

Abstract
Notch is a highly conserved cell-cell communication mechanism that regulates development, tissue homeostasis and repair. Recent studies indicate that Notch plays a key role in kidney development by establishing proximal tubular epithelial cell fate and cell type specification in the renal collecting system. Notch signalling is markedly reduced in the adult kidney, however, increased Notch signalling has been noted in both acute and chronic kidney injury. Increased glomerular epithelial Notch signaling has been associated with albuminuria and glomerulosclerosis, while tubular epithelial Notch activation might cause altered repair and fibrosis development. Here, we review the role of Notch signalling in the kidney during development as well as in acute and chronic injury.
Keywords: Notch signalling, development, repair, regeneration, kidney fibrosis, chronic kidney disease, renal cell cancer
The Notch signalling pathway
The Notch signalling pathway is an evolutionarily conserved cell-cell communication mechanism. It is present in all metazoans and functions as one of the major pathways that determine cell identity during development (1, 2). There are four receptors in mammals, Notch1-4, and 5 different ligands, Delta-like1, 3, 4 (Dll1,3,4), Jagged1 and 2 (Jag1,2)(3). Both receptors and ligands are single-pass type I transmembrane receptor proteins (3, 4). Notch is produced in the endoplasmic reticulum as a 300 kDa precursor protein that is cleaved by a furin-like convertase in the trans-Golgi compartment to an intra- and extracellular domain (3). The extracellular portion of Notch undergoes extensive N- and O-linked glycosylation during synthesis and secretion (5). The Fringe family of glycosyltransferases catalyze O-fucose elongation by adding N-acethylglucosamines on specific EGF-like repeats. This modification alters the responsiveness of the receptor to the different ligands (5, 6). Vertebrate Notch is constitutively cleaved in the Golgi and is reassembled into a functional heterodimeric receptor at the cell surface. Notch signalling is triggered by ligand binding which in turn initiates a series of proteolytic cleavages by metalloproteases of the ADAM family (S2 site) and finally the γ-secretase complex (S3 site) (Fig1) (7, 8). The functional γ-secretase complex has four principal transmembrane components: Presenilin, Nicastrin, Aph1 and Pen2. S3 cleavage induces endocytosis of Notch’s extracellular portion along with the ligand by the signal-sending cell and the subsequent release of the Notch intracellular domain (NICD) which then translocates to the nucleus of the signal-receiving cell (9). Upon NICD’s nuclear translocation, co-repressors associated with Rbpj (also referred to as CSL or CBF1), a binding partner of NICD, are displaced, and a transcriptionally active complex consisting of NICD, Rbpj and Mastermind (Maml) assembles, thus leading to the activation of Notch target genes (10). The response to Notch signalling varies greatly between different cell types – for example, Notch promotes cell proliferation in certain cells while inducing apoptosis in others (1, 11-13). Its ability to elicit different responses may be at least partially attributed to a crosstalk with other pathways. For example several lines of evidence support an interaction between the Notch and NFkβ pathways (14). In addition, cellular response likely depends on cell type specific enhancers that are responsive to Notch regulation in a given cell. Interestingly, despite a high homology between the various receptors, even the role of each isoform can be markedly different (15-17). The best characterized target genes are the bHLH (basic helix-loop-helix) genes mainly belonging to the Hes and Hey family, which in turn function as transcription factors (18, 19). Signal termination usually occurs by ubiquitin-ligase mediated NICD degradation, facilitated by Maml and involves cyclin-dependent kinase-8, which phosphorylates NICD (20). This core signal transduction pathway used in most Notch-dependant processes is known as the canonical Notch signalling pathway.
Figure 1. Canonical Notch signalling.

The signal-sending cell presents the ligand, belonging to the DSL (Delta, Serrate, Lag-2) family to the signal-receiving cell, expressing a Notch receptor on its cell surface. This leads to a sequential cleavage of Notch through ADAM metalloproteinases at site S2 and the ɣ-secretase complex at site S3. The released extracellular domain of Notch is endocytosed along with the ligand, whereas the cleaved intracellular Notch (NICD) translocates to the nucleus where it interacts with its binding partner Rbpj, thus driving target gene (for example Hes, Hey) transcription.
Patients with Notch pathway mutations
Notch is a very tightly regulated signaling mechanism with multiple special features of the signalling pathway. For example Notch activation not only needs cell-cell communication, making it short-distanced, but also occurs without a signal amplification step; i.e. a single receptor is cleaved which travels to the nucleus without amplification step. Therefore, it is not surprising that even subtle genetic alterations of the pathway could have significant phenotypic consequences.
Mutations of JAGGED1 or NOTCH2 (21, 22) have been described to be causally linked to Alagille syndrome (AGS). Most cases of AGS are caused by JAGGED1 mutations, whereas only a small subset can be attributed to NOTCH2 mutations. The estimated prevalence of AGS is 1 in every 100,000 live births and it is inherited in an autosomal dominant pattern. AGS is a multiorgan disorder which is mainly diagnosed by liver abnormalities, usually presenting with hepatic bile duct paucity and cholestasis(22). Cardiac, skeletal and opthtalmologic abnormalities are also fairly common in patients with AGS. Renal involvements in patients with AGS are well described in the pediatric literature. The spectrum of renal abnormalities in AGS are wide ranging and include renovascular disease, renal tubular acidosis, tubulointerstitial nephritis, and renal dysplasia/hypoplasia (23). In addition, a peculiar pattern of glomerular lesion; also called “mesangiolipidosis” has been described. This is characterised by the presence of mesangial and extramembranous lipid inclusions (24, 25). The significance and specificity of these deposits are not fully established. It is interesting to note that mice haploinsufficient for Jagged1 show no significant phenotypic abnormalities, indicating that other modifier genes may contribute to the human AGS phenotype (26).
Notch signalling and the classic developmental paradigms
Notch is involved in the development of diverse tissues in almost all species where it exhibits a myriad of effects (2, 27-29). Notch’s effects can be categorised into a few, classic patterns. For example Notch can provide an “inductive signal” by promoting the development of a given cell type through inducion of positively acting regulatory molecules. Thus expression of Notch can induce specific cell fate in neighbouring cells. An example of this occurs in Drosophila wing development, where loss of Notch signalling eliminates wing margin and wing tissue, while ectopic Notch signalling results in extra wing tissue development(30). In this model, loss of Notch signalling leads to a loss of a given cell type while excessive Notch signalling has the reverse effect. Another pattern of Notch signalling is to restrict a given cell fate i.e. “inhibitory” Notch signalling. Initially cells that share a special cell fate both send and receive Notch signals, known as “mutual” inhibitory Notch signalling. Later, one cell commits to the specialized fate and inhibits surrounding cells from adopting this fate, a phenomenon called “lateral” inhibitory Notch signalling. In this situation, failure of Notch signalling results in cells adopting the special cell fate, while excessive Notch signalling prevents the differentiation of these cells. As an example of lateral inhibitrion through Notch takes place in Drosophila neurogenesis, where Notch inhibits neurogenesis. Excessive Notch expression inhibits neuronal differentiation, while its dearth is associated with an excessive neurogenic phenotype (31-33).
Notch and kidney development
The renal epithelium is a secondary epithelium formed by reciprocally inductive interactions between two different mesenchymal precursor tissues(34); the ureteric bud (UB), which gives rise to the collecting duct system, and the metanephric mesenchyme (MM), which gives rise to the other renal epithelial cells (from podocytes to distal tubular epithelial cells)(35, 36). Glial cell line-derived neurotrophic factor is secreted by the mesenchyme and attracts the ureteric bud toward the mesenchyme(37-41). The attracted ureteric bud in turn secretes Wnt9b and induces the mesenchyme(42). Upon this induction by the ureteric bud, mesenchymal cells secrete Wnt4. Wnt4, fibroblast growth factor 8 (Fgf8) are known to play essential roles in the mesenchymal-to-epithelial transition (MET) (43). The MM then undergoes condensation followed by segmentation resulting in structures, called comma and S-shape bodies (44) (Fig2). The proximal segments of the S-shaped bodies will become future podocytes (glomerular epithelial cells), the mid-sections give rise to the proximal tubules and loop of Henle and the distal segments differentiate into distal tubules (Fig2).
Figure 2. Notch plays role in proximal tubule specification and establishing the cell composition of the collecting duct.
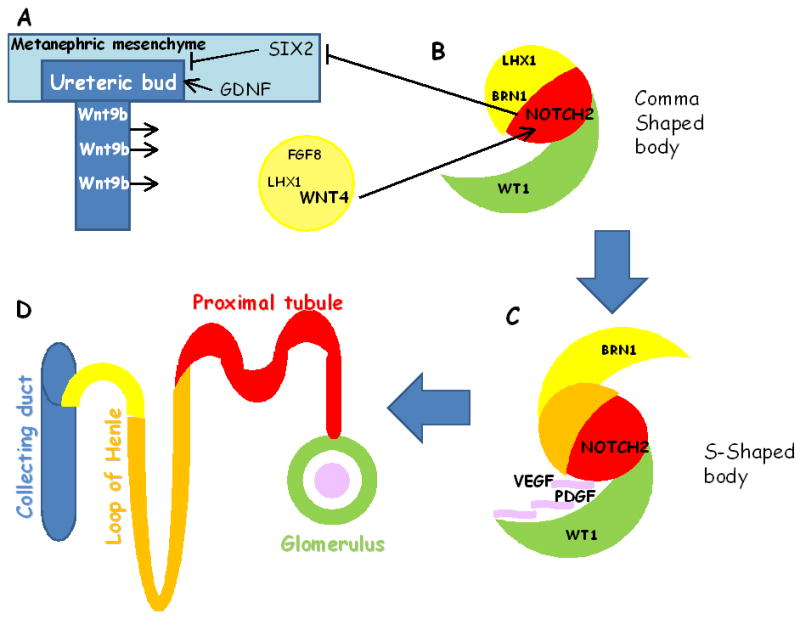
The renal epithelium is formed by reciprocally inductive interactions between two different mesenchymal precursor tissues; the ureteric bud (UB) and the metanephric mesenchyme (MM). Sine oculis homeobox domain 2 (SIX2) suppreses tubulogenesis in the mesenchyme. Glial cell line-derived neurotrophic factor is secreted by the MM attracts the UB toward the mesenchyme. The UB secretes Wnt9b and induces the mesenchyme to epithelial (MET) condensation via inducing Fgf8, Lhx1 and Wnt4. After MET, the nascent epithelial renal vesicles (RV) form a proximo-distal axis, where the surface facing the cortex is distal (yellow) and the medullary surface is proximal (green). The proximal segment of the S-shaped body is thought to give rise to the future glomerulus and proximal tubule, Wt1 and Notch2 plays key role in defining these segments. Endothelial cells are attracted by vascular endothelial growth factor (VEGF) and platalet derived growth factorβ (PDGFβ). (D) The mature nephron is further subdivided to glomerulus (green), proximal tubule (red), the the loop of Henle (orange), distal convoluted tubule (yellow), which connects to the collecting duct (blue). The scheme adapted and modified from Schedl et al (34).
Notch signalling molecules are expressed throughout kidney development, however, the reports addressing the exact location of different Notch ligands and receptors are somewhat controversial. Receptor (Notch1, 2) expression has been described in pretubular aggregates as well as in developing glomerular and tubular epithelial cells. The ligands Delta1 and Jagged1 are expressed in early pretubular aggregates as well (45-47). It is not clear whether Notch1 is expressed in the prospective proximal tubules during development. Notch2 is expressed in the primitive proximal tubules with faint co-localization in podocyte progenitors. Later on, Jagged1 seems to be expressed in the collecting duct and glomerular epithelial cells. Endothelial cells appear to express Jagged1 and Dll4(48-51).
Functional studies performed by Cheng et al.(52) showed a key role for Notch signalling during nephrogenesis. Mouse metanephroi cultured in the presence of a γ-secretase inhibitor a blocker of Notch receptor cleavage that precludes its activation and nuclear translocation (see Figure 2), caused a severe deficiency in proximal tubules and glomeruli. Further substantiating these findings Wang et al. showed that mice with genetic deletion for subunits of the Notch processing γ-secretase complex (Presenilin 1, 2) have severe defects in nephrogenesis and virtually no comma- and S-shaped bodies or mature glomeruli(53). Mice with a hypomorphic allele for Notch2, Notch2del1 or mice with a targeted deletion of Notch2 from the MM showed a similar phenotype(54). Early kidney development, ureteric bud migration, branching and mesenchymal aggregate formation occurred normally, however glomeruli did not form, nor did podocytes or proximal tubules appear. Interestingly, the glomerular vascularization defect in the Notch2del1 mice phenocopied glomerular abnormalities observed in mice with PDGFβ and VEGF mutations(54). The development of distal tubules and collecting ducts was not compromised in these animals. While Notch1 ectopic expression could rescue the Notch2 null phenotype, Notch1 deletion from the metanephric mesenchyme alone lead to no detectable developmental abnormalities (15). Later studies indicated that although the Notch2 receptor is dominant (its loss in the MM prior to the onset of kidney development results in complete nephron loss) Notch1 may also contribute and it becoming more apparent when Notch2 levels are limited (55). Studies by Cheng et al. suggest that Notch1 may be a weaker activator of the target promoters of Notch2, possibly owing to tertiary structure differences, and that its endogenous levels could be below the threshold required to activate Notch2 targets. These studies suggest a Notch mediated proximal fate determination (proximal tubular epithelium and podocyte), an attractive idea considering the lateral inhibition mechanisms of Notch in a variety of biological processes.
Recent studies further clarified Notch’s role in kidney development and somewhat contradict the original observations. First of all it appears that the initial proximodistal axis is still established even when Notch2 deleted, and it is likely mediated by an Lhx1-dependent mechanism. In addition Bonagio et al. also showed that cells can adopt a podocyte fate despite the absence of Notch signalling(56) and that the lack of podocyte development might have occurred secondary to a lack of proximal tubule specification in their models. Furthermore experiments from the Nishinakamura group showed that genetic overexpression of Notch2 in the embryonic kidney did not induce proximal fate specification, but lead to the depletion of the Six2 positive progenitor pool through premature differentiation toward renal tubules via ectopic Wnt4 activation. The result could be consistent with a model where Notch2 activation does not directly lead to proximal fate specification, but rather is necessary for the stabilization of the proximal fate (57, 58).
We can presume that Jagged1 is the Notch2 ligand during development as mice with compound heterozygous mutations for Jagged1 and Notch2 phenocopy the homozygous Notch2del1 mice.
In addition to Notch2’s role in proximal-distal axis establishment and development of proximal renal epithelium(60) studies indicate a role for Notch in the development of the collecting duct. In this nephron segment (which develops from the UB), two different cell types are intertwined: principal cells that are mainly involved in solute and water absorption and intercalated cells that regulate the acid-base homeostasis(61, 62). Jeong et al. very elegantly showed that Notch signalling in the developing collecting duct is involved in determining the ratio of principal and intercalated cells (63). They demonstrated that inactivation of Mind-bomb1, a protein expressed in the ligand-expressing cells and required for efficient Notch activation, led to an increase in intercalated cell number and a decrease in principal cell number in the developing collecting ducts. Given that intercalated cells are involved in regulating the acid-base homeostasis and principal cells in salt and water homeostasis, Mind-bomb1 deficient mice presented with increased urinary production, decreased urinary osmolality, sodium wasting and a severe urinary concentrating defect, similar to nephrogenic diabetes insipidus(63).
Moreover, studies performed in the zebrafish pronephros system by the Drummond(64) and Jiang (65) groups showed that single and multi-ciliated cell (MCC) fate is controlled by Notch signalling as well. In the zebrafish pronephros single ciliated cells possess the morphology of typical transport epithelial cells, and multiciliated cells that, by several criteria, appear specialized for fluid propulsion. These cell types are distributed in a “salt-and-pepper” fashion in the pronephros, showing some similarities to principal and intercalated cells in the mammalian kidney. Jagged2 is expressed in MCCs and Notch3 is expressed in pronephric epithelial cells. Morpholino knockdown of either Jagge 2 or Notch3, dramatically expanded ciliogenic gene expression, whereas ion transporter expression was lost, indicating that pronephric cells are transfated to MCCs. The reciprocal relationship between the ligand and receptor expressing cells during development give rise to two different cell types next to eachother. In summary, studies indicate that Notch signalling is involved in establishing the cellular composition of the collecting duct system during development. It is not clear whether Notch plays any role in regulating expression of ciliary genes in the mammalian kidney. Studies, however, suggest that Notch might regulate oriented cell division and reduced Notch signaling during development can induce cyst formation.
Role of Notch beyond development; Acute kidney injury, repair and regeneration
Compared to development the expression of Notch receptors and ligands is much decreased in the mature kidney. A few tubular epithelial and most probably endothelial cells, however, remain positive for Notch ligand or receptor expression (66-68). Further studies will be needed to better characterise the exact expression pattern and location of these cells.
Adult organ injury forces regeneration and repair activity leading to the expression of many different developmental signaling pathways (69). The kidney has a tremendous capacity for repair, following different types of acute injury (acute kidney injury, AKI). Experimentally AKI can be induced by short term hypoxia (ischemia, by clamping of the renal arteries) or injection of compounds that are directly toxic to tubular epithelial cells (for example gentamycin or mercury chloride). Proximal tubular epithelial cells are particularly susceptible to ischemic injury as they have the highest metabolic rate. Under such conditions the renal epithelium generally repairs and kidney function returns to normal. In the case of AKI, tubular epithelial cells re-differentiate after an initial phase of de-differentiation and proliferation, thus replacing cells that underwent apoptosis during injury (70). Hence it is not surprising that in the setting of injury, Notch (47), is reactivated, and could be responsible for cellular differentiation, proliferation and repair. This idea is supported by studies performed by Kobayashi et al. who observed increased expression of Delta1, Notch2 and the downstream target Hes1 using a rat ischemia reperfusion injury model (71). Co-culture of Delta1-expressing stromal cells and renal tubular epithelial cells lead to an increased proliferation rate of the latter. From this study the authors concluded that Notch2 expression might play a role in tubular epithelial cell regeneration during AKI. Studies by the Rosenberg group using the same rat model of ischemia reperfusion indicate that Dll4 blockage, perhaps via influencing endothelial cells, interferes with repair after ischemia reperfusion injury (72). On the other hand Bielesz et al observed activation of the Jagged1/Notch1/HeyL genes in the folic acid precipitation induced acute kidney injury(67) model. There was some increase in the expression of Notch2 and Hes1 in this model, but the failed to see regulation of Dll molecules. Treatment of animals with the pharmacological blocker of the ɣ-secretase complex had no effect on the rise and decline of serum creatinine and blood urea nitrogen induced by folic acid. However, an investigation into Notch’s functional role using mice with targeted genetic deletion has not been performed. In summary, while the Notch activation is observed in different models of acute kidney injury, its functional contribution to injury, repair and regeneration requires further elucidation.
Chronic kidney disease and fibrosis
Kidney fibrosis is the final common pathway leading to chronic kidney disease (CKD). Proper kidney function depends on the correct cellular interaction of over 20 different cell types. (2). In fibrosis this complex architectural is changed and the cell-cell interaction balance is altered. It is characterized by accumulation of collagen, activated myofibroblasts, inflammatory cells as well as loss of microvasculature and epithelial cells.
The first evidence suggesting a role for Jagged1/Notch1 regulation in chronic kidney disease and fibrosis came from early gene expression profiling studies performed on mouse models of fibrosis using the unilateral ureteral obstruction model (73). Gene array studies performed on control and diseased human kidneys (68, 74) confirmed the regulation of the Notch signalling pathway in chronic kidney disease (CKD). Using in-situ hybridization, Walsh and co-workers showed an increase of Jagged1 and Hes1 expression as well as their co-expression in tubules from kidneys of diabetic nephropathy patients. Notch pathway based gene clustering showed that diabetic nephropathy samples can be grouped together thus distinguishing themselves from control living donor samples and kidneys with minimal change disease, respectively. Furthermore, a recent study by Murea et al. showed increased levels of Notch pathway protein expression not only in diabetic nephropathy samples, but also in other chronic kidney disease samples associated with kidney fibrosis (Figure 2) (68). The severity of fibrosis correlated with the expression level of cleaved Notch1 in the tubulointerstitium, suggesting that Notch pathway activation might be a common downstream mechanism in a range of kidney diseases.
Further evidence for the role of Notch in fibrosis is emerging from a recently performed study by Bielesz et al. In this study the authors performed comprehensive pharmacological, genetic in-vivo and in-vitro experiments to analyze the role of Notch in renal tubules during kidney fibrosis(67). They reported that γ-secretase inhibitor treatment significantly alleviated fibrosis development in two different mouse models of fibrosis; induced by injection of folic acid (FA) or by unilateral ureteral obstruction (UUO). As the mice with systemic deletion of Notch related genes die early during development the authors had to use mice where Notch related proteins could be deleted in a cell type specific manner. Mice where canonical Notch signalling (using Rbpjfloxed mice) is deleted specifically from the proximal tubule (using PEPCKcre mice) did not show any significant developmental phenotype. Using the FA model of fibrosis, Bielesz et al. also demonstrated that proximal tubular epithelial cell-specific genetic deletion of the canonical Notch signalling effector; Rbpj (using the PEPCKcre/Rbpjflox/flox) lead to a decreased expression fibrosis markers, such as collagen1a1, smooth muscle actin and vimentin. In concordance with these findings, conditional inducible expression of Notch1 intracellular domain (NICD) in a tubular epithelial cell-specific manner induced rapid TIF development in mice. Notch induced the expression of collagen and smooth muscle actin transcripts and promoted macrophage influx similar to human chronic kidney disease and fibrosis. Transforming growth factor beta (TGF) regulates the expression of Notch ligand Jagged1 in renal epithelial cells (74-76). Moreover Notch expression in cultured human and rat tubular epithelial cells is a strong inducer of epithelial-to-mesenchmyal transition (EMT) (75). However, in vivo expression of NICD did not regulate EMT transcriptional profile, indicating that the Notch induced fibrosis development is independent of EMT. Transcriptome analysis performed on NICD over-expressing and control mice showed increased expression of collagen, proliferation- and inflammation-related genes. Similar to Notch’s developmental role in mature tubular epithelial cells Notch also strongly induced the expression of Wnt4 and proximal tubule marker cadherin 6 (58, 77). On the other hand markers of terminal differentiation such as solute carriers were decreased, suggesting a less differentiated cell state in NICD over-expressing mice. The authors concluded that sustained Notch activation induces a failed differentiation in epithelial cells and that incomplete epithelial cell differentiation along with prolonged proliferation and growth factor production results in a vicious circle, activating other pathways and mechanisms, which in turn perpetuate the process and eventually lead to CKD and TIF development (70).
Recent studies indicate a broader role for Notch regulating fibrosis. Increased expression of Notch was noted in patients and animal models of systemic sclerosis. Expression of Notch induced fibrosis development, while inhibition of Notch using genetic (anti-sense method) or pharmacological inhibitors (GSI) reduced fibrosis development. Similarly studies inducate that Notch expression contributes to peritoneal fibrosis development.
The role of Notch in glomerular disease
Whereas Notch activity is indispensible for glomerular development, Notch is almost completely absent from healthy adult glomeruli. Recent gene expression studies performed in our and other laboratories showed increased levels of Notch receptors and ligands in different glomerular diseases (68, 74, 78). Antibody-based expression analysis using kidney samples from 10 different disease groups showed increased expression of cleaved Notch1, Notch2 and Jagged1 in the glomerulus in different proteinuric nephropathies. More importantly, statistical analysis showed that the degree of kidney disease, measured by the amount of albuminuria and glomerulosclerosis, correlated with the level of Notch1, 2 and Jagged1 expression.
Studies performed in the zebrafish pronephros system inducate that the Notch, HeyL, Foxc2 regulatory network plays critical role in podocyte specification. Walsh and co-workers demonstrated that in mice the expression of Notch transcriptional targets decrease in developing podocytes after cell fate determination is complete, suggesting that podocyte development and differentiation is dependent on a temporal fine-tuning of Notch pathway activity (74). They further highlighted that ectopic expression of NICD in developing podocytes, using a transgenic mouse model, caused glomerulosclerosis along with proteinuria. Disease development was paralleled by a loss of mature podocyte features. Simultaneous conditional inactivation of Rbpj, the downstream transcriptional partner of Notch (Figure 1), rescued the described phenotype, highlighting the role of Notch silencing in the podocyte maturation process and maintenance of a functional glomerular filtration barrier.
In a recent study, Lasagni et al. looked at the role of Notch activation in putative renal progenitors in the setting of glomerular disease (79). Performing in vitro experiments, they first showed that Notch activation in renal progenitors induced cell proliferation, whereas Notch pathway silencing is required for progenitor cell differentiation towards the podocyte lineage. However, a prolonged activation of Notch signalling in these cells induced mitotic catastrophe and cell death. Accordingly, Notch pathway inhibition using a γ-secretase inhibitor in the adriamycin-induced mouse model of glomerulosclerosis alleviated proteinuria and diminished podocyte loss. However, prolonged Notch pathway inhibition throughout the regenerative phases of glomerular injury resulted in increased proteinuria and more severe glomerulosclerosis, suggesting a beneficial role of Notch during later stages of renal regeneration. This model would be highly consistent with a Notch regulated stem cell niche in the kidney, however the existance of resident renal stem cells in the kidney is highly questionable.
Functional studies performed by Niranjan et al. using transgenic and knockout animal models showed the deleterious effects of sustained Notch signalling in podocytes(66). Transgenic inducible expression of NICD in podocytes induced rapid development of albuminuria followed by glomerulosclerosis and death of the animals. Mechanistically, inducible podocyte-specific NICD expression led to p53 expression and subsequent apoptosis. In line with these findings, podocyte-specific genetic deletion of Rbpj, the transcription partner of Notch, protected mice from proteinuria and podocyte loss in the streptozotocin mouse model of diabetes. Moreover pharmacological inhibition of Notch signalling through the application of a γ-secretase inhibitor ameliorated proteinuria in the puromycin aminonucleosid rat model of glomerular disease. Similarly, studies performed in a diabetic rat model replicated these findings, as long term use of a gamma secretase inhibitor protected rats from diabetes induced albuminuria and glomerulosclerosis development (80, 81). Mechanistic studies indicate that Notch interacts with both the VEGF and the TGF pathways in the glomerulus. In another study, the application of γ-secretase inhibitors ameliorated renal damage in a model of lupus nephritis (82). However, the authors speculated that the main effect was most likely attributable to an interference with the immune system rather than to a direct renal effect. In summary, evidence is mounting that there is increased expression of Notch in different glomerular disorders. Sustained activation of Notch podocytes induce severe renal damage, while inhibition of Notch signalling showed benefit in multiple glomerular disease models.
Renal Cell Cancer
There are several sub-types of renal cell cancer (RCC) the majority of the cases (83%) of RCC’s are of clear cell type (clear cell renal cell cancer; CCRCC), where about 11-18% belong to the group of chromophil or papillary RCC. Chromophobe (CRCC) is the least common and seems to have a better prognosis compared to other RCC types. The most common genetic mutations that occur in RCC are mutations in the tumor suppressor genes VHL (von Hippel Lindau protein) and TSC (tuberous sclerosis complex)(83-87). Tissue microarray analyses showed that there is significantly increased Notch1 expression in chromophobe renal cell carcinoma (CRCC)(48). In another study high Jagged1 expression in RCC samples was statistically linked to reduced overall and disease-free survival, especially at the early stage. As Notch interacts both with the HIF/VHL(88) and the TSC(89) pathways and is a strong inducer of proliferation in tubular epithelial cells, these findings may not be very surprising(67). The Axelsson group showed that the Notch signalling cascade is constitutively active in human CCRCC cell lines independently of the VHL/HIF pathway(90). Notch signalling blockade resulted in attenuated proliferation and restrained anchorage-independent growth of CCRCC cell lines. Treatment of nude mice with an inhibitor of Notch signalling potently inhibited growth of xenotransplanted CCRCC cells. Moreover, the growth of primary CCRCC cells was attenuated upon Notch inhibition. However, it is interesting to note, that transgenic expression of ICNotch1 in tubular epithelial cells did not directly induce RCC development in mice(67). Therefore it is possible that Notch plays role in RCC progression rather than initiation.
Analysis of a rare form of renal cell cell cancer; the human type 1 papillary renal cell cancer (PRCC) indicated that there is reduced Notch signaling in this type of cancer. This was evident as gene expression analysis indicated that at least one Notch target (Hey1) was reduced while increased expression and nuclear localization of KyoT3/FHL1B was noted. KyoT/FHL1B is a splice variant of which is known to act as an inhibitor of the canonical Notch signalling pathway(91). Therefore these observations could be consistent with the notion that canonical Notch signalling pathway suppresses type 1 PRCC development. Consistently, mice with developmental deletion of Notch signalling (Notch1,2,or Rbp) presented with papillary microadenoma formation(59).
Therapeutic potential
In addition to the emerging role of Notch signalling in chronic kidney Notch signalling, is known to be involved in many oncological diseases such as T-cell lymphoblastic leukaemia (92, 93), colon cancer (48) as well as tumor angiogenesis (94). Therefore Notch gained significance as a therapeutic target in the field of oncology. Furthermore, its unique circuitry makes Notch signalling pathway an attractive therapeutic target. In this context, ligand binding followed by γ-secretase mediated cleavage is an important step at which Notch pathway activity can be pharmacologically modified. In addition due to its ability to cleave Amyloidβ precursors, the γ-secretase complex received attention as a potential therapeutic target for Alzheimer’s disease as well (95, 96). Many small molecular compounds targeting the ɣ-secretase complex have been developed (96, 97) and being tested in phase I-III human trials. Although very promising, pharmacologic Notch pathway inhibition through application of non-specific γ-secretase inhibitors showed many undesired side effects. Most importantly, the application of non-selective γ-secretase inhibitors cause intestinal goblet cell hyperplasia, as inhibition of Notch in intestinal progenitor cells drives differentiation towards goblet cell fate and inhibits enterocyte cell specification (98). Recent work, however, suggests that selective blocking of Notch1 or Notch2 by therapeutic antibody targeting reduces gut toxicity (99). Similarly concomitant administration of glucocorticoids might also rescue from GI toxicity(100). The avaialability of ɣ-secretase inhibitors makes Notch an attractive therapeutic candidate. However, further studies will be needed to determine, whether these are clinically applicable strategies.
Figure 3. Expression of Notch1 in healthy and diseased human kidney.

Representative immunostaining images showing cleaved Notch1 stainings on control and chronic kidney diseased (CKD) human kidneys. Scale bars: 50 μm
Figure 4. Increased expression of Notch in kidney induces fibrosis development.

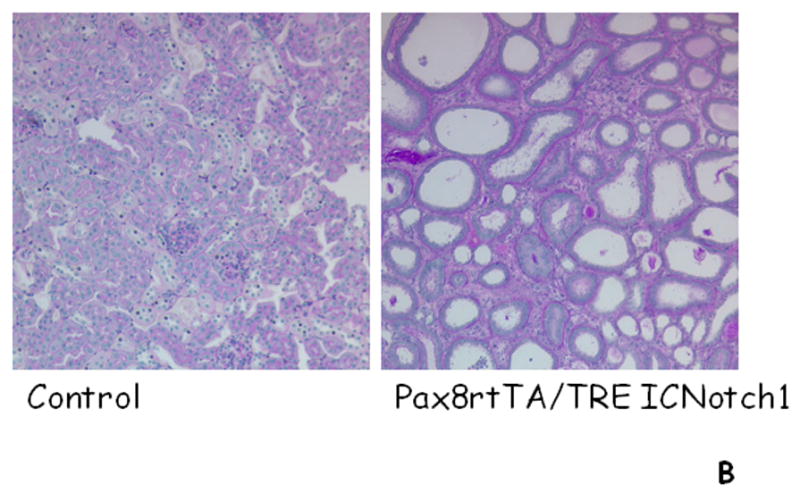
A. In the adult kidney very few tubular epithelial cells expression Notch1 and Jagged1. In injury expression of Notch1 and Jagged1 is increased. Notch activation induces proliferationa and failed differentiation of tubular epithelial cells. In addition, epithelial Notch activation induces fibroblast proliferation and myofibroblast activation. B. Representative histological images of control mouse kidneys and from mice with tubule specific Notch1 expression (Pax8rtTA/TREICNotch1).
Acknowledgments
This work was supported by grants from the National Institute of Health to KS (R01DK076077) and by the Juvenile Diabetes Research Foundation.
Footnotes
Authors contribution statement
KS and YS jointly wrote and edited the manuscript and produced the accompanying figures.
Teaching materials
Powerpoint slides of the figures from this review are supplied as supporting information in the online version of this article.
There is no conflict of interest.
References
- 1.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 2.Sharma S, Sirin Y, Susztak K. The story of Notch and chronic kidney disease. Curr Opin Nephrol Hypertens. 20:56–61. doi: 10.1097/MNH.0b013e3283414c88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilagan MX, Kopan R. SnapShot: notch signaling pathway. Cell. 2007;128:1246. doi: 10.1016/j.cell.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signalingpathways. Proc Natl Acad Sci U S A. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Moloney DJ, Stanley P. Fringe modulation of Jagged1-induced Notch signaling requires the action of beta 4galactosyltransferase-1. Proc Natl Acad Sci U S A. 2001;98:13716–13721. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, Selkoe DJ, Kopan R, Goate AM. Cell surface presenilin-1 participates in the gamma-secretase-like proteolysis of Notch. J Biol Chem. 1999;274:36801–36807. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 8.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 9.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 10.Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 11.Qi R, An H, Yu Y, Zhang M, Liu S, Xu H, Guo Z, Cheng T, Cao X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323–8329. [PubMed] [Google Scholar]
- 12.Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81–94. doi: 10.1016/j.ydbio.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6:881–888. doi: 10.1038/ni1234. [DOI] [PubMed] [Google Scholar]
- 14.Shin HM, Minter LM, Cho OH, Gottipati S, Fauq AH, Golde TE, Sonenshein GE, Osborne BA. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. Embo J. 2006;25:129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134:801–811. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrow D, Guha S, Sweeney C, Birney Y, Walshe T, O’Brien C, Walls D, Redmond EM, Cahill PA. Notch and vascular smooth muscle cell phenotype. Circ Res. 2008;103:1370–1382. doi: 10.1161/CIRCRESAHA.108.187534. [DOI] [PubMed] [Google Scholar]
- 17.Tien AC, Rajan A, Bellen HJ. A Notch updated. J Cell Biol. 2009;184:621–629. doi: 10.1083/jcb.200811141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steidl C, Leimeister C, Klamt B, Maier M, Nanda I, Dixon M, Clarke R, Schmid M, Gessler M. Characterization of the human and mouse HEY1, HEY2, and HEYL genes: cloning, mapping, and mutation screening of a new bHLH gene family. Genomics. 2000;66:195–203. doi: 10.1006/geno.2000.6200. [DOI] [PubMed] [Google Scholar]
- 19.Leimeister C, Bach A, Woolf AS, Gessler M. Screen for genes regulated during early kidney morphogenesis. Dev Genet. 1999;24:273–283. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<273::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Callahan R, Egan SE. Notch signaling in mammary development and oncogenesis. J Mammary Gland Biol Neoplasia. 2004;9:145–163. doi: 10.1023/B:JOMG.0000037159.63644.81. [DOI] [PubMed] [Google Scholar]
- 21.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 23.Habib R, Dommergues JP, Gubler MC, Hadchouel M, Gautier M, Odievre M, Alagille D. Glomerular mesangiolipidosis in Alagille syndrome (arteriohepatic dysplasia) Pediatr Nephrol. 1987;1:455–464. doi: 10.1007/BF00849254. [DOI] [PubMed] [Google Scholar]
- 24.Alagille D, Estrada A, Hadchouel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. doi: 10.1016/s0022-3476(87)80153-1. [DOI] [PubMed] [Google Scholar]
- 25.Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975;86:63–71. doi: 10.1016/s0022-3476(75)80706-2. [DOI] [PubMed] [Google Scholar]
- 26.Nyfeler Y, Kirch RD, Mantei N, Leone DP, Radtke F, Suter U, Taylor V. Jagged1 signals in the postnatal subventricular zone are required for neural stem cell self-renewal. Embo J. 2005;24:3504–3515. doi: 10.1038/sj.emboj.7600816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 28.Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16:633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Sebring A, Esch JJ, Kraus ME, Vorwerk K, Magee J, Carroll SB. Integration of positional signals and regulation of wing formation and identity by Drosophila vestigial gene. Nature. 1996;382:133–138. doi: 10.1038/382133a0. [DOI] [PubMed] [Google Scholar]
- 31.Nye JS, Kopan R, Axel R. An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells. Development. 1994;120:2421–2430. doi: 10.1242/dev.120.9.2421. [DOI] [PubMed] [Google Scholar]
- 32.Nye JS, Kopan R. Developmental signaling. Vertebrate ligands for Notch. Curr Biol. 1995;5:966–969. doi: 10.1016/s0960-9822(95)00189-8. [DOI] [PubMed] [Google Scholar]
- 33.Kopan R, Turner DL. The Notch pathway: democracy and aristocracy in the selection of cell fate. Curr Opin Neurobiol. 1996;6:594–601. doi: 10.1016/s0959-4388(96)80090-0. [DOI] [PubMed] [Google Scholar]
- 34.Schedl A. Renal abnormalities and their developmental origin. Nat Rev Genet. 2007;8:791–802. doi: 10.1038/nrg2205. [DOI] [PubMed] [Google Scholar]
- 35.Costantini F, Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell. 18:698–712. doi: 10.1016/j.devcel.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies JA. Morphogenesis of the metanephric kidney. ScientificWorldJournal. 2002;2:1937–1950. doi: 10.1100/tsw.2002.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vega QC, Worby CA, Lechner MS, Dixon JE, Dressler GR. Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc Natl Acad Sci U S A. 1996;93:10657–10661. doi: 10.1073/pnas.93.20.10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 39.Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 41.Durbec P, Marcos-Gutierrez CV, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M, et al. GDNF signalling through the Ret receptor tyrosine kinase. Nature. 1996;381:789–793. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- 42.Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell. 2005;9:283–292. doi: 10.1016/j.devcel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 43.Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 44.Majumdar A, Vainio S, Kispert A, McMahon J, McMahon AP. Wnt11 and Ret/Gdnf pathways cooperate in regulating ureteric branching during metanephric kidney development. Development. 2003;130:3175–3185. doi: 10.1242/dev.00520. [DOI] [PubMed] [Google Scholar]
- 45.Chen L, Al-Awqati Q. Segmental expression of Notch and Hairy genes in nephrogenesis. Am J Physiol Renal Physiol. 2005;288:F939–952. doi: 10.1152/ajprenal.00369.2004. [DOI] [PubMed] [Google Scholar]
- 46.Piscione TD, Wu MY, Quaggin SE. Expression of Hairy/Enhancer of Split genes, Hes1 and Hes5, during murine nephron morphogenesis. Gene Expr Patterns. 2004;4:707–711. doi: 10.1016/j.modgep.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 47.McCright B. Notch signaling in kidney development. Curr Opin Nephrol Hypertens. 2003;12:5–10. doi: 10.1097/00041552-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Aparicio LM, Villaamil VM, Gallego GA, Cainzos IS, Campelo RG, Rubira LV, Estevez SV, Mateos LL, Perez JL, Vazquez MR, et al. Expression of Notch1 to -4 and their Ligands in Renal Cell Carcinoma: A Tissue Microarray Study. Cancer Genomics Proteomics. 8:93–101. [PubMed] [Google Scholar]
- 49.Meehan B, Appu S, St Croix B, Rak-Poznanska K, Klotz L, Rak J. Age-related properties of the tumour vasculature in renal cell carcinoma. BJU Int. 107:416–424. doi: 10.1111/j.1464-410X.2010.09569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun S, Du R, Gao J, Ning X, Xie H, Lin X, Liu J, Fan D. Expression and clinical significance of Notch receptors in human renal cell carcinoma. Pathology. 2009;41:335–341. doi: 10.1080/00313020902885003. [DOI] [PubMed] [Google Scholar]
- 51.Rae FK, Stephenson SA, Nicol DL, Clements JA. Novel association of a diverse range of genes with renal cell carcinoma as identified by differential display. Int J Cancer. 2000;88:726–732. doi: 10.1002/1097-0215(20001201)88:5<726::aid-ijc7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 52.Cheng HT, Miner JH, Lin M, Tansey MG, Roth K, Kopan R. Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development. 2003;130:5031–5042. doi: 10.1242/dev.00697. [DOI] [PubMed] [Google Scholar]
- 53.Wang P, Pereira FA, Beasley D, Zheng H. Presenilins are required for the formation of comma- and S-shaped bodies during nephrogenesis. Development. 2003;130:5019–5029. doi: 10.1242/dev.00682. [DOI] [PubMed] [Google Scholar]
- 54.McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Herzlinger D, Weinmaster G, Jiang R, Gridley T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development. 2001;128:491–502. doi: 10.1242/dev.128.4.491. [DOI] [PubMed] [Google Scholar]
- 55.Surendran K, Boyle S, Barak H, Kim M, Stomberski C, McCright B, Kopan R. The contribution of Notch1 to nephron segmentation in the developing kidney is revealed in a sensitized Notch2 background and can be augmented by reducing Mint dosage. Dev Biol. 337:386–395. doi: 10.1016/j.ydbio.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonegio RG, Beck LH, Kahlon RK, Lu W, Salant DJ. The fate of Notch-deficient nephrogenic progenitor cells during metanephric kidney development. Kidney Int. 79:1099–1112. doi: 10.1038/ki.2010.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishinakamura R, Uchiyama Y, Sakaguchi M, Fujimura S. Nephron progenitors in the metanephric mesenchyme. Pediatr Nephrol. doi: 10.1007/s00467-011-1806-0. [DOI] [PubMed] [Google Scholar]
- 58.Fujimura S, Jiang Q, Kobayashi C, Nishinakamura R. Notch2 activation in the embryonic kidney depletes nephron progenitors. J Am Soc Nephrol. 21:803–810. doi: 10.1681/ASN.2009040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Surendran K, Selassie M, Liapis H, Krigman H, Kopan R. Reduced Notch signaling leads to renal cysts and papillary microadenomas. J Am Soc Nephrol. 21:819–832. doi: 10.1681/ASN.2009090925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129:1075–1082. doi: 10.1242/dev.129.4.1075. [DOI] [PubMed] [Google Scholar]
- 61.Madsen KM, Tisher CC. Structural-functional relationship along the distal nephron. Am J Physiol. 1986;250:F1–15. doi: 10.1152/ajprenal.1986.250.1.F1. [DOI] [PubMed] [Google Scholar]
- 62.Nielsen S, Kwon TH, Christensen BM, Promeneur D, Frokiaer J, Marples D. Physiology and pathophysiology of renal aquaporins. J Am Soc Nephrol. 1999;10:647–663. doi: 10.1681/ASN.V103647. [DOI] [PubMed] [Google Scholar]
- 63.Jeong HW, Jeon US, Koo BK, Kim WY, Im SK, Shin J, Cho Y, Kim J, Kong YY. Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J Clin Invest. 2009;119:3290–3300. doi: 10.1172/JCI38416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Pathak N, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134:1111–1122. doi: 10.1242/dev.02806. [DOI] [PubMed] [Google Scholar]
- 65.Ma M, Jiang YJ. Jagged2a-notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genet. 2007;3:e18. doi: 10.1371/journal.pgen.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 67.Bielesz B, Sirin Y, H, S, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, et al. Epithelial Notch Signaling Regulates Interstitial Fibrosis Development in Kidneys of Mice and Man. 2010 doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murea M, Park JK, Sharma S, Kato H, Gruenwald A, Niranjan T, Si H, Thomas DB, Pullman JM, Melamed ML, et al. Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int. doi: 10.1038/ki.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lazzeri E, Mazzinghi B, Romagnani P. Regeneration and the kidney. Curr Opin Nephrol Hypertens. 19:248–253. doi: 10.1097/MNH.0b013e32833680dc. [DOI] [PubMed] [Google Scholar]
- 70.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi T, Terada Y, Kuwana H, Tanaka H, Okado T, Kuwahara M, Tohda S, Sakano S, Sasaki S. Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int. 2008;73:1240–1250. doi: 10.1038/ki.2008.74. [DOI] [PubMed] [Google Scholar]
- 72.Gupta S, Li S, Abedin MJ, Wang L, Schneider E, Najafian B, Rosenberg M. Effect of Notch activation on the regenerative response to acute renal failure. Am J Physiol Renal Physiol. 298:F209–215. doi: 10.1152/ajprenal.00451.2009. [DOI] [PubMed] [Google Scholar]
- 73.Morrissey J, Guo G, Moridaira K, Fitzgerald M, McCracken R, Tolley T, Klahr S. Transforming growth factor-beta induces renal epithelial jagged-1 expression in fibrotic disease. J Am Soc Nephrol. 2002;13:1499–1508. doi: 10.1097/01.asn.0000017905.77985.4a. [DOI] [PubMed] [Google Scholar]
- 74.Walsh DW, Roxburgh SA, McGettigan P, Berthier CC, Higgins DG, Kretzler M, Cohen CD, Mezzano S, Brazil DP, Martin F. Co-regulation of Gremlin and Notch signalling in diabetic nephropathy. Biochim Biophys Acta. 2008;1782:10–21. doi: 10.1016/j.bbadis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 75.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. Embo J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Niimi H, Pardali K, Vanlandewijck M, Heldin CH, Moustakas A. Notch signaling is necessary for epithelial growth arrest by TGF-beta. J Cell Biol. 2007;176:695–707. doi: 10.1083/jcb.200612129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng HT, Kopan R. The role of Notch signaling in specification of podocyte and proximal tubules within the developing mouse kidney. Kidney Int. 2005;68:1951–1952. doi: 10.1111/j.1523-1755.2005.00627.x. [DOI] [PubMed] [Google Scholar]
- 78.Niranjan T, Murea M, Susztak K. The pathogenic role of notch activation in podocytes. Nephron Exp Nephrol. 2009;111:e73–79. doi: 10.1159/000209207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, Peired A, Ronconi E, Becherucci F, Bani D, et al. Notch Activation Regulates Differentially Renal Progenitors Proliferation and Differentiation Toward the Podocyte Lineage in Glomerular Disorders. Stem Cells. doi: 10.1002/stem.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin CL. Modulation of Notch-1 signaling alleviates VEGF-mediated diabetic nephropathy. Diabetes. 2010 doi: 10.2337/db09-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ahn SH, Susztak K. Getting a notch closer to understanding diabetic kidney disease. Diabetes. 59:1865–1867. doi: 10.2337/db10-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Teachey DT, Seif AE, Brown VI, Bruno M, Bunte RM, Chang YJ, Choi JK, Fish JD, Hall J, Reid GS, et al. Targeting Notch signaling in autoimmune and lymphoproliferative disease. Blood. 2008;111:705–714. doi: 10.1182/blood-2007-05-087353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peruzzi B, Athauda G, Bottaro DP. The von Hippel-Lindau tumor suppressor gene product represses oncogenic beta-catenin signaling in renal carcinoma cells. Proc Natl Acad Sci U S A. 2006;103:14531–14536. doi: 10.1073/pnas.0606850103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14:90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding M, Cui S, Li C, Jothy S, Haase V, Steer BM, Marsden PA, Pippin J, Shankland S, Rastaldi MP, et al. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006;12:1081–1087. doi: 10.1038/nm1460. [DOI] [PubMed] [Google Scholar]
- 86.Patel U, Simpson E, Kingswood JC, Saggar-Malik AK. Tuberose sclerosis complex: analysis of growth rates aids differentiation of renal cell carcinoma from atypical or minimal-fat-containing angiomyolipoma. Clin Radiol. 2005;60:665–673. doi: 10.1016/j.crad.2005.01.009. discussion 663-664. [DOI] [PubMed] [Google Scholar]
- 87.Walker C. Molecular genetics of renal carcinogenesis. Toxicol Pathol. 1998;26:113–120. doi: 10.1177/019262339802600113. [DOI] [PubMed] [Google Scholar]
- 88.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 89.Karbowniczek M, Zitserman D, Khabibullin D, Hartman T, Yu J, Morrison T, Nicolas E, Squillace R, Roegiers F, Henske EP. The evolutionarily conserved TSC/Rheb pathway activates Notch in tuberous sclerosis complex and Drosophila external sensory organ development. J Clin Invest. 120:93–102. doi: 10.1172/JCI40221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sjolund J, Johansson M, Manna S, Norin C, Pietras A, Beckman S, Nilsson E, Ljungberg B, Axelson H. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest. 2008;118:217–228. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liang L, Zhang HW, Liang J, Niu XL, Zhang SZ, Feng L, Liang YM, Han H. KyoT3, an isoform of murine FHL1, associates with the transcription factor RBP-J and represses the RBP-J-mediated transactivation. Biochim Biophys Acta. 2008;1779:805–810. doi: 10.1016/j.bbagrm.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 92.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 93.Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6:347–359. doi: 10.1038/nrc1880. [DOI] [PubMed] [Google Scholar]
- 94.Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–1087. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- 95.Wolfe MS. Therapeutic strategies for Alzheimer’s disease. Nat Rev Drug Discov. 2002;1:859–866. doi: 10.1038/nrd938. [DOI] [PubMed] [Google Scholar]
- 96.Wolfe MS, Esler WP, Das C. Continuing strategies for inhibiting Alzheimer’s gamma-secretase. J Mol Neurosci. 2002;19:83–87. doi: 10.1007/s12031-002-0015-5. [DOI] [PubMed] [Google Scholar]
- 97.Tsai JY, Wolfe MS, Xia W. The search for gamma-secretase and development of inhibitors. Curr Med Chem. 2002;9:1087–1106. doi: 10.2174/0929867023370185. [DOI] [PubMed] [Google Scholar]
- 98.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 99.Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 464:1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 100.Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C, Homminga I, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]