

Abstract
In recent years, Chikungunya virus (CHIKV) was responsible for epidemic outbreaks in intertropical regions. Although acquired immunity has been shown to be crucial during CHIKV infection in both humans and mice, their exact role in the control of CHIKV infection remains unclear. In this study, wild-type (WT), CD4−/−, and B cell (μMT) knockout mice were infected with CHIKV. Sera were taken at different days postinfection and measured for anti-CHIKV Ab levels. Isotype and neutralizing capacity of these Abs were assessed in vitro, and specific linear epitopes were mapped. Viremia in CHIKV-infected μMT mice persisted for more than a year, indicating a direct role for B cells in mediating CHIKV clearance. These animals exhibited a more severe disease than WT mice during the acute phase. Characterization of CHIKV-specific Abs revealed that anti-CHIKV Abs were elicited early and targeted epitopes mainly at the C terminus of the virus E2 glycoprotein. Furthermore, CD4−/− mice could still control CHIKV infection despite having lower anti-CHIKV Ab levels with reduced neutralizing capacity. Lastly, pre-existing natural Abs in the sera of normal WT mice recognized CHIKV and were able to partially inhibit CHIKV. Taken together, natural and CHIKV infection–induced specific Abs are essential for controlling CHIKV infections.
Introduction
Chikungunya virus (CHIKV) is an arthropod-borne alphavirus transmitted by Aedes mosquitoes, namely, A. aegypti and A. albopictus (1). Infected patients experience development of Chikungunya fever, characterized mainly by polyarthralgia (2), febrile illness, maculopapular rashes, myalgia, headache, edema of the extremities, and gastrointestinal complaints (3, 4). Patients may also experience development of neurologic complications and in some extreme cases, death has been reported (5, 6). Currently, CHIKV is endemic in Africa, India, and many parts of Asia (7), with occasional sporadic outbreaks (8, 9). The lack of herd immunity in countries surrounding these endemic areas presents imminent risks for the spread of large-scale outbreaks. Hence, CHIKV remains a public threat that should not be ignored.
Anti-CHIKV immunity is poorly understood, and most studies have focused on innate immunity, particularly in elucidating the roles of type I IFN and their related antiviral pathways (10–18). Although these studies have demonstrated the importance of type I IFNs in restricting virus replication during the acute phase of infection, their effects are insufficient for the complete elimination of virus in infected hosts. Moreover, CHIKV has been reported to persist in the tissues and organs in animal model studies even after viremia has subsided, and when levels of IFN-α/β have returned to normal (10, 14, 19). Nevertheless, CHIKV in the organs was shown to be eliminated progressively over time (20). Furthermore, persistently high levels of viremia with no signs of joint inflammation were observed in infected RAG2−/− mice that have no T and B cells (20). These observations strongly suggest that adaptive immunity has a crucial role in controlling and eliminating CHIKV after the initial IFN-α/β and other innate immune responses have subsided. It was further revealed that CD4+, but not CD8+, T cells play a major role in mediating the severity of joint inflammation, although both subsets have no role in the control of CHIKV replication and dissemination (20). In addition, passive transfer of anti-CHIKV Abs was able to clear CHIKV infection in mice (21, 22). These observations indicated that Abs could be the main effectors in anti-CHIKV immunity.
In this article, CHIKV infections in B cell–deficient (μMT) mice (23) were investigated to clearly define the role of B cells in anti-CHIKV immunity. We characterized the Ab response elicited during the infection and demonstrated that the breadth of protective Ab response is dependent on CD4+ T cells. Remarkably, we also uncovered a role for natural Abs that are present in the sera of uninfected mice to control early CHIKV infection.
Materials and Methods
Mice
Six-week-old female wild-type (WT), μMT, and CD4−/− C57BL/6J mice were used. All mice were bred and kept under specific pathogen-free conditions in the Biological Resource Centre, Agency for Science, Technology and Research, Singapore. In all experiments, age- and sex-matched WT and deficient mice were used. All experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC: 120714) of the Agency for Science, Technology and Research, Singapore, in accordance with the guidelines of the Agri-Food and Veterinary Authority and the National Advisory Committee for Laboratory Animal Research of Singapore.
Virus
CHIKV SGP11 isolate, previously described (13), was used for all experiments. Viruses were further propagated in C6/36 cells and purified by ultracentrifugation (24) before in vivo infections. Virus titer was determined by standard plaque assays using Vero-E6 cells (13).
Virus infection and evaluation of disease
Mice were infected as previously described (18, 20, 24). Typically, 106 PFUs CHIKV SGP11 particles (in 50 μl PBS) were inoculated in the s.c. region of the ventral side at the right hind footpad toward the ankle. Viremia (assessed by viral RNA quantification) was monitored daily starting from 24 h postinfection until 8 d postinfection (dpi), and subsequently at every alternate day until 38 dpi. Viremia was assessed again at 79 and 402 dpi. Joint swelling at the footpad was scored daily from 0 to 19 dpi (18, 20, 24).
Viral RNA extraction and quantification
Ten microliters blood was collected from the tail vein and diluted in 120 μl PBS containing 10 μl citrate-phosphate-dextrose solution (Sigma-Aldrich). Viral RNA was extracted with the QIAamp Viral RNA Kit (Qiagen) following manufacturer’s instructions. Viral RNA copies were quantified by quantitative RT-PCR (18). Viral RNA copies were estimated from a standard curve generated using serial dilutions of CHIKV negative-sense nsP1 RNA transcripts as reported previously (22).
Ab quantification and isotyping
Ab titers were assessed by a virion-based ELISA (24–26). CHIKV-coated (106 virions/well in 50 μl PBS) polystyrene 96-well MaxiSorp plates (Nunc) were blocked with PBS containing 0.05% Tween 20 (PBST) and 5% w/v nonfat milk for 1.5 h at 37°C. Sera from normal or infected groups were heat inactivated and serially diluted in Ab diluent (0.05% PBST + 2.5% w/v nonfat milk). One hundred microliters of diluted sera was added into each well and incubated for 1 h at 37°C. HRP-conjugated goat anti-mouse IgG, IgG1, IgG2b, IgG2c, IgG3, and IgM Abs were used. IgG2c was tested instead of IgG2a because only the IgG2c gene is present in C57BL/6 mice (27). Total IgG and IgM quantification assays were performed using sera from respective animals diluted at 1:2000 and 1:100, respectively. Pooled sera were used for IgG1, IgG2b, IgG2c, and IgG3 isotyping. All HRP-conjugated Abs were from Santa Cruz, except for IgG3 (Southern Biotech). ELISA assays were developed by TMB substrate (Sigma-Aldrich) and terminated by Stop reagent (Sigma-Aldrich). Absorbance was measured at 450 nm. CHIKV-specific Ab isotypes are expressed as Ab titer that is defined as the lowest dilution required for a detectable signal above control naive pooled sera.
Detection of CHIKV-specific natural Abs
CHIKV-specific naturally occurring Abs from naive mice sera were assessed by Western blot, immunofluorescence (Supplemental Fig. 1), and virion-based ELISA assays (24–26). CHIKV-coated (106 virions/well in 50 μl PBS) polystyrene 96-well MaxiSorp plates (Nunc) were blocked with PBST and 5% w/v nonfat milk for 1.5 h at 37°C. Sera from WT or μMT mice were heat-inactivated and serially diluted in Ab diluent (0.05% PBST + 2.5% w/v nonfat milk). One hundred microliters of diluted sera was added into each well and incubated for 1 h at 37°C. HRP-conjugated goat anti-mouse IgG (Santa Cruz) and IgM (Santa Cruz) were used. ELISA assays were developed by TMB substrate (Sigma-Aldrich) and terminated by Stop reagent (Sigma-Aldrich). Absorbance was measured at 450 nm, and Ab titers were determined by detectable signals above the mean background signal + 3 SD.
Sero-neutralization assay
Neutralizing activity of Abs was tested in a fluorescence-based cell infection assay in HEK293T cells as described previously (24–26). In brief, CHIKV was incubated with diluted heat-inactivated mouse sera at a multiplicity of infection of 10 for 2 h at 37°C with gentle agitation (350 rpm). Virus–Ab mixtures were then added to HEK293T cells seeded in a 96-well plate (104 cells/well) and incubated for another 1.5 h at 37°C. Virus–Ab overlays were subsequently removed and fresh DMEM medium supplemented with 5% FBS was replenished. Cells were incubated for 6 h at 37°C before fixation with 4% paraformaldehyde followed by immunofluorescence staining and quantification using the Cellomics ArrayScan V (Thermo Fisher Scientific, Waltham, MA). Percentage of infectivity was calculated according to this equation: % Infectivity = 100 × (% responder from sero-neutralization group/% responder from virus infection group). All data presented are representative of two independent experiments with similar results.
Epitope determination and structural localization
Peptide-based ELISA was performed using heat-inactivated pooled sera from infected mice to screen for B cell epitopes using overlapping synthetic 18-mer biotinylated-peptides (Mimotopes) (24, 25). Streptavidin-coated 96-well plates (Nunc) were blocked with 1% w/v sodium caseinate (Sigma-Aldrich) in 0.1% PBST for 1 h at room temperature. Peptides were dissolved in DMSO (15 μg/ml), diluted 1:1000 in 0.1% PBST, and coated onto the plates (100 μl/well). Heat-inactivated pooled sera were diluted 1:500 in 0.1% PBST and 100 μl diluted sera were added into each well and incubated for 1 h at 37°C. HRP-conjugated goat anti-mouse IgG Abs (Santa Cruz) diluted 1:10,000 in Ab diluent were used to detect the bound Abs. Reactions were developed and absorbance was measured accordingly. Pools of five peptides were first used for initial screening. Positive peptide pools were determined as absorbance values greater than mean ± 6 SD values. Peptides from positive pools were then screened individually, and those with absorbance values greater than mean ± 6 SD of the noninfected controls were plotted. Results were expressed as percentage of Ab recognition within the CHIKV structural proteome.
Computational modeling
Structural data of the E1, E2, and E3 glycoproteins were retrieved from Protein Data Bank (PDB) (ID: 3N42) and visualized using the UCSF CHIMERA software (28). Structures of Capsid sequences were predicted using individual I-TASSER queries (29) and visualized using UCSF CHIMERA software.
Statistical analysis
Data are presented as mean ± SD. Differences between groups and controls were analyzed using Mann–Whitney U test. Statistics were performed with GraphPad Prism 5.04.
Results
B cells and Abs are essential for the control of CHIKV infection
CHIKV was inoculated at the joint footpad of WT and μMT mice to test for the role of B cells and Abs (23). In WT mice, CHIKV infection developed very rapidly, with high levels of viral RNA detected in the circulation at 2 dpi. It was observed that CHIKV infection peaked at 2 dpi and cleared by 10 dpi (Fig. 1A). In contrast, viremia developed more rapidly in μMT mice, reaching a peak of infection with viral load 3 logs higher than that of WT mice (Fig. 1A). After a strong reduction in CHIKV replication, infection became chronic with high levels of viral RNA copies still detectable at 79 dpi (Fig. 1A). Furthermore, viral RNA of 2.75 × 105 copies/μl could still be detected in one of the six infected μMT mice that survived up to 402 dpi (data not shown). The increase in viremia and the prolonged presence of CHIKV in the blood circulation might play a role in the exacerbated disease severity observed in the joint footpad of μMT mice during the peak of the acute phase (Fig. 1B). These observations clearly demonstrated that B cells and Abs are necessary for the control of viral replication.
FIGURE 1.

B cells mediate virus clearance and disease pathology in CHIKV infection. (A) Viremia and (B) joint swelling observed in both μMT (n = 6) and WT mice (n = 5). (C) Total CHIKV-specific IgG and IgM in pooled sera of WT infected mice (n = 3–5) at 2, 4, 6, and 9 dpi in replicates of four. (D) Total CHIKV-specific IgG and IgM Abs were measured in WT mice (n = 5) at 3, 15, and 26 dpi. (E) Total CHIKV-specific IgG and IgM in pooled sera of naive (n = 5) and WT infected mice (n = 5) at 402 dpi. This assay was done in quadruplicates. All IgM and IgG titers were determined at 1:100 and 1:2000 dilutions, respectively. (F) Pooled sera from CHIKV-infected WT mice (n = 5) were neutralizing against CHIKV infection. Percentage infectivity was normalized against virus-infected samples. All data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.005 by Mann–Whitney U, one-tailed test.
To further characterize the role of Abs induced by CHIKV infection, we quantified CHIKV-specific Ab responses using a CHIKV virion-based ELISA (26). Anti-CHIKV Ab responses were followed through different disease phases: the acute phase (5–10 dpi when joint swelling is prominent; Fig. 1C), the convalescent to early chronic phase (11–30 dpi; Fig. 1D), and the late chronic phase (30 dpi onward; Fig. 1E). Total IgG and IgM were first tested in pooled sera from five CHIKV-infected mice at 2, 4, 6, and 9 dpi (Fig. 1C). CHIKV-specific IgM was detected as early as 2 dpi, and production peaked at 6 dpi, before declining (Fig. 1C). Contrary to IgM, CHIKV-specific IgG was detected starting at 6 dpi, and their levels increased substantially and remained constant even after CHIKV has been cleared from the circulation (Fig. 1D). Interestingly, both IgG and IgM could still be detected up to 402 dpi at a level 2- to 3-fold lower than the peak (Fig. 1E). Sera taken at 15 dpi showed potent neutralizing activity against CHIKV infection in an in vitro cell-based assay, with >60% reduction in infectivity (p < 0.005) at different dilutions (Fig. 1F).
By comparing the viral kinetics, it was observed that at 1 dpi, μMT mice already had a significantly higher viral load than the WT mice (Fig. 1A). Moreover, IgM production is normally observed only after 24 h even with strong polyclonal activation (30). Therefore, the difference in viral load observed in this study between the WT and μMT mice is not due to the IgM induced by the infection. One possible explanation is the presence of natural Abs in the sera of naive WT animals that cross-reacted with CHIKV as seen in other viral infections (30). To test this hypothesis, we assayed low serial dilutions of sera collected from naive WT and μMT mice in the same CHIKV virion-based ELISA system for IgM (Fig. 2A) and IgG quantification (Fig. 2B). WT mice, but not μMT mice, had very low levels of IgM detected. However, these Abs failed to detect CHIKV by both immunofluorescence and Western blot assays (Supplemental Fig. 1). Nevertheless, these Abs were neutralizing as sera from WT mice diluted at 1:20 were able to reduce CHIKV infection significantly by ∼40% when compared with sera from naive μMT mice (p < 0.05; Fig. 2C).
FIGURE 2.

Presence of natural Abs against CHIKV in the serum of naive WT mice. Presence of naturally occurring Abs against CHIKV is found in the sera of naive WT mice (n = 5), and these Abs can be of either (A) IgM or (B) IgG. Presence of natural Abs against CHIKV was determined by detectable signals above the mean background signal + 3 SD (dotted lines). (C) Pooled sera from naive WT mice (n = 5) were neutralizing against CHIKV infection in an in vitro neutralization assay. Percentage infectivity was normalized to sera isolated from μMT mice. All data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.005 by Mann–Whitney U or unpaired t test, one-tailed test.
Isotype profile of anti-CHIKV Abs
Isotyping of CHIKV-specific Abs from pooled sera collected during the early convalescent phase of 15 dpi from WT mice revealed that the major isotype was IgG2c (Fig. 3A). This observation supports earlier reports that IgG2c is the major class of anti-CHIKV Abs present in CHIKV-infected mice (10). Because IgG2c production is driven mainly by IFN-γ (31), a cytokine that is induced during CHIKV infection in mice (10, 20), we went on to determine the isotype profile of IgG in IFN-γ–deficient mice at 15 dpi (Fig. 3B). In contrast, a modest reduction in total IgG titer, and notably only in IgG2c, was observed in these animals, suggesting that IFN-γ is not the main driver of isotype selection during CHIKV infection in mice.
FIGURE 3.

CD4+ T cell CHIKV-specific Abs (IgG). Isotype IgG2c is the dominant isotype of CHIKV-specific IgG Abs in pooled sera from (A) CD4−/− mice (n = 5) and from (B) IFN-γ−/− mice (n = 5) collected at 15 dpi. Pooled sera from WT mice (n = 5) were also isotyped in parallel. Production of (C) CHIKV-specific IgG Abs and (D) CHIKV-specific IgM Abs was significantly reduced in CD4−/− mice (n = 5). (E) Pooled sera from animals diluted 1:500 in serum-free DMEM medium were neutralizing against CHIKV, with the neutralizing capacity of the WT sera being significantly stronger. Percentage infectivity was normalized against virus-infected samples. CHIKV-specific Ab isotypes are expressed as Ab titer that is defined as the lowest dilution required for a detectable signal above control naive pooled sera. All data are presented as mean ± SD. *p < 0.05, ***p < 0.005 by Mann–Whitney U test.
CD4+ T cells promote a stronger production of CHIKV-specific Abs
We have previously shown that in the absence of CD4+ T cells, CHIKV viremia was still resolved in mice (20). This observation was surprising because CD4+ T cells are important for Ab production (32, 33). To determine whether neutralizing Abs were produced in the absence of CD4+ T cells, we infected CD4−/− mice with CHIKV. Clearly, anti-CHIKV IgG and IgM Abs were still being produced (Fig. 3C, 3D), albeit at a significantly lower titer (p < 0.05) when compared with WT animals. Furthermore, IgG isotyping of the sera from CHIKV-infected CD4−/− mice showed reduced levels of IgG2c, IgG1, and IgG2b Abs at 15 dpi (Fig. 3A). Despite the reduced titers, these Abs were still neutralizing (∼40% reduction; p < 0.005) against CHIKV infection, although with less efficiency than the Abs from WT animals (p < 0.005; Fig. 3E).
B cell linear epitopes by anti-CHIKV Abs
To identify linear epitopes recognized by anti-CHIKV Abs, peptide-based ELISA assays using a peptide library (Mimotopes) consisting of 18-mer overlapping, biotinylated peptides covering the whole CHIKV structural proteome (24, 25) were performed with pooled sera using an optimized serum dilution of 1:500. Peptides were screened individually, and those with an absorbance greater than the mean ± 6 SD of the noninfected controls are presented (Fig. 4A, 4B). CHIKV-specific sera from CHIKV-infected WT animals recognized 28 different epitope-containing peptides: 4 in Capsid, 4 in E1, 19 in E2, and 1 in E3 glycoproteins (Fig. 4A). No recognizable linear epitopes were found in the 6K protein. Most of these CHIKV-specific Abs detected epitopes localized in the E2 glycoprotein, which concord to previous investigations with CHIKV-infected patients’ plasma (Fig. 4C) (24, 25). However, the prominent epitope-containing peptides identified were P399 and P400, followed by P351, P382, and P391 in the E2 glycoprotein (Fig. 4A, 4B). It is interesting to note that P351 contain part of the linear “E2EP3” peptide previously reported to be a major epitope recognized by plasma samples obtained from both CHIKV-infected human patients and macaques (24, 25). When E2EP3 was tested, it was also strongly recognized (Fig. 4B). P399 and P400, which are located at the C terminus of the E2 glycoprotein, had the strongest detection signal (>12% total IgG Abs when results were expressed as percentage of Ab recognition for the entire CHIKV structural proteome; percentage Ab recognition is defined as the percentage of total anti-CHIKV IgG specific for a particular peptide). In contrast, sera from CHIKV-infected CD4−/− mice recognized only four epitope-containing peptides within the E2 glycoprotein (P379, P398, P399, and E2EP3; Fig. 4B). Annotation and the precise location of the individual epitope-containing peptides recognized by the anti-CHIKV Abs are illustrated in Table I. Positions of the amino acid sequences recognized by mouse Abs within the CHIKV structural proteins are illustrated (Fig. 4C). Epitopes identified from CHIKV-infected human patients’ plasma (24, 25) are also illustrated for comparative purposes (Fig. 4C, Table II). Position of the E2EP3 peptide (polypeptide sequence 2800–2818) is highlighted in red (Fig. 4C).
FIGURE 4.
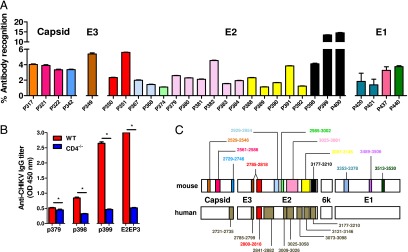
Mapping of CHIKV B cell epitopes within the CHIKV structural proteome with CHIKV-infected mice sera. Pooled sera at 15 dpi from CHIKV-infected (A) WT mice (n = 5) and (B) CD4−/− mice (n = 5) were screened. Peptides plotted are significantly above the mean ± 6 SD values of the noninfected sera. Sera from noninfected mice were used as negative controls. (C) Regions of identified B cell epitopes from mouse and human CHIKV-specific Abs. The numbers correspond to the position of the amino acid of the identified peptides along the CHIKV viral genome. E2EP3 peptide is highlighted in red. Percentage of Ab recognition was calculated according to this equation: % Ab recognition = 100 × (OD values from individual peptide group/sum of OD values from all peptide groups). All data are presented as mean ± SD. *p < 0.05 by Mann–Whitney U, two-tailed test.
Table I. Annotation of the individual peptides for mapping of CHIKV B cell epitopes within the CHIKV structural proteome.
| Viral Ags | Peptide Annotation | Polypeptide Sequencea |
|---|---|---|
| Capsid | P317 | 2529–2546 |
| P321 | 2561–2578 | |
| P322 | 2569–2586 | |
| P342 | 2729–2746 | |
| E3 | P349 | 2785–2799 |
| E2 | P350 | 2800–2810 |
| P351 | 2801–2818 | |
| P367 | 2929–2946 | |
| P368 | 2937–2954 | |
| P374 | 2985–3002 | |
| P379 | 3025–3042 | |
| P380 | 3033–3050 | |
| P381 | 3041–3058 | |
| P382 | 3049–3066 | |
| P383 | 3057–3074 | |
| P384 | 3065–3081 | |
| P388 | 3097–3114 | |
| P389 | 3105–3122 | |
| P390 | 3113–3130 | |
| P391 | 3121–3138 | |
| P392 | 3129–3146 | |
| P398 | 3177–3194 | |
| P399 | 3185–3202 | |
| P400 | 3193–3210 | |
| E1 | P420 | 3353–3370 |
| P421 | 3361–3378 | |
| P437 | 3489–3506 | |
| P440 | 3513–3530 |
The numbers correspond to the amino acid positions along the CHIKV viral genome. The first amino acid from nsP1 is annotated as 1.
Table II. Comparison of identified CHIKV B cell epitopes between human and mouse, within the CHIKV structural proteome.
| Ags | Identified B Cell Epitope (Human)a | Amino Acidb | Identified B Cell Epitope (Mouse)a | Amino Acidb |
|---|---|---|---|---|
| Capsid | KDIVTKITPEGAEEW | 2721–2735 | AVPQQKPRRNRKNKKQKQ | 2529–2546 |
| PPKKKPAQKKKKPGRRERMCMKIEND | 2561–2586 | |||
| PEGAEEWSLAIPVMCLLA | 2729–2746 | |||
| E3 | LLQASLTCSPHRQRR | 2785–2799 | LLQASLTCSPHRQRR | 2785–2799 |
| E2 | STKDNFNVYKATRPYLAHC | 2800–2818 | STKDNFNVYKATRPYLAHC | 2800–2818 |
| TDGTLKIQVSLQIGIKTDDSHDWTKLRYMDNHMPADAERAGL | 2841–2882 | HHDPPVIGREKFHSRPQHGKELPCST | 2929–2954 | |
| LTTTDKVINNCKVDQCHA | 3009–3026 | GNVKITVNGQTVRYKCNC | 2985–3002 | |
| HAAVTNHKKWQYNSPLVPRNAELGDRKGKIHIPF | 3025–3058 | HAAVTNHKKWQYNSPLVPRNAELGDRKGKIHIPFPLANVTCRVPKARNPTVTYGKNQ | 3025–3081 | |
| PTVTYGKNQVIMLLYPDHPTLLSYRN | 3073–3098 | RNMGEEPNYQEEWVMHKKEVVLTVPTEGLEVTWGNNEPYKYWPQLSTNGT | 3097–3146 | |
| PTEGLEVTWGNNEPYKYWPQLSTNGT | 3121–3146 | LLSMVGMAAGMCMCARRRCITPYELTPGATVPFL | 3177–3210 | |
| LLSMVGMAAGMCMCARRRCITPYELTPGATVPFL | 3177–3210 | |||
| 6K | ||||
| E1 | DKNLPDYSCKVFTGVYPFMWGGAYCF | 3353–3378 | ||
| RTPESKDVYANTQLVLQR | 3489–3506 | |||
| HVPYSQAPSGFKYWLKER | 3513–3530 |
Regions of B cell epitopes found that are common to both human and mouse are underlined.
The numbers correspond to the amino acid positions along the CHIKV viral genome. The first amino acid from nsP1 is annotated as 1.
Linear epitope-containing sequences were next mapped onto predicted three-dimensional structures of the Capsid proteins (Fig. 5A) or available crystal structures of the E3 (Fig. 5B), E2 (Fig. 5C), and E1 (Fig. 5D) glycoproteins (PDB no. 3N42).
FIGURE 5.

Localization of identified CHIKV B cell epitopes within CHIKV structural proteome. Schematic representation of identified B cell epitopes in (A) Capsid, (B) E3, (C) E2, and (D) E1 proteins. Epitopes in the E3, E2, and E1 proteins were located based on the structural data obtained from PDB records: 3N42. Epitopes in the Capsid protein were located based on the structures predicted via the I-TASSER server. Number corresponds to the position of the amino acid of the identified peptides along the CHIKV viral genome. All representations are shown in frontal view except for the E1 glycoprotein peptides, and peptides 2729–2746 of capsid protein, which are shown in the back view.
Discussion
In this study, we demonstrated the functional roles of B cells and Abs in a mouse model of CHIKV infection. Upon CHIKV infection, mice usually display a bimodal phase of joint inflammation, with the shorter and less severe phase observed at 2 dpi, whereas the longer and more severe phase was observed during the peak of the acute phase at 5–6 dpi (10, 20). We have previously identified CD4+ T cells as the major effector for maximal joint inflammation during the peak of the acute phase, but not in viral clearance (20). In WT mice, CHIKV infection induced the production of anti-CHIKV Abs that led to the rapid clearance of CHIKV and resulted in less severe joint inflammation during the peak of the acute phase, when compared with μMT mice. Anti-CHIKV Abs persisted for a long time. Interestingly, IgM could be detected for more than a year postinfection. Unusual long-term persistence for IgM was also reported in CHIKV patients (34). We also demonstrated that IgM Abs that are able to recognize CHIKV are present in the sera of naive WT mice. These Abs likely recognize conformational epitopes (25) because reactivity to CHIKV proteins was not detected by Western blot or by immunofluorescence. Natural Abs of IgM, IgG, and IgA isotypes were previously shown to be present both in human subjects (35, 36) and in mice (35). They were shown to be capable of providing early protection and controlling dissemination of infectious pathogens via direct neutralization, activation of the complement system, as well as enhancing the immune responses in secondary lymphoid organs (37). Our data clearly suggest that these natural Abs are neutralizing against CHIKV and control viral replication in CHIKV-infected animals during the early phase of infection. Studies are under way to determine whether such Abs exist in naive human populations.
We have recently demonstrated in CD4−/− mice that viremia was cleared as efficiently as in WT mice (20). This was surprising because CD4+ Th cells interact with B cells to enhance Ab production, induce class switching, and promote affinity maturation (32, 33). Therefore, in this study, we sought to characterize the specific anti-CHIKV Ab response in both WT and CD4−/− mice. Interestingly, we observed that both CHIKV-specific total IgG and IgM were significantly reduced in infected CD4−/− mice. Despite this significant difference in Ab levels, sera from both groups were capable of reducing CHIKV infectivity in in vitro neutralization assays, although sera from CD4−/− mice exhibited a reduced neutralizing capacity. This led to the conclusion that CD4+ T cells control the breadth of Ab response, but nevertheless in their absence, anti-CHIKV Abs against the E2 glycoprotein are still induced and are important in the antiviral protection during CHIKV primary infection. In fact, we have shown that anti-CHIKV Abs from infected CD4−/− mice, targeting just four epitope-containing regions of the E2 glycoprotein, were able to fully protect against CHIKV infection as efficiently as the WT mice, suggesting that these epitope-containing regions are major targets of CD4+ T cell–independent protective Ab responses. Nevertheless, it is plausible that CD4+ T cells could still be required for the long-term persistence of anti-CHIKV Abs. In addition, the limited influence of CD4+ T cells in the development of protective anti-CHIKV Abs could explain why CHIKV infections are not more frequent or severe in regions where CHIKV and HIV coinfection occurs (38–43). Several CHIKV endemic regions are found in areas where HIV transmissions are prevalent, such as India and the sub-Saharan African nations (38–42). Furthermore, HIV/CHIKV coinfections have been previously reported in northern Tanzania (43). The resolution of CHIKV infection in CD4−/− mice would strongly imply that HIV patients with low CD4+ T cell counts (44) could still resolve CHIKV infection effectively.
Previous studies have shown that CHIKV-specific CD4+ T cells were induced by CHIKV infection, and the specific CD4+ T cells were the major producers of IFN-γ (20). Th1 cells have been shown to skew the production of IgG2c Abs by B cells through their production of IFN-γ (31). However, this does not apply to CHIKV infection as we have shown that CHIKV-specific IgG2c Abs were only marginally reduced in IFN-γ−/− mice at 15 dpi. This finding suggests that other alternative IFN-γ–independent pathways are responsible for IgG2c production and will warrant further studies to decipher the mechanism.
The demonstration of an essential role for B cells and Abs in controlling CHIKV infection together with the identification of B cell epitopes recognized by protective Abs further advocate the mouse model as an important preclinical tool for diagnosis and vaccine development.
Acknowledgments
We thank Carla Claser (Singapore Immunology Network) for technical expertise and assistance provided in the animal experiments. We also express gratitude to Cindy Phua (Mutant Mouse Collection, Singapore Immunology Network) for assistance in breeding of and providing the mutant mice used in this study.
This work was supported by the Singapore Immunology Network, Agency for Science, Technology and Research. F.-M.L. is supported by a Yong Loo Lin School of Medicine, National University of Singapore postgraduate scholarship. T.-H.T. is supported by an Agency for Science, Technology and Research postgraduate scholarship. W.W.L.L. is supported by a National University of Singapore Graduate School for Integrative Sciences and Engineering postgraduate scholarship.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The online version of this article contains supplemental material.
- CHIKV
- Chikungunya virus
- dpi
- days postinfection
- PBST
- PBS containing 0.05% Tween 20
- PDB
- Protein Data Bank
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Her Z., Kam Y. W., Lin R. T. P., Ng L. F. P. 2009. Chikungunya: a bending reality. Microbes Infect. 11: 1165–1176 [DOI] [PubMed] [Google Scholar]
- 2.Robinson M. C. 1955. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952-53. I. Clinical features. Trans. R. Soc. Trop. Med. Hyg. 49: 28–32 [DOI] [PubMed] [Google Scholar]
- 3.Borgherini G., Poubeau P., Staikowsky F., Lory M., Le Moullec N., Becquart J. P., Wengling C., Michault A., Paganin F. 2007. Outbreak of chikungunya on Reunion Island: early clinical and laboratory features in 157 adult patients. Clin. Infect. Dis. 44: 1401–1407 [DOI] [PubMed] [Google Scholar]
- 4.Lakshmi V., Neeraja M., Subbalaxmi M. V. S., Parida M. M., Dash P. K., Santhosh S. R., Rao P. V. L. 2008. Clinical features and molecular diagnosis of Chikungunya fever from South India. Clin. Infect. Dis. 46: 1436–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandak N. H., Kashyap R. S., Kabra D., Karandikar P., Saha S. S., Morey S. H., Purohit H. J., Taori G. M., Daginawala H. F. 2009. Neurological complications of Chikungunya virus infection. Neurol. India 57: 177–180 [DOI] [PubMed] [Google Scholar]
- 6.Ganesan K., Diwan A., Shankar S. K., Desai S. B., Sainani G. S., Katrak S. M. 2008. Chikungunya encephalomyeloradiculitis: report of 2 cases with neuroimaging and 1 case with autopsy findings. AJNR Am. J. Neuroradiol. 29: 1636–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers A. M., Logue C. H. 2007. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J. Gen. Virol. 88: 2363–2377 [DOI] [PubMed] [Google Scholar]
- 8.Renault P., Balleydier E., D’Ortenzio E., Bâville M., Filleul L. 2012. Epidemiology of Chikungunya infection on Reunion Island, Mayotte, and neighboring countries. Med. Mal. Infect. 42: 93–101 [DOI] [PubMed] [Google Scholar]
- 9.Chua K. B. 2010. Epidemiology of chikungunya in Malaysia: 2006-2009. Med. J. Malaysia 65: 277–282 [PubMed] [Google Scholar]
- 10.Gardner J., Anraku I., Le T. T., Larcher T., Major L., Roques P., Schroder W. A., Higgs S., Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 84: 8021–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gardner C. L., Burke C. W., Higgs S. T., Klimstra W. B., Ryman K. D. 2012. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology 425: 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werneke S. W., Schilte C., Rohatgi A., Monte K. J., Michault A., Arenzana-Seisdedos F., Vanlandingham D. L., Higgs S., Fontanet A., Albert M. L., Lenschow D. J. 2011. ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLoS Pathog. 7: e1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Her Z., Malleret B., Chan M., Ong E. K. S., Wong S. C., Kwek D. J. C., Tolou H., Lin R. T. P., Tambyah P. A., Rénia L., Ng L. F. 2010. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol. 184: 5903–5913 [DOI] [PubMed] [Google Scholar]
- 14.Labadie K., Larcher T., Joubert C., Mannioui A., Delache B., Brochard P., Guigand L., Dubreil L., Lebon P., Verrier B., et al. 2010. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Invest. 120: 894–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schilte C., Couderc T., Chretien F., Sourisseau M., Gangneux N., Guivel-Benhassine F., Kraxner A., Tschopp J., Higgs S., Michault A., et al. 2010. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 207: 429–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Couderc T., Chrétien F., Schilte C., Disson O., Brigitte M., Guivel-Benhassine F., Touret Y., Barau G., Cayet N., Schuffenecker I., et al. 2008. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 4: e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudd P. A., Wilson J., Gardner J., Larcher T., Babarit C., Le T. T., Anraku I., Kumagai Y., Loo Y. M., Gale M., Jr., et al. 2012. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J. Virol. 86: 9888–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teng T. S., Foo S. S., Simamarta D., Lum F. M., Teo T. H., Lulla A., Yeo N. K. W., Koh E. G. L., Chow A., Leo Y. S., et al. 2012. Viperin restricts chikungunya virus replication and pathology. J. Clin. Invest. 122: 4447–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teo T. H., Lum F. M., Lee W. W. L., Ng L. F. P. 2012. Mouse models for Chikungunya virus: deciphering immune mechanisms responsible for disease and pathology. Immunol. Res. 53: 136–147 [DOI] [PubMed] [Google Scholar]
- 20.Teo T. H., Lum F. M., Claser C., Lulla V., Lulla A., Merits A., Rénia L., Ng L. F. P. 2013. A pathogenic role for CD4+ T cells during Chikungunya virus infection in mice. J. Immunol. 190: 259–269 [DOI] [PubMed] [Google Scholar]
- 21.Couderc T., Khandoudi N., Grandadam M., Visse C., Gangneux N., Bagot S., Prost J. F., Lecuit M. 2009. Prophylaxis and therapy for Chikungunya virus infection. J. Infect. Dis. 200: 516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee C. Y., Kam Y. W., Fric J., Malleret B., Koh E. G. L., Prakash C., Huang W., Lee W. W. L., Lin C., Lin R. T. P., et al. 2011. Chikungunya virus neutralization antigens and direct cell-to-cell transmission are revealed by human antibody-escape mutants. PLoS Pathog. 7: e1002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitamura D., Roes J., Kühn R., Rajewsky K. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350: 423–426 [DOI] [PubMed] [Google Scholar]
- 24.Kam Y. W., Lum F. M., Teo T. H., Lee W. W. L., Simarmata D., Harjanto S., Chua C. L., Chan Y. F., Wee J. K., Chow A., et al. 2012. Early neutralizing IgG response to Chikungunya virus in infected patients targets a dominant linear epitope on the E2 glycoprotein. EMBO Mol. Med. 4: 330–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kam Y. W., Lee W. W. L., Simarmata D., Harjanto S., Teng T. S., Tolou H., Chow A., Lin R. T. P., Leo Y. S., Rénia L., Ng L. F. 2012. Longitudinal analysis of the human antibody response to Chikungunya virus infection: implications for serodiagnosis and vaccine development. J. Virol. 86: 13005–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kam Y. W., Simarmata D., Chow A., Her Z., Teng T. S., Ong E. K. S., Rénia L., Leo Y. S., Ng L. F. P. 2012. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J. Infect. Dis. 205: 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin R. M., Brady J. L., Lew A. M. 1998. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J. Immunol. Methods 212: 187–192 [DOI] [PubMed] [Google Scholar]
- 28.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- 29.Roy A., Kucukural A., Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5: 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson J., Melchers F. 1973. Induction of immunoglobulin M synthesis and secretion in bone-marrow-derived lymphocytes by locally concentrated concanavalin A. Proc. Natl. Acad. Sci. USA 70: 416–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr T. A., Brown S., Mastroeni P., Gray D. 2009. B cell intrinsic MyD88 signals drive IFN-gamma production from T cells and control switching to IgG2c. J. Immunol. 183: 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vitetta E. S., Fernandez-Botran R., Myers C. D., Sanders V. M. 1989. Cellular interactions in the humoral immune response. Adv. Immunol. 45: 1–105 [DOI] [PubMed] [Google Scholar]
- 33.Swain S. L., McKinstry K. K., Strutt T. M. 2012. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 12: 136–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malvy D., Ezzedine K., Mamani-Matsuda M., Autran B., Tolou H., Receveur M. C., Pistone T., Rambert J., Moynet D., Mossalayi D. 2009. Destructive arthritis in a patient with chikungunya virus infection with persistent specific IgM antibodies. BMC Infect. Dis. 9: 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ochsenbein A. F., Zinkernagel R. M. 2000. Natural antibodies and complement link innate and acquired immunity. Immunol. Today 21: 624–630 [DOI] [PubMed] [Google Scholar]
- 36.Herzenberg L. A., Kantor A. B. 1993. B-cell lineages exist in the mouse. Immunol. Today 14: 79–83, discussion 88–90 [DOI] [PubMed] [Google Scholar]
- 37.Ochsenbein A. F., Fehr T., Lutz C., Suter M., Brombacher F., Hengartner H., Zinkernagel R. M. 1999. Control of early viral and bacterial distribution and disease by natural antibodies. Science 286: 2156–2159 [DOI] [PubMed] [Google Scholar]
- 38.Mayer K. 2011. The evolving Indian AIDS epidemic: hope & challenges of the fourth decade. Indian J. Med. Res. 134: 739–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Cock K. M., Jaffe H. W., Curran J. W. 2012. The evolving epidemiology of HIV/AIDS. AIDS 26: 1205–1213 [DOI] [PubMed] [Google Scholar]
- 40.Thiboutot M. M., Kannan S., Kawalekar O. U., Shedlock D. J., Khan A. S., Sarangan G., Srikanth P., Weiner D. B., Muthumani K. 2010. Chikungunya: a potentially emerging epidemic? PLoS Negl. Trop. Dis. 4: e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ravi V. 2006. Re-emergence of chikungunya virus in India. Indian J. Med. Microbiol. 24: 83–84 [DOI] [PubMed] [Google Scholar]
- 42.Sergon K., Yahaya A. A., Brown J., Bedja S. A., Mlindasse M., Agata N., Allaranger Y., Ball M. D., Powers A. M., Ofula V., et al. 2007. Seroprevalence of Chikungunya virus infection on Grande Comore Island, union of the Comoros, 2005. Am. J. Trop. Med. Hyg. 76: 1189–1193 [PubMed] [Google Scholar]
- 43.Hertz J. T., Munishi O. M., Ooi E. E., Howe S., Lim W. Y., Chow A., Morrissey A. B., Bartlett J. A., Onyango J. J., Maro V. P., et al. 2012. Chikungunya and dengue fever among hospitalized febrile patients in northern Tanzania. Am. J. Trop. Med. Hyg. 86: 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roederer M., Dubs J. G., Anderson M. T., Raju P. A., Herzenberg L. A., Herzenberg L. A. 1995. CD8 naive T cell counts decrease progressively in HIV-infected adults. J. Clin. Invest. 95: 2061–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]