10.1182/blood-2011-01-331074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen J, Fu X, Wang Y, Ling M, McMullen B, Kulman J, et al. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010;115:706–12. doi: 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lancellotti S, De Filippis V, Pozzi N, Peyvandi F, Palla R, Rocca B, et al. Formation of methionine sulfoxide by peroxynitrite at position 1606 of von Willebrand factor inhibits its cleavage by ADAMTS-13: A new prothrombotic mechanism in diseases associated with oxidative stress. Free Radic Biol Med. 2010;48:446–56. doi: 10.1016/j.freeradbiomed.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 72.De Filippis V, Lancellotti S, Maset F, Spolaore B, Pozzi N, Gambaro G, et al. Oxidation of Met1606 in von Willebrand Factor is a Risk Factor for Thrombotic and Septic Complications in Chronic Renal Failure. Biochem J. 2011 Nov 18; doi: 10.1042/BJ20111798. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 73.Hulstein JJ, de Groot PG, Silence K, Veyradier A, Fijnheer R, Lenting PJ. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood. 2005;106:3035–42. doi: 10.1182/blood-2005-03-1153. [DOI] [PubMed] [Google Scholar]
- 74.Hulstein JJ, Lenting PJ, de Laat B, Derksen RH, Fijnheer R, de Groot PG. beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood. 2007;110:1483–91. doi: 10.1182/blood-2006-10-053199. [DOI] [PubMed] [Google Scholar]
- 75.de Laat B, de Groot PG, Derksen RH, Urbanus RT, Mertens K, Rosendaal FR, et al. Association between beta2-glycoprotein I plasma levels and the risk of myocardial infarction in older men. Blood. 2009;114:3656–61. doi: 10.1182/blood-2009-03-212910. [DOI] [PubMed] [Google Scholar]
- 76.De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand Factor: An Emerging Target in Stroke Therapy. Stroke. 2011 Dec 15; doi: 10.1161/STROKEAHA.111.628867. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kleinschnitz C, De Meyer SF, Schwarz T, Austinat M, Vanhoorelbeke K, Nieswandt B, et al. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. 2009;113:3600–3. doi: 10.1182/blood-2008-09-180695. [DOI] [PubMed] [Google Scholar]
- 78.De Meyer SF, Schwarz T, Deckmyn H, Denis CV, Nieswandt B, Stoll G, et al. Binding of von Willebrand factor to collagen and glycoprotein Ibalpha, but not to glycoprotein IIb/IIIa, contributes to ischemic stroke in mice--brief report. Arterioscler Thromb Vasc Biol. 2010;30:1949–51. doi: 10.1161/ATVBAHA.110.208918. [DOI] [PubMed] [Google Scholar]
- 79.Chen J, Reheman A, Gushiken FC, Nolasco L, Fu X, Moake JL, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121:593–603. doi: 10.1172/JCI41062. [DOI] [PMC free article] [PubMed] [Google Scholar]