

Abstract
Insufficient insulin secretion and insulin resistance are hallmarks of diabetes. Recent studies indicate that insulin plays an important role in maintaining the glomerular filtration barrier. Mima et al. report that glomeruli of diabetic and obese rats suffer from insulin resistance and altered insulin signaling. Protein kinase C inhibitors are able to overcome insulin resistance, offering new hopes for the treatment of the condition.
In 2007, diabetes had affected 23.6 million people in the United States, and the number was expected to increase significantly. The disease is characterized by hyperglycemia in the setting of either inadequate insulin secretion or insulin resistance. Obesity is one of the major risk factors for type 2 diabetes. Diabetic nephropathy is the number one cause of kidney failure in the United States.1 Histologically, diabetic nephropathy is characterized by glomerular basement membrane thickening and mesangial expansion followed by nodular sclerosis (Figure 1c and d). Clinically, micro- and, later, macroalbuminuria is observed, followed by a decline in renal function. In diabetes, all three layers of the glomerular filtration barrier are affected: glomerular endothelial cells, mesangial cells, and glomerular epithelial cells (or podocytes). Endothelial-cell dysfunction is a common theme in all diabetic complications; at the same time, decreased podocyte numbers have been shown to be a strong predictor of albuminuria and decline in renal function.2, 3 Since podocytes are the major source of vascular endothelial growth factor (VEGF) in glomeruli, and endothelial cells express the VEGF receptor, the cross-talk between podocytes and endothelial cells is likely to be abnormal in diabetes and might contribute to development of diabetic nephropathy.4, 5 Podocytes are particularly vulnerable to apoptosis in the setting of hyperglycemia, which could set up a vicious cycle causing both podocyte depletion and endothelial dysfunction.3
Figure 1.
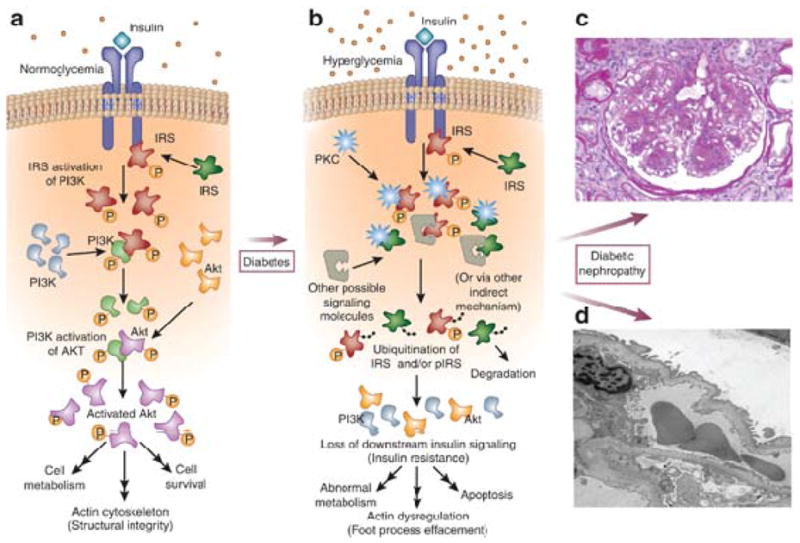
Diabetes-induced changes to insulin signaling within the glomerulus and their downstream consequences. (a) Schematic representation of insulin-mediated intracellular signaling cascade under normal physiological conditions. (b) Changes to insulin-mediated signaling due to protein kinase C and other second messengers in the setting of hyperglycemia, with increased ubiquitination of insulin receptor substrate and subsequent development of insulin resistance. (c) Representative light microscopic (Periodic–acid Schiff-stained) image of a human diabetic sample, showing nodular sclerosis and mesangial expansion. (d) Electron microscopic image of diabetic nephropathy showing foot process effacement and thickening of the glomerular basement membrane. IRS, insulin receptor substrate; pIRS, phosphorylated IRS; PI3K, phosphatidylinositol-3-kinase; PKC, protein kinase C.
Insulin is a hormone that binds to its receptor, a tyrosine kinase, which then phosphorylates insulin receptor substrates (IRSs), the most well studied of which is IRS1.6 IRSs then bind to phosphatidylinositol-3′-kinase (PI3K) or growth factor receptor-bound (GRB) proteins, which recruit many other downstream targets, such as Akt, glycogen synthase kinase 3 (GSK3), endothelial nitric oxide synthase (eNOS), Ras, extracellular signal-regulated kinase (ERK), and protein kinase C (PKC) to elicit wide-ranging effects including but not limited to glucose uptake, glycogenesis and lipogenesis, and cellular proliferation7 (Figure 1a). PKC represents a group of serine/threonine kinases that are activated by a variety of chemical signals besides insulin, including calcium, diacylglycerol, phosphatidylserine, and phorbol esters; the downstream effects are again wide-ranging. Some isoforms of PKC appear to have downregulatory effects on insulin signaling, such as PKC-β, which suppresses the insulin-induced activation of Akt and eNOS via direct phosphorylation of IRS1.8 Recent genetic studies indicate an association between PKC polymorphisms and the development of diabetic end-stage kidney disease.9 Additionally, certain isoforms of PKC, including PKC-β, are persistently activated by hyperglycemia, suggesting that there is perhaps overstimulation of PKC in diabetes. Therefore it has been suggested that PKC plays a critical role in the development of diabetic complications, and inhibitors of the PKC pathway are currently being investigated as potential cures for diabetic complications.
Interestingly, insulin also seems to play a role in regulating the glomerular filtration barrier. Welsh and colleagues recently reported that podocyte-specific deletion of the insulin receptor leads to the rapid development of albuminuria and glomerulosclerosis even in the setting of normoglycemia.10, 11 Since insulin is a major prosurvival factor for cells, the absence of insulin signaling probably resulted in podocyte death, contributing to the phenotype. While these studies highlight the key role of insulin signaling in diabetic nephropathy, yet, in most rodent models with type 1 diabetes (when insulin levels are almost undetectable), we do not observe rapid development of albuminuria and glomerulosclerosis. In addition, although insulin resistance is almost universally observed in patients with type 2 diabetes, there is never a complete loss of insulin signaling in patients.
To better understand insulin signaling in the glomerulus, Mimaet al.12 (this issue) used two genetically distinct rat models of diabetes and obesity. Their initial experiments showed that insulin induces IRS1, Akt, eNOS, and GSK3a phosphorylation both in glomeruli and in tubules; however, the relative degree of IRS1, Akt, and GSK3 phosphorylation was decreased only in diabetic and obese glomeruli, not in the tubules. This suggests that insulin resistance specifically affects the IRS PI3 K signaling pathwayin glomeruli. Perhaps just as importantly, Mimaet al.12 also showed that eNOS phosphorylation follows a similar glomerulus-specific trend of depressed stimulation after insulin, which could implicate the glomerulus as the initial target of dysfunction in diabetic nephropathy. Ruboxistaurin, a specific inhibitor of PKC-β, however, can increase active Akt, eNOS, and GSK3 in diabetic and obese rats to almost normal levels after insulin, suggesting that PKC-β may be responsible for the abnormal molecular signaling in insulin resistance.
Mima et al.12 also found a significant decrease in protein levels of IRS1, but not in mRNA levels of IRS1, in diabetic glomeruli of rats compared with their controls. They also found that diabetic rats have increased levels of ubiquitin-associated IRS1, suggesting that diabetes leads to abnormal signaling that steers IRS1 toward degradation. It seems that IRS1 and PKC may be the culprits behind insulin resistance in diabetes, as both IRS1 overexpression and PKC inhibitor treatment were individually capable of partially overcoming the elevated-glucose-induced suppression of Akt and eNOS in glomerular endothelial cells in vitro.
Putting all this together, in diabetes and perhaps in metabolic syndrome, blunted effects of insulin can be observed in the glomerulus. The precise mechanism by which insulin resistance ultimately results in proteinuria and fibrosis is being elucidated, as are the players necessary to mediate these changes. The research from Mima et al.12 suggests that glomerular endothelial cells exhibit changes in downstream insulin signaling that occur via a decrease in the availability of IRS1; the effects of this particular change are unknown, and whether or not this is necessary for the development of diabetic nephropathy is unclear. Future studies should evaluate the consequences of IRS1 depletion specifically within glomerular endothelial and other cells in vivo. Also, although insulin-related second messengers showed decreased phosphorylation in diabetic and obese rats, these results paint an unclear picture regarding in precisely which cell types these changes occur. Lastly, given the effect of PKC inhibitors on levels of other second messengers, there is still much to be said about what happens upstream of IRS and whether there are other unidentified players.
The studies performed by Mimaet al.12 nevertheless represent an important step forward in understanding the development of insulin resistance in the kidney and perhaps ultimately the pathway to end-stage kidney fibrosis. Diabetes and hyperglycemia probably have multiple effects on cell signaling and, when combined with other genetic factors, result in varying degrees of abnormal kidney function. At least on the basis of what we know currently, PKC inhibitors have shown promise as therapeutic tools to combat the effects of diabetes. Although there are still many questions regarding the steps that lead to diabetic nephropathy, we now have several new places to begin our search for answers.
Acknowledgments
We thank David Thomas for the histological images and members of the Susztak laboratory for valuable discussion. The work was supported by National Institutes of Health grants DK076077 and DK087635.
Footnotes
Disclosure
All the authors declared no competing interests.
References
- Collins AJ, Foley RN, Gilbertson DT, Chen SC. The state of chronic kidney disease ESRD, and morbidity and mortality in the first year of dialysis. Clin J Am Soc Nephrol. 2009;4(Suppl 1):S5–S11. doi: 10.2215/CJN.05980809. [DOI] [PubMed] [Google Scholar]
- Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- Cooper ME, Vranes D, Youssef S, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–2239. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Way KJ, Katai N, King GL. Protein kinase C and the development of diabetic vascular complications. Diabet Med. 2001;18:945–959. doi: 10.1046/j.0742-3071.2001.00638.x. [DOI] [PubMed] [Google Scholar]
- Ma RC, Tam CH, Wang Y, et al. Genetic variants of the protein kinase C-beta 1 gene and development of end-stage renal disease in patients with type 2 diabetes. JAMA. 2010;304:881–889. doi: 10.1001/jama.2010.1191. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Hale LJ, Eremina V, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–340. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Coward RJ. Podocytes, glucose and insulin. Curr Opin Nephrol Hypertens. 2010;19:379–384. doi: 10.1097/MNH.0b013e32833ad5e4. [DOI] [PubMed] [Google Scholar]
- Mima A, Ohshiro Y, Kitada M, et al. Glomerular-specific protein kinase C-β-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79:883–896. doi: 10.1038/ki.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]