

Abstract
To understand better how different genomic regions may confer pathogenicity for the coxsackievirus B (CVB), two intratypic CVB1 variants and a number of recombinant viruses were studied. Sequencing analysis showed 23 nucleotide changes between the parental non-pathogenic CVB1N and the pathogenic CVB1Nm. Mutations present in CVB1Nm were more conserved than those in CVB1N when compared to other CVB sequences. Inoculation in C3H/HeJ mice showed that the P1 region is critical for pathogenicity in murine pancreas and heart. The molecular determinants of disease for these organs partially overlap. Several P1 region amino acid differences appear to be located in the decay accelerating factor (DAF) footprint CVBs. CVB1N and CVB1Nm interacted with human CAR, but only CVB1N seemed to interact with human DAF, as determined using soluble receptors in a plaque reduction assay. However, the murine homologue Daf-1 did not interact with any virus assessed by haemagglutination. The results of this study suggest that an unknown receptor interaction with the virus play an important role in the pathogenicity of CVB1Nm. Further in vivo studies may clarify this issue.
Keywords: pancreatitis, myocarditis, DAF, CAR, mice
Introduction
Coxsackieviruses (CVs) belong to the genus Enterovirus within the Picornaviridae family [Pallansch and Roos, 2001]. Soon after their initial isolation, CVs were divided into subgroups A and B (CVA and CVB) according to their pathogenicity in suckling mice [Hyypia et al., 1993]. Based on its molecular characteristics, the CVB subgroup, which includes six serotypes, is currently classified as human enterovirus B [Fauquet et al., 2005; Oberste, 2008]. CVs cause commonly subclinical infection, but occasionally they cause significant disease, especially in the central nervous system (CNS) and muscle tissues, with neonates and immunocompromised patients being particularly susceptible [Pallansch and Roos, 2001]. CVBs cause the majority of cases of enterovirus-related viral myocarditis [Kim et al., 2001], a condition that has been linked to dilated cardiomyopathy [Feldman and McNamara, 2000]. Although the role of CVB as an etiologic agent of insulin-dependent diabetes mellitus is still controversial, this virus infects pancreatic acinar cells leading to widespread necrosis, fulminant pancreatitis and subsequent pancreatic insufficiency [Whitton et al., 2005]. Importantly, CVB1 was the predominant enterovirus isolated in the USA among young infants with severe disease in 2007 [MMRW, 2008; Wikswo et al., 2009] and continued to be the most common serotype detected in 2008 [MRRW, 2010].
Picornaviruses are small, icosahedral non-enveloped animal viruses. In many cases, their detailed atomic structures have been determined [Filman et al., 1998; Fry et al., 2003; Hadfield et al., 1997; Hendry et al., 1999; Hogle et al., 1985; Luo et al., 1987; Muckelbauer et al., 1995; Rossmann et al., 1985; Verdaguer et al., 2000]. The capsid is comprised of 60 copies each of four viral proteins, VP1, VP2, VP3 and VP4, forming an icosahedral shell of about 300 Å in diameter, which is filled with a positive-sense, single-stranded RNA genome. The surface topology of the capsid includes a depression called the “canyon” located near the five-fold symmetry axes. Genetic and structural studies have revealed that the canyon is the receptor-binding site for many of these viruses [Belnap et al., 2000; Colonno et al., 1988; Kolatkar et al., 1999; Olson et al., 1993; Rossmann et al., 2002]. When a receptor molecule binds within the canyon, it dislodges a “pocket factor”, probably a lipid molecule residing within a pocket immediately below the canyon surface [Rossmann, 1994]. The absence of the hydrophobic pocket factor destabilizes the virus and initiates a transition to altered “A” particles and virion uncoating [Curry et al., 1996].
CVBs interact with at least two receptor proteins. All CVBs use the coxsackievirus-adenovirus receptor (CAR), a 46-kDa protein that binds within the canyon [Bergelson et al., 1997; He et al., 2001; Tomko et al., 1997]. Although an interaction with CAR is all that is required for transition to the A-particle and subsequent uncoating [Milstone et al., 2005], some CVBs can interact with an additional 70-kDa molecule called the decay-accelerating factor (DAF, also known as CD-55). DAF is a complement regulatory protein that is expressed commonly on most cell surfaces [Bergelson et al., 1995; Shafren et al., 1995]. DAF binds to the virus capsid outside the canyon [Hafenstein et al., 2007] and contributes to the efficient infection of polarized epithelial cells [Coyne and Bergelson, 2006; Shieh and Bergelson, 2002]. Haemagglutination seems to be associated with the use of DAF although there are enterovirus strains with DAF affinity that do not haemagglutinate [Pasch et al., 1999; Powell et al., 1998; Powell et al., 1999; Spiller et al., 2000].
Since CVB infection in mice resembles human infection [Cihakova et al., 2004], the murine model has been used extensively in pathogenicity studies [Pallansch and Roos, 2001]. Important factors for determining the outcome of CVB-induced murine disease include: age, inoculation route [Bopegamage et al., 2005], mouse strain [Cihakova et al., 2004], nutritional status [Beck et al., 2003] and virus genotype [Chapman et al., 1997]. In addition, it has been suggested that the 5′UTR stem loop II (SLII) structure [Dunn et al., 2000; Lee et al., 1997] plays a major role in myocarditis [Dunn et al., 2003; Stadnick et al., 2004]. Furthermore, SLII has been found to determine virulence in CVB1-inoculated newborn mice [Rinehart et al., 1997]. Studies have demonstrated that single mutation at VP2-165 (in the “puff” loop) [Knowlton et al., 1996; Stadnick et al., 2004] or at VP1-155 [Cameron-Wilson et al., 1998] are important for the myocarditic phenotype. VP1-129 and, to a lesser extent, VP4-16 in CVB4 have been implicated as major pathogenic determinants of pancreatic disease [Chapman et al., 1997]. Although multiple nucleotides throughout the genome have been identified as pathogenic determinants, the pathogenic phenotype also depends on the particular intratypic or intertypic CVB variant [Pallansch and Roos, 2001]. Nevertheless, relatively few studies have compared molecular determinants using the same CV strain in different organs.
In order to identify determinants of disease and clarify the mechanism(s) involved, the pathogenic properties of two CVB1 viruses and the intratypic recombinant viruses obtained from them were studied. Sequence analysis showed 23 nucleotide differences between the parental non-pathogenic CVB1N and the pathogenic CVB1Nm. The results obtained suggest that the molecular determinants for pancreas and heart disease may differ. Mapping the CVB1 sequence-equivalent residues to the known structure of the CVB3-DAF complex showed that several amino acid changes in the P1 region are located within the human DAF binding site. Although both viral variants interacted with human CAR, only CVB1N was able to interact with human DAF. The results of this study suggest that an unknown murine receptor interaction with the virus play an important role in the pathogenicity of CVB1Nm.
Materials and Methods
Cells
HeLa cells (American Type Culture Collection) were maintained as monolayers in minimal essential medium (MEM), supplemented with 10% fetal calf serum, 2 mM L-glutamine, 25.5 mM sodium bicarbonate, and 50 μg/ml gentamicin. HeLa cells were used in transfections, virus propagation, determination of growth curves and plaque assays.
Virus
An infectious CVB1 cDNA clone (CVB1N) was generously provided by Dr. A. Nomoto, University of Tokyo, Japan [Iizuka et al., 1991]. The cDNA engineered behind the T7 promoter was in vitro transcribed after linearization with XbaI. The resultant RNA was transfected into HeLa cells to produce infectious virus, which was purified partially by pelleting through a 30% sucrose cushion as described previously [Rinehart et al., 1997].
To develop a pathogenic variant from this virus, a previously described protocol was used [Hufnagel et al., 1995]. Briefly, 4-week-old severe combined immunodeficiency (SCID) mice were intraperitoneally (i.p.) inoculated with 50 μl of Hanks’ balanced salt solution (HBSS) containing 104 PFU of CVB1N and sacrificed at 8 weeks post-infection (p.i.). The virus was isolated several times from heart or pancreas tissue homogenates after a single passage in SCID mice. One heart-isolated viral strain, CVB1Nm, showed a different plaque phenotype and was chosen for further studies. The CVB3 variant used in this study has been described previously [Hafenstein et al., 2007]. The CVB5 (Faulkner strain) was obtained from the Center for Disease Control (CDC), Atlanta, USA.
Cloning of a CVB1N-derived myocarditic variant and construction of chimeric cDNAs
The viral variant CVB1Nm was plaque-purified twice and semi-purified as described above. The viral RNA was isolated and subjected to RT-PCR as described previously [Rinehart et al., 1997]. Products obtained from nucleotides 1 to 3,448, 1,571 to 5,135 and 4,916 to polyA were cloned into pGEM-T vector (Promega). These clones were used to construct chimeric cDNAs between CVB1N and CVB1Nm by digestion of the parental cDNAs with restriction enzymes that recognize unique sites. Infectious viruses were obtained from the chimeric cDNAs as described above.
Sequence analysis
The full sequence of CVB1Nm was determined using an AmpliTaq FS dye terminator cycle sequencing kit (Applied Biosystems) and an ABI Prism 377 DNA sequencer and was compared to the published CVB1N sequence [Iizuka et al., 1987]. Both DNA strands of at least two different samples were sequenced.
To perform a comparative analysis of the mutations in the 5′UTR nucleotidic sequence, CVB1N segments were aligned with the corresponding sequences of other human enterovirus B (NCBI taxid:138949). Segments of 51 nts from the 5′UTR, in which each of the mutations was centered (at position 26), were used to obtain up to 100 sequences using BLASTn from the NCBI database. These segments were then aligned using Clustal X and the frequency of the mutations was calculated [Altschul et al., 1997; Thompson et al., 1994]. The amino acid changes in the VPs were analyzed by a similar procedure. Segments of 21 amino acids of the encoded polyprotein, in which the mutation was centered at position 11, were used to obtain up to 100 sequences using BLASTp and then aligned using NCBI COBALT [Papadopoulos and Agarwala, 2007].
Plaque forming assay
The plaque assay was performed in HeLa cells as described previously [Rinehart et al., 1997]. Briefly, confluent HeLa cells cultured in 6-well plates were incubated for 1 h (37 ºC, 5% CO2) with 20 PFU/well of each virus (three plates for each strain). After removing the medium, infected cells were covered with 0.8% agarose and cultured for 48 h. Prior to observation, the cells were fixed with paraformaldehyde and stained with 1% crystal violet.
One-step growth curves
A one-step growth curve with a multiplicity of infection (MOI) of 10 for each virus was determined in HeLa cells as described previously [Rinehart et al., 1997]. The monolayer was harvested and the virus titre was determined by plaque assay. Each time interval was assayed in duplicate.
Animal inoculation
Weanling (3-week-old) wild-type male C3H/HeJ mice (Jackson Laboratory, Bar Harbor, Maine) were inoculated i.p. with 50 μl of HBSS containing 104 PFU of each of the parental or recombinant viruses. At 5 or 10 days p.i., the pancreas and heart from 5 to 10 mice were collected from two separate experiments and processed for infectivity studies or fixed in 10% neutral buffered formalin, respectively, as described previously [Gomez et al., 1996]. All animal procedures were approved by the UNLP Animal Care and Use Committee.
Histopathology studies
Samples were embedded in paraffin, oriented along their longest axis and at least three 5-μm sections were stained with haematoxylin and eosin for histopathological examination. Pancreatitis and myocarditis were graded blindly by two pathologists on a scale of 0 to 4 using a whole longitudinal section of the organ: a score of 0 corresponded to the absence of cellular infiltration or necrosis, 1 to minimal inflammation (1 to 5 foci), 2 to mild inflammation (less than 25% of the pancreas section affected or 6 to 10 foci in the heart), 3 to moderate inflammation (25 to 50% of the pancreas section affected or 11 to 20 foci in the heart) and 4 to severe inflammation (more than 50% of the pancreas tissue section showed infiltration or necrosis or more than 20 foci in the heart).
Mutation mapping
The site of each mutation was established using equivalent CVB1 residues based on sequence alignment with the known structure of the closely related CVB3 [Muckelbauer et al., 1995]. Receptor footprints were obtained from cryo-electron microscopy (cryo-EM) of the reconstruction structures of CVB3 interacting with CAR [He et al., 2001] and of CVB3 (RD strain) interacting with the cellular co-receptor DAF [Hafenstein et al., 2007].
Plaque reduction assays
Human DAF was expressed in Pichia pastoris as a C-terminally 6xHis-tagged protein, as described previously [He et al., 2002]. The DAF construct consisted of the full-length ectodomain containing SCR-1, -2, -3, and -4 (amino acids 1 to 254) but lacking the S/T-rich linker domain and the glycosylphosphatidylinositol anchor. The CAR ectodomain protein (D1D2) used in the plaque reduction assays was provided by Paul Freimuth (Brookhaven National Laboratory) and produced and purified as described previously [He et al., 2001].
Haemagglutination studies
Human (Type 0 Rh-) and murine (BALB/c mice) erythrocytes were collected in 15 mM EDTA (final concentration) and washed three times with 50 volumes of PBS. A 0.5% suspension of washed erythrocytes was made in PBS. Viral dilutions were performed by adding 1 ml of sucrose-gradient purified CVB serotypes to 100 ml of PBS. Equal volumes of serially diluted virus and erythrocytes were mixed in V-bottom 96-well plates (Costar Bio-Rad, USA) and incubated at 4 °C for 4 h. CVB1N, CVB1Nm and CVB5 (positive control) were tested.
Statistics
Data were expressed as the mean ± S.E.M. and were analysed by one-way analysis of variance (ANOVA) followed by a Bonferroni multiple comparison test to determine significant differences between groups. P values <0.05 were considered statistically significant.
Results
Sequence analysis
To obtain viral variants, CVB1N was inoculated into SCID mice and viral isolations were obtained from various tissues. One of the heart tissue isolations presented a different plaque phenotype (Figure 1) and was chosen for further studies. This variant, CVB1Nm, was cloned and sequenced completely. Sequencing analysis showed 23 nucleotide changes from the parental strain CVB1N (Table I). Many of these mutations mapped to regions previously reported to affect pathogenicity, including two mutations in SLII, eight amino acid changes and one amino acid insertion in the P1 region, and three amino acid changes in the viral polymerase 3D.
Figure 1. Schematic representation of CVB1 parental and recombinant viruses, viral titres and pathology score.
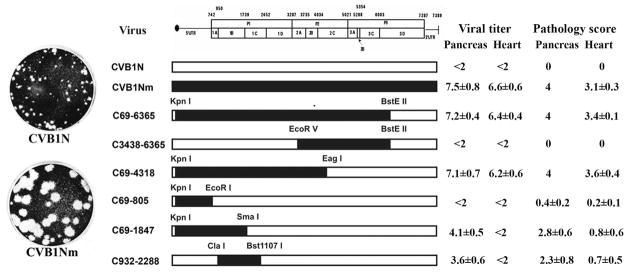
The restriction sites used to obtain the constructs are noted above the margins of the chimeric segments. CVB1N segments are shown as open bars while CVB1Nm segments are shown as solid bars. The pathology score was obtained as described in the text. Viral titres are expressed as log10 TCID50±SE per gram of tissue; <2 indicates that the infectivity was below the limit of the sensitivity assay. All determinations were performed using tissues corresponding to the C3H/HeJ mice shown in Figure 4.
Table I.
Nucleotide, predicted coding differences and their location between CVB1N and CVB1Nm
| Viral region | nta | Nucleotide differences
|
Amino acid changeb | Location | |
|---|---|---|---|---|---|
| CVB 1N | CVB 1Nm | ||||
| 5′ UTR | 35 | - | G | ||
| 5′ UTR | 118 | U | C | SL II | |
| 5′ UTR | 133 | U | A | SL II | |
| 5′ UTR | 184 | C | - | ||
| 5′ UTR | 606 | A | G | ||
| VP4 | 888 | C | U | ||
| VP4 | 919 | A | G | I→V, 60 | |
| VP2 | 1443 | U | A | D→E, 165 | DAF Footprint E-F loop “Puff” VP2 (antigenic site 2A) |
| VP3 | 1836 | G | A | ||
| VP3 | 1916 | A | C | N→T, 60 | DAF footprint neighboring area β knob VP3 (antigenic site 3A) |
| VP3 | 1972 | A | G | R→G, 79 | B-C loop VP3 (antigenic site 3B) |
| VP1 | 2690 | A | C | N→T, 80 | Fivefold B-C loop VP1 (antigenic site 1) |
| VP1 | 2770 | - | CUG | L, 107 | Below the pocket |
| VP1 | 2824 | G | C | E→Q, 125 | Vicinity of fivefold D-E loop VP1 |
| VP1 | 2850 | C | U | ||
| VP1 | 2914 | A | G | T→A, 155 | Vicinity of CAR Footprint |
| VP1 | 3272 | A | G | N→S, 274 | DAF footprint C-terminus of VP1 (antigenic site 3A) |
| 2B | 3816 | U | C | ||
| 2B | 3873 | A | G | ||
| 3D | 6133 | G | A | V→I, 78 | |
| 3D | 6953 | G | A | C→Y, 351 | |
| 3D | 7231 | GC | CG | A→R, 444 | |
Nucleotide positions are relative to the sequence of CVB1N.
The predicted changes are given relative to the mature viral protein.
Each mutation, was placed in the center of a selected protein (or RNA) segment and used as query sequence against the human enterovirus B data base (taxid:138949) (see methods). The resulting sequences were then aligned and analyzed. The frequency of occurrence was determined according to the identity of the residue of interest, based on whether the identity was that found in CVB1N, CVB1Nm or other virus.
The analysis of nucleotide identity at the positions of interest in the 5′UTR showed that, in general, the nucleotides corresponding to CVB1Nm were found more frequently than those corresponding to the parental CVB1N. In particular at nt 35 in the loop I “cloverleaf”, the CVB1Nm nucleotide was found with a frequency of 89% versus 11% for the deletion found in CVB1N. At nts 118 and 133, both located in the stem loop II, the frequency was 49% and 61%, respectively for CVB1Nm whereas the frequency was 4% and 9%, respectively for CVB1N. Located between loops III and IV at nt 606, the frequency of the mutant nucleotide was 74%. However, at nt 118, both the G corresponding to CVB1Nm and the gap for CVB1N were found with similar frequencies. Finally, the deletion of C at nt 184 in the mutant, was not found in the database (Figure 2A).
Figure 2. Sequencing studies.
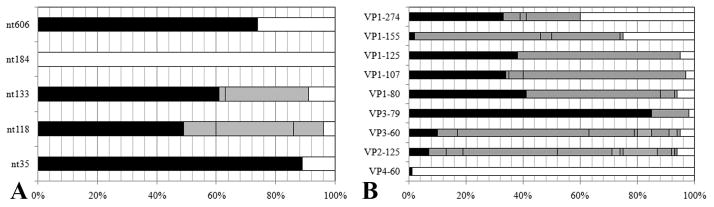
A: Bar chart showing frequency of identical nucleotides (including gaps) of the 5′UTR (A) and identical amino acid residues for the capsid proteins (B) from alignments with other human enterovirus B (taxid:138949). The frequency of matching identity is shown for CVB1Nm (black), CVB1N (white), with unmatched identity indicated in grey.
A similar analysis was performed to compare the amino acid sequence of the P1 region with other human enterovirus B. The results for VP1 showed that for position 80, 107 and 125, those residues in CVB1Nm were common, with frequencies greater than 33%. However, the same position for the residue in the parental strain was found in less than 7% of the sequences. At position 274 both residues were found at similar frequencies. In contrast, at position 155 the residue found in CVB1N was frequent but that of CVB1Nm was rare. In VP3 the frequency of the mutant residue at position 80 (10%) was higher than that of the parental residue (5%). The CVB1Nm identity of the residue at VP3-79 is highly conserved (85%). Mutation at VP3-60 and VP2-165 showed low frequencies for both the parental and the mutant. On the other hand, the residue at VP4-60 is highly conserved and matched that found in CVB1N; the mutant residue was not found (Figure 2B). Taken together, these results indicate that mutations present in CVB1Nm are closer to the consensus sequence than CVB1N, from a comparison with other human enterovirus B sequences.
Growth capabilities of recombinant viruses on HeLa cells
To identify determinants of disease and to clarify the mechanism(s) involved, several intratypic recombinant viruses were obtained. The restriction enzymes used and mutations included in each recombinant virus are shown in Figure 1.
The growth capabilities of recombinant and parental viruses were studied in HeLa cells. Enhanced replication was observed in the CVB1Nm, C69-805, C69-4318 and C69-1847 intratypic recombinant viruses (Figure 3). In contrast, the kinetics of the C932-2288 recombinant virus was similar to that of the parental virus CVB1N (Figure 3). These results suggest that the 5′UTR of CVB1Nm is responsible for the enhanced replication observed in cell culture.
Figure 3. Comparative studies on HeLa cells.
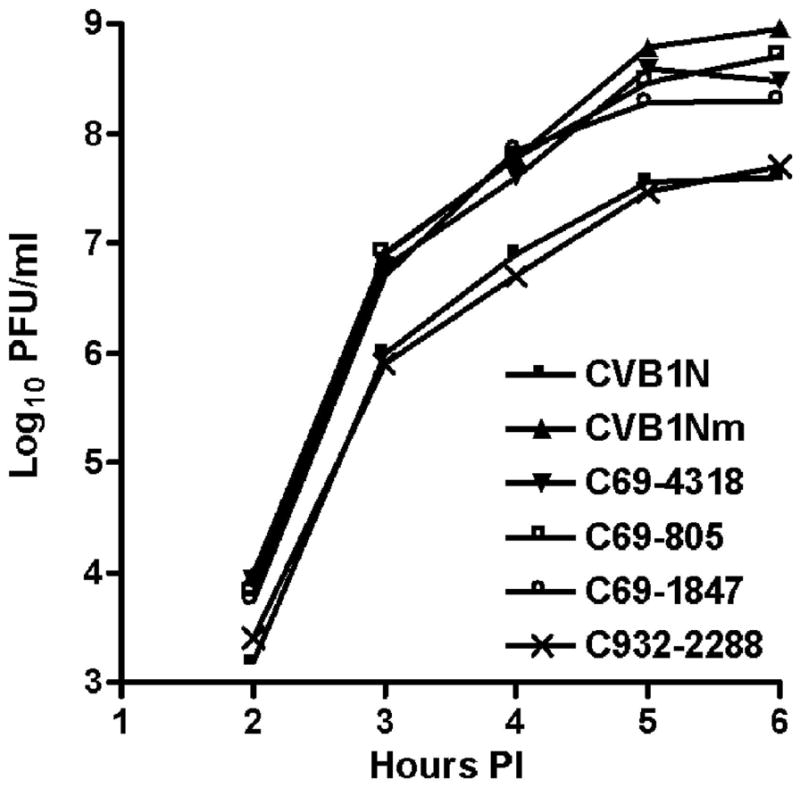
One-step growth curves with a multiplicity of infection (MOI) of 10 for each virus were performed on HeLa cells. At the indicated time points after infection, cells were washed with 0.1 ml of PBS, harvested and freeze-thawed three times. The live viral particles were counted using a plaque-forming assay. P<0.05 after comparison of CVB1N and C932-2288 with CVB1Nm and the remaining recombinant viruses. p.i.= post-inoculation.
Pathogenic properties in mice
To characterize the phenotype of CVB1N, CVB1Nm and the intratypic recombinant viruses in vivo, viruses were inoculated separately into C3H/HeJ weanling mice. Histological examination of samples from the CVB1N-infected mice (Figure 4A–B) showed no difference from mock-infected mice (Figure 4M–N). In contrast, CVB1Nm induced severe pancreatitis and myocarditis (Figure 4C–D) with moderate mortality (<20%). CVB1Nm viral titres in the pancreas and heart were 7.5±0.8 and 6.6±06, respectively (Figure 2). Recombinant viruses C69-6365 and C69-4318 (Figure 4E–F) displayed the same pathogenic phenotype as CVB1Nm. Pancreatic and cardiac viral titres for C69-6365 and C69-4318 (7.2±0.4 and 6.4±0.4, respectively for the former recombinant virus; 7.1±0.7 and 6.2±0.6, respectively for the latter recombinant virus) were similar to those obtained for CVB1Nm (Figure 1). However, recombinant virus C3438-6365 was not pathogenic (data not shown) and the viral titres in both organs analysed were below the assay detection limit (Figure 1), suggesting that the genomic region comprising nucleotides 69 to 3438 is important for the pathogenic properties observed after CVB1Nm inoculation. Recombinant virus C69-805 induced a small number of lesions in the pancreas and heart (Figure 4G–H), indicating that the 5′UTR is not critical for pathogenicity in the CVB1Nm variant. Recombinant viruses C69-1847 (Figure 4I–J) and C932-2288 induced only a few small, scattered necrotic foci in the myocardium (Figure 4L) but induced more severe pathological changes in the pancreas (Figure 4K). Viral titres correlated with the histopathology results. The data are summarized in Figure 1. These results suggest that major determinants for CVB1Nm pathogenicity are located in the P1 region and that mutations in the 5′UTR and 3C do not contribute to disease. Furthermore, mutations in VP1 are necessary for myocarditis and contribute to pancreatitis. In contrast, mutations located in VP2, 3 and 4 directly contribute to pancreatitis but do not induce myocarditis.
Figure 4. Representative histology of murine pancreas and heart after inoculation with CVB1N, CVB1Nm and intratypic recombinant viruses.

Ten days after inoculation of weanling male C3H/HeJ with these viruses, the pancreas and heart were harvested and processed for routine staining with haematoxylin and eosin. Representative sections from the pancreas (right panels) and myocardium (left panels) of mice inoculated with CVB1N (A–B), CVB1Nm (C–D), C69-4318 (E–F), C69-805 (G–H), C69-1847 (IJ), and C932-2288 (K–L) are shown. Scale bars: 125 μm.
Mutation mapping
The amino acid sequences of the P1 regions of CVB3 and CVB1Nm share more than 80% similarity and probably have similar secondary structures and characteristics, including receptor usage. The known structure of CVB3 (pdb code 1COV) was used to approximate the location of the sequence-equivalent residues corresponding to the nine mutations in the CVB1Nm P1 region. Mutation VP4-60 is located at the internal surface of the capsid near the three-fold symmetry axes, whereas mutation VP1-107, an L insertion, is buried within the capsid protein shell, located about 10Å beneath the canyon adjacent to the pocket. All of the remaining seven mutations map to the external surface of the virus capsid. Mutations at VP2-165, VP3-60 and VP1-274 are located within the DAF footprint. Mutations at VP1-155 and VP3-79 are adjacent to viral residues known to interact with DAF (Figure 5A) [He et al., 2002]. In contrast, none of the mutations present in the P1 region of CVB1Nm are located within the CAR binding site or predicted to interact with CAR, as suggested by the 3-D structure (Figure 5B). Residue VP2-165 has been reported to be a possible contact residue with CAR, based on its distance from the fitted CAR structure bound within the viral ‘canyon’ [He et al., 2001]. However, CVB1Nm VP2-165 maps to a location on the opposite side of the puff, with the top of the puff sterically blocking the direct access of VP2-165 to the canyon (Figure 5B).
Figure 5.
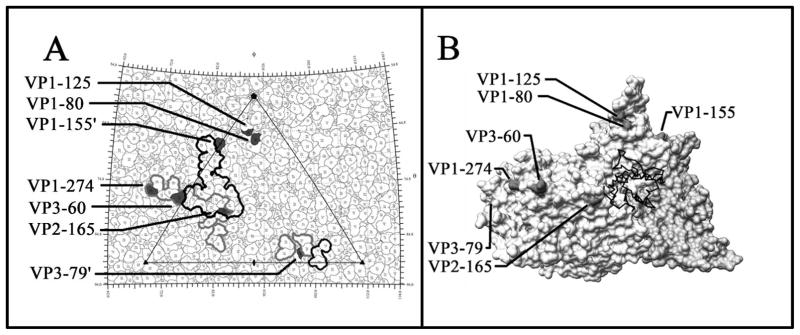
A: The viral surface is represented as a quilt of amino acids and shown as a stereographic projection, where the polar angles θ and ϕ represent latitude and longitude, respectively [Xiao and Rossmann, 2006]. The viral icosahedral asymmetric unit is indicated by the triangular boundary. The DAF footprint for echoviruses (a grey outline) was plotted using the equivalent CVB3 residues based on multiple sequence alignments [Chenna et al., 2003] according to the DAF contact residues for EV7 [He et al., 2002] and EV12 [Bhella et al., 2004]. EV and CVB3 have a sequence identity of 61% for VP1, 70% for VP2 and 67% for VP3. The DAF footprint on CVB3 is outlined in black. Mutations are indicated in black. VP1-155 and VP3-79 are highlighted in neighbouring asymmetric units. Five of the seven amino acid changes, which map to the viral surface, are located within areas of CVB3 that are known to interact with DAF. ‘ Denotes symmetry-related amino acids. B: This figure was obtained using Chimera [Pettersen et al., 2004; Sanner et al., 1996]. One protomer of the CVB3 surface rendered with a symmetry-related copy of VP3 to show the canyon and CAR binding region, relative to the location of the CVB1Nm surface residue changes. None of the mutations are within the CAR binding site. The closest residue, VP2-165, is located opposite the puff at a distance from the canyon.
Receptor binding properties
Capsid surface topology dictates receptor recognition and makes a major contribution to the tropism of the virus. Since the P1 structural region is likely to be responsible for the pathogenic phenotype and seven of the mutations map to the capsid surface, receptor usage was investigated. One of the mutations was within the known CAR binding footprint of CVB3 and four of the mutations detected in CVB1Nm mapped within or adjacent to the DAF footprint. Therefore, plaque reduction assays using CVB1N, CVB1Nm, CVB3 (positive control) alone or pre-incubated with either soluble hCAR or DAF were performed to characterize receptor usage [Hafenstein et al., 2007]. More than 2-log reductions in plaque number were observed when each virus was pre-incubated with soluble hCAR compared to virus alone (Table II, P<0.05). In contrast, an approximately 1-log reduction was observed when CVB1N, but not CVB1Nm, was pre-incubated with soluble DAF (Table II, P<0.05). These results suggest that the attenuated parental CVB1N may have an affinity for DAF that has been lost by the pathogenic CVB1Nm.
Table II.
CVB1 interaction with soluble human CAR and DAF evaluated by reduction plaque assays.
| Virus | +hCAR | +hDAF |
|---|---|---|
|
| ||
| CVB1N | 2 log reductions* | 1 log reductions* |
| CVB1Nm | 2 log reductions* | 0 log reductions |
| CVB3 | 2 log reductions* | 0 log reductions |
P<0.05; hCAR (human coxsackievirus-adenovirus receptor); hDAF (human decay-accelerating factor).
It has been reported that CVBs that bind the human DAF receptor do not bind murine DAF [Spiller et al., 2000]. In addition, although some CVBs with affinity for DAF do not haemagglutinate, haemagglutination has been associated with the use of DAF [Powell et al., 1998; Powell et al., 1999; Spiller et al., 2000]. Since the CVB1Nm mutant was developed in SCID mice, haemagglutination assays with human or murine erythrocytes incubated with purified CVB1N or CVB1Nm were performed in order to explore whether the mutations have a role in haemagglutination and thus, DAF affinity. The CVB5 Faulkner strain with known haemagglutination properties was employed as a positive control. Both CVB1N and CVB5 agglutinated human erythrocytes, but CVB1Nm failed to show any haemagglutination activity. As expected, none of the viruses agglutinated murine erythrocytes (Table III).
Table III.
CVB1N and CVB1Nm hemagglutination properties.
| Virus | Cells | Hemagglutination |
|---|---|---|
|
| ||
| CVB1N | hRBC | + |
| mRBC | − | |
|
| ||
| CVB1NM | hRBC | − |
| mRBC | − | |
|
| ||
| CVB5 | hRBC | + |
| mRBC | − | |
hRBC (human red blood cells); mRBC (murine red blood cells).
Discussion
A few genomic differences can lead to significant changes in the phenotype of RNA viruses [Domingo et al., 2006]. The CVB1Nm sequence revealed 23 nt changes compared to the CVB1N sequence, five of which were silent changes. Of special interest were those mutations located in the 5′UTR, particularly in and downstream of the SLII region, as this region has been associated with CVB3 pathogenic determinants of myocarditis [Chapman et al., 1997] and CVB1 virulence [Rinehart et al., 1997; Zhong et al., 2008]. Since nucleotide changes in CVB1Nm allowed for a more conserved sequence than CVB1N with respect to other CVB sequences, these mutations could underlie the non-attenuated phenotype of CVB1Nm. In addition, the studies performed on HeLa cells with the parental and recombinant viruses demonstrated that the presence of the 5′UTR of CVB1Nm led to enhanced replication. Two cellular RNA-binding proteins are known to interact with the SLII of VP1: the translation factor eIF-2α interacts with nucleotide regions 97 to 182 and 510 to 629 [del Angel et al., 1989], whereas the polypyrimidine tract-binding protein interacts with nucleotide regions 70 to 288, 443 to 539 and 630 to 730 [Hellen et al., 1994]. Mutations in the 5′UTR of CVB1Nm may influence viral replication efficiency in a cell type-specific fashion by affecting viral RNA translation and/or synthesis, a major determinant of virulence [Sonenberg and Dever, 2003].
Although several studies have characterized the molecular determinants of disease following infection with intratypic or intertypic CVB variants, relatively few have compared the molecular determinants of disease in the main target organs using the same virus. The pancreas and heart pathogenicity studies showed that although several chimeric viruses were pathogenic for the pancreas, only a few induced myocarditis and none of them induced myocarditis in the absence of pancreatitis. In addition, those viruses that achieved high titres in the pancreas were also virulent in the heart, whereas those that achieved intermediate titres in the pancreas induced an intermediate level of pancreatitis and virtually no myocarditis. Based on the chimeric studies, the determinants that are related to cardiovirulence also include those related to producing high viral titres in the pancreas. Furthermore, it is likely that more than one determinant accounts for cardiopathogenicity, because one change led to dramatic attenuation in myocarditis without an attendant reduction in pancreatitis. However, the opposite was not observed, as a reduction in pancreatitis was always concomitant with a reduction in myocarditis, as in previous studies [Kallewaard et al., 2009; Tracy et al., 2000].
Characterization of the chimeric viruses showed that the molecular determinants of pancreatitis and myocarditis in CVB1Nm-infected C3H/HeJ mice were also located in the P1 region. Although the P1 region had been implicated previously by a comparison of CVA9 and CVB3 [Harvala et al., 2005; Harvala et al., 2002], this is the first study to conclude that the P1 region contains determinants of pathogenicity using a single parental virus CVB1 and its variant isolated from a single passage in mice.
A number of antibody escape mutants of CVB3 that cause cardiac and pancreatic disease have been characterized previously [Stadnick et al., 2004], in which (i) a VP2-158 K to R mutation and a VP3-60 E to G lead to attenuation of the virus with respect to myocarditis but with only a partial reduction in pancreatitis; (ii) the presence of two mutations in the 5′UTR (a C-to-U transition at nucleotide 119 in SLII and a C-to-U transition at nucleotide 609) that were associated with enhanced pancreatic and cardiac disease. These authors also suggested that the escape mutants’ P1 mutations affected the virus–DAF interaction based on data extrapolated from the EV7–DAF interaction. A number of pathogenic CVB3 antibody escape mutants became attenuated in a murine model [Stadnick et al., 2004]. In contrast, the present study used a non-pathogenic variant of CVB1 that became pathogenic after a single passage in SCID mice. Given these different approaches, it is interesting that both studies found that mutations in similar genomic areas had a greater effect on myocarditis than pancreatitis in the murine model. This similarity suggests the existence of ‘hot spots’ along the genome that influence the pathogenic phenotype of CVs, whether the virus is an antibody escape mutant generated in vitro or a murine-adapted virus. Comparison of genomic sequences obtained from the data bank showed that this could also apply to human-isolated viruses.
The mapping of the equivalent CVB1 residues was based on the known structure of CVB3. However there may be slight differences in the topography of the actual CVB1 puff structure that may affect the predicted interactions between receptor residues and CVB1Nm VP2-165. Most of the CVB1Nm mutations mapped to the DAF-binding site, instead of the CAR binding site. Although all CVB viruses require CAR to initiate a productive infection, affinity for the DAF co-receptor appears to be more variable [Shieh and Bergelson, 2002]. On the other hand, it has been reported previously that CVBs that use human DAF as a receptor do not bind murine DAF [Spiller et al., 2000]. In this regard, the results obtained here showed that although CVB1N agglutinated human erythrocytes, neither CVB1N nor CVB1Nm haemagglutinated murine red blood cells. Furthermore, soluble hCAR significantly reduced the number of plaques induced by both viruses, whereas soluble DAF reduced only the number of CVB1N plaques and to a lower extent. These results suggest that both CVB1N and CVB1Nm variants use CAR, but that only CVB1N may use DAF. However, since some CVBs that do not haemagglutinate have DAF affinity, these data should be interpreted with caution. Furthermore, mutations in CVB1N may result in a viral variant that interacts differently with murine CAR, although the possibility of an interaction with another unidentified receptor cannot be excluded and has also been suggested by others [Orthopoulos et al., 2004].
The L insertion at VP1-107 of CVB1Nm is not directly related to the CAR or DAF footprint as it is located between the α-helix A and β-strand D, adjacent to the pocket beneath the canyon [Muckelbauer et al., 1995]. It has been suggested that the drug-binding cavity itself and not the putative pocket factor is crucial for capsid dynamics and infection of rhinoviruses [Katpally and Smith, 2007]. Consequently, this amino acid insertion may alter the local structure, affecting the capsid dynamics.
In summary, the major determinants of CVB1’s pathogenicity in the murine model are located in the P1 region and may differ according to the organ. Furthermore, critical residues on the viral surface map to the known DAF binding site, although the murine homologue Daf-1 does not seem to interact with any virus. The results obtained here suggest that an unknown murine receptor interaction with the virus plays an important role in the pathogenicity of CVB1Nm. Further studies may clarify this issue.
Acknowledgments
This work was supported by Grants PICT 13768 (National Agency for the Promotion of Science and Technology, PIP 5102/04 from the National Council of Scientific and Technical Research (CONICET, Argentina), CABBIO 04-19, x592 from National University of La Plata (RMG) and NIH 1 K22 Al 079271 (SH). VR and RMG are researchers and JOC and CJG are fellows from CONICET, Argentina.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck MA, Levander OA, Handy J. Selenium deficiency and viral infection. J Nutr. 2003;133:1463S–1467S. doi: 10.1093/jn/133.5.1463S. [DOI] [PubMed] [Google Scholar]
- Belnap DM, McDermott BM, Jr, Filman DJ, Cheng N, Trus BL, Zuccola HJ, Racaniello VR, Hogle JM, Steven AC. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc Natl Acad Sci USA. 2000;97:73–78. doi: 10.1073/pnas.97.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Bergelson JM, Mohanty JG, Crowell RL, St John NF, Lublin DM, Finberg RW. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55) J Virol. 1995;69:1903–1936. doi: 10.1128/jvi.69.3.1903-1906.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhella D, Goodfellow IG, Roversi P, Pettigrew D, Chaudhry Y, Evans DJ, Lea SM. The structure of echovirus type 12 bound to a two-domain fragment of its cellular attachment protein decay-accelerating factor (CD 55) J Biol Chem. 2004;279:8325–8332. doi: 10.1074/jbc.M311334200. [DOI] [PubMed] [Google Scholar]
- Bopegamage S, Kovacova J, Vargova A, Motusova J, Petrovicova A, Benkovicova M, Gomolcak P, Bakkers J, van Kuppeveld F, Melchers WJ, Galama JM. Coxsackie B virus infection of mice: inoculation by the oral route protects the pancreas from damage, but not from infection. J Gen Virol. 2005;86:3271–3280. doi: 10.1099/vir.0.81249-0. [DOI] [PubMed] [Google Scholar]
- Cameron-Wilson CL, Pandolfino YA, Zhang HY, Pozzeto B, Archard LC. Nucleotide sequence of an attenuated mutant of coxsackievirus B3 compared with the cardiovirulent wildtype: assessment of candidate mutations by analysis of a revertant to cardiovirulence. Clin Diagn Virol. 1998;9:99–105. doi: 10.1016/s0928-0197(98)00008-7. [DOI] [PubMed] [Google Scholar]
- Chapman NM, Ramsingh AI, Tracy S. Genetics of coxsackievirus virulence. Curr Top Microbiol Immunol. 1997;223:227–258. doi: 10.1007/978-3-642-60687-8_11. [DOI] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihakova D, Sharma RB, Fairweather D, Afanasyeva M, Rose NR. Animal models for autoimmune myocarditis and autoimmune thyroiditis. Methods Mol Med. 2004;102:175–193. doi: 10.1385/1-59259-805-6:175. [DOI] [PubMed] [Google Scholar]
- Colonno RJ, Condra JH, Mizutani S, Callahan PL, Davies ME, Murcko MA. Evidence for the direct involvement of the rhinovirus canyon in receptor binding. Proc Natl Acad Sci USA. 1988;85:5449–5453. doi: 10.1073/pnas.85.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Curry S, Chow M, Hogle JM. The poliovirus 135S particle is infectious. J Virol. 1996;70:7125–7131. doi: 10.1128/jvi.70.10.7125-7131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Angel RM, Papavassiliou AG, Fernandez-Tomas C, Silverstein SJ, Racaniello VR. Cell proteins bind to multiple sites within the 5′ untranslated region of poliovirus RNA. Proc Natl Acad Sci USA. 1989;86:8299–8303. doi: 10.1073/pnas.86.21.8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E, Martin V, Perales C, Grande-Pérez A, García-Arriaza J, Arias A. Viruses as quasispecies: biological implications. Curr Top Microbiol Immunol. 2006;299:51–82. doi: 10.1007/3-540-26397-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JJ, Bradrick SS, Chapman NM, Tracy SM, Romero JR. The stem loop II within the 5′ nontranslated region of clinical coxsackievirus B3 genomes determines cardiovirulence phenotype in a murine model. J Infect Dis. 2003;187:1552–1561. doi: 10.1086/374877. [DOI] [PubMed] [Google Scholar]
- Dunn JJ, Chapman NM, Tracy S, Romero JR. Genomic determinants of cardiovirulence in coxsackievirus B3 clinical isolates: localization to the 5′ nontranslated region. J Virol. 2000;74:4787–4794. doi: 10.1128/jvi.74.10.4787-4794.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. VIIIth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press; 2005. Virus Taxonomy. [Google Scholar]
- Feldman AM, McNamara D. Myocarditis. N Engl J Med. 2000;343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- Filman DJ, Wien MW, Cunningham JA, Bergelson JM, Hogle JM. Structure determination of echovirus 1. Acta Crystallogr D Biol Crystallogr. 1998;54:1261–1272. doi: 10.1107/s0907444998002790. [DOI] [PubMed] [Google Scholar]
- Fry EE, Knowles NJ, Newman JW, Wilsden G, Rao Z, King AM, Stuart DI. Crystal structure of Swine vesicular disease virus and implications for host adaptation. J Virol. 2003;77:5475–5486. doi: 10.1128/JVI.77.9.5475-5486.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez RM, Rinehart JE, Wollmann R, Roos RP. Theiler’s murine encephalomyelitis virus-induced cardiac and skeletal muscle disease. J Virol. 1996;70:8926–8933. doi: 10.1128/jvi.70.12.8926-8933.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield AT, Lee W, Zhao R, Oliveira MA, Minor I, Rueckert RR, Rossmann MG. The refined structure of human rhinovirus 16 at 2.15 A resolution: implications for the viral life cycle. Structure. 1997;5:427–441. doi: 10.1016/s0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- Hafenstein S, Bowman VD, Chipman PR, Bator Kelly CM, Lin F, Medof ME, Rossmann MG. Interaction of decay-accelerating factor with coxsackievirus B3. J Virol. 2007;81:12927–12935. doi: 10.1128/JVI.00931-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvala H, Kalimo H, Bergelson J, Stanway G, Hyypia T. Tissue tropism of recombinant coxsackieviruses in an adult mouse model. J Gen Virol. 2005;86:1897–1907. doi: 10.1099/vir.0.80603-0. [DOI] [PubMed] [Google Scholar]
- Harvala H, Kalimo H, Dahllund L, Santti J, Hughes P, Hyypia T, Stanway G. Mapping of tissue tropism determinants in coxsackievirus genomes. J Gen Virol. 2002;83:1697–1706. doi: 10.1099/0022-1317-83-7-1697. [DOI] [PubMed] [Google Scholar]
- He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, Kuhn RJ, Anderson CW, Freimuth P, Rossmann MG. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat Struct Biol. 2001;8:874–878. doi: 10.1038/nsb1001-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Lin F, Chipman PR, Bator CM, Baker TS, Shoham M, Kuhn RJ, Medof ME, Rossmann MG. Structure of decay-accelerating factor bound to echovirus 7: a virus-receptor complex. Proc Natl Acad Sci USA. 2002;99:10325–10329. doi: 10.1073/pnas.152161599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Pestova TV, Litterst M, Wimmer E. The cellular polypeptide p57 (pyrimidine tract-binding protein) binds to multiple sites in the poliovirus 5′ nontranslated region. J Virol. 1994;68:941–950. doi: 10.1128/jvi.68.2.941-950.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry E, Hatanaka H, Fry E, Smyth M, Tate J, Stanway G, Santti J, Maaronen M, Hyypia T, Stuart D. The crystal structure of coxsackievirus A9: new insights into the uncoating mechanisms of enteroviruses. Structure. 1999;7:1527–1538. doi: 10.1016/s0969-2126(00)88343-4. [DOI] [PubMed] [Google Scholar]
- Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 A resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- Hufnagel G, Chapman N, Tracy S. A non-cardiovirulent strain of coxsackievirus B3 causes myocarditis in mice with severe combined immunodeficiency syndrome. Eur Heart J. 1995;16(Suppl O):18–19. doi: 10.1093/eurheartj/16.suppl_o.18. [DOI] [PubMed] [Google Scholar]
- Hyypia T, Kallajoki M, Maaronen M, Stanway G, Kandolf R, Auvinen P, Kalimo H. Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res. 1993;27:71–78. doi: 10.1016/0168-1702(93)90113-2. [DOI] [PubMed] [Google Scholar]
- Iizuka N, Kuge S, Nomoto A. Complete nucleotide sequence of the genome of coxsackievirus B1. Virol. 1987;156:64–73. doi: 10.1016/0042-6822(87)90436-3. [DOI] [PubMed] [Google Scholar]
- Iizuka N, Yonekawa H, Nomoto A. Nucleotide sequences important for translation initiation of enterovirus RNA. J Virol. 1991;65:4867–4873. doi: 10.1128/jvi.65.9.4867-4873.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallewaard NL, Zhang L, Chen JW, Guttenberg M, Sanchez MD, Bergelson JM. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe. 2009;6:91–98. doi: 10.1016/j.chom.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katpally U, Smith TJ. Pocket factors are unlikely to play a major role in the life cycle of human rhinovirus. J Virol. 2007;81:6307–6315. doi: 10.1128/JVI.00441-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Hufnagel G, Chapman NM, Tracy S. The group B coxsackieviruses and myocarditis. Rev Med Virol. 2001;11:355–368. doi: 10.1002/rmv.326. [DOI] [PubMed] [Google Scholar]
- Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber S. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol. 1996;70:7811–7818. doi: 10.1128/jvi.70.11.7811-7818.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolatkar PR, Bella J, Olson NH, Bator CM, Baker TS, Rossmann MG. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 1999;18:6249–6259. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Maull E, Chapman N, Tracy S, Gauntt C. Genomic regions of coxsackievirus B3 associated with cardiovirulence. J Med Virol. 1997;52:341–347. [PubMed] [Google Scholar]
- Luo M, Vriend G, Kamer G, Minor I, Arnold E, Rossmann MG, Boege U, Scraba DG, Duke GM, Palmenberg AC. The atomic structure of Mengo virus at 3.0 A resolution. Science. 1987;235:182–191. doi: 10.1126/science.3026048. [DOI] [PubMed] [Google Scholar]
- Milstone AM, Petrella J, Sanchez MD, Mahmud M, Whitbeck JC, Bergelson JM. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J Virol. 2005;79:655–660. doi: 10.1128/JVI.79.1.655-660.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MMWR. Increased detections and severe neonatal disease associated with coxsackievirus B1 infection--United States, 2007. Morb Mortal Wkly Rep. 2008;57:553–556. [PubMed] [Google Scholar]
- MMWR. Increased detections and severe neonatal disease associated with coxsackievirus B1 infection--United States, 2007. Morb Mortal Wkly Rep. 2010;59:1577–1580. [PubMed] [Google Scholar]
- Muckelbauer JK, Kremer M, Minor I, Diana G, Dutko FJ, Groarke J, Pevear DC, Rossmann MG. The structure of coxsackievirus B3 at 3.5 A resolution. Structure. 1995;3:653–667. doi: 10.1016/s0969-2126(01)00201-5. [DOI] [PubMed] [Google Scholar]
- Oberste MS. Comparative genomics of the coxsackie B viruses and related enteroviruses. Curr Top Microbiol Immunol. 2008;323:33–47. doi: 10.1007/978-3-540-75546-3_2. [DOI] [PubMed] [Google Scholar]
- Olson NH, Kolatkar PR, Oliveira MA, Cheng RH, Greve JM, McClelland A, Baker TS, Rossmann MG. Structure of a human rhinovirus complexed with its receptor molecule. Proc Natl Acad Sci USA. 1993;90:507–511. doi: 10.1073/pnas.90.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthopoulos G, Triantafilou K, Triantafilou M. Coxsackie B viruses use multiple receptors to infect human cardiac cells. J Med Virol. 2004;74:291–299. doi: 10.1002/jmv.20184. [DOI] [PubMed] [Google Scholar]
- Pallansch MA, Roos RP. Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses, and Newer Enteroviruses. In: Fields BN, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, Knipe DM, editors. Virology. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 589–630. [Google Scholar]
- Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23:1073–1079. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- Pasch A, Kupper JH, Wolde A, Kandolf R, Selinka HC. Comparative analysis of virus-host cell interactions of haemagglutinating and non-haemagglutinating strains of coxsackievirus B3. J Gen Virol. 1999;80:3153–3158. doi: 10.1099/0022-1317-80-12-3153. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Powell RM, Schmitt V, Ward T, Goodfellow I, Evans DJ, Almond JW. Characterization of echoviruses that bind decay accelerating factor (CD55): evidence that some haemagglutinating strains use more than one cellular receptor. J Gen Virol. 1998;79:1707–1713. doi: 10.1099/0022-1317-79-7-1707. [DOI] [PubMed] [Google Scholar]
- Powell RM, Ward T, Goodfellow I, Almond JW, Evans DJ. Mapping the binding domains on decay accelerating factor (DAF) for haemagglutinating enteroviruses: implications for the evolution of a DAF-binding phenotype. J Gen Virol. 1999;80:3145–3152. doi: 10.1099/0022-1317-80-12-3145. [DOI] [PubMed] [Google Scholar]
- Rinehart JE, Gomez RM, Roos RP. Molecular determinants for virulence in coxsackievirus B1 infection. J Virol. 1997;71:3986–3991. doi: 10.1128/jvi.71.5.3986-3991.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann MG. Viral cell recognition and entry. Protein Sciences. 1994;3:1712–1725. doi: 10.1002/pro.5560031010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, Hecht HJ, Johnson JE, Kamer G, Luo M, Mosser AG, et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, He Y, Kuhn RJ. Picornavirus-receptor interactions. Trends Microbiol. 2002;10:324–331. doi: 10.1016/s0966-842x(02)02383-1. [DOI] [PubMed] [Google Scholar]
- Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J Virol. 1995;69:3873–3877. doi: 10.1128/jvi.69.6.3873-3877.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JT, Bergelson JM. Interaction with decay-accelerating factor facilitates coxsackievirus B infection of polarized epithelial cells. J Virol. 2002;76:9474–9480. doi: 10.1128/JVI.76.18.9474-9480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Dever TE. Eukaryotic translation initiation factors and regulators. Curr Opinion Struct Biol. 2003;13:56–63. doi: 10.1016/s0959-440x(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Spiller OB, Goodfellow IG, Evans DJ, Almond JW, Morgan BP. Echoviruses and coxsackie B viruses that use human decay-accelerating factor (DAF) as a receptor do not bind the rodent analogues of DAF. J Infect Dis. 2000;181:340–343. doi: 10.1086/315210. [DOI] [PubMed] [Google Scholar]
- Stadnick E, Dan M, Sadeghi A, Chantler JK. Attenuating mutations in coxsackievirus B3 map to a conformational epitope that comprises the puff region of VP2 and the knob of VP3. J Virol. 2004;78:13987–14002. doi: 10.1128/JVI.78.24.13987-14002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RP, Xu R, Philipson L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA. 1997;94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S, Hofling K, Pirruccello S, Lane PH, Reyna SM, Gauntt CJ. Group B coxsackievirus myocarditis and pancreatitis: connection between viral virulence phenotypes in mice. J Med Virol. 2000;62:70–81. doi: 10.1002/1096-9071(200009)62:1<70::aid-jmv11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Verdaguer N, Blaas D, Fita I. Structure of human rhinovirus serotype 2 (HRV2) J Mol Biol. 2000;300:1179–1194. doi: 10.1006/jmbi.2000.3943. [DOI] [PubMed] [Google Scholar]
- Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol. 2005;3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]
- Wikswo ME, Khetsuriani N, Fowlkes AL, Zheng X, Penaranda S, Verma N, Shulman ST, Sircar K, Robinson CC, Schmidt T, Schnurr D, Oberste MS. Increased activity of Coxsackievirus B1 strains associated with severe disease among young infants in the United States, 2007–2008. Clin Infect Dis. 2009;49:e44–51. doi: 10.1086/605090. [DOI] [PubMed] [Google Scholar]
- Xiao C, Rossmann MG. Interpretation of electron density with stereographic roadmap projections. J Struct Biol. 2007;158:182–187. doi: 10.1016/j.jsb.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Li X, Zhao W, Tong L, Liu J, Wu S, Lin L, Zhang Z, Tian Y, Zhang F. Mutations at nucleotides 573 and 579 within 5′-untranslated region augment the virulence of coxsackievirus B1. Virus Res. 2008;135:255–259. doi: 10.1016/j.virusres.2008.04.012. [DOI] [PubMed] [Google Scholar]