

Abstract
Research efforts on the human immunodeficiency virus (HIV) integrase have resulted in two approved drugs. However, co-infection of HIV with Mycobacterium tuberculosis and other microbial and viral agents has introduced added complications to this pandemic, requiring favorable drug-drug interaction profiles for antiviral therapeutics targeting HIV. Cytochrome P450 (CYP) and uridine 5'-diphospho-glucuronosyltransferase (UGT) are pivotal determining factors in the occurrence of adverse drug-drug interactions. For this reason, it is important that anti-HIV agents, such as integrase inhibitors, possess favorable profiles with respect to CYP and UGT. We have discovered a novel HIV integrase inhibitor (compound 1) that exhibits low nM antiviral activity against a diverse set of HIV-1 isolates, and against HIV-2 and the simian immunodeficiency virus (SIV). Compound 1 displays low in vitro cytotoxicity and its resistance and related drug susceptibility profiles are favorable. Data from in vitro studies revealed that compound 1 was not a substrate for UGT isoforms and that it was not an inhibitor or activator of key CYP isozymes.
Keywords: Anti-HIV, Integrase, Resistance, Drug-drug interactions, CYP/UGT profiles
1. Introduction
Although the global therapeutic response to HIV/AIDS has seen tangible progress, this viral pandemic nevertheless continues to ravage both the US and worldwide communities (Trono et al., 2010). Moreover, co-infection of HIV with tuberculosis (TB) and other microbial and viral agents has taken the pandemic to an elevated level of seriousness (Dye and Williams, 2010; Russell et al., 2010), which has created a critical need for favorable drug-drug interactions for therapeutics targeting HIV and associated co-infections (Josephson, 2010; Kiang et al., 2005). Thus, it is vital that anti-HIV agents, such as integrase inhibitors, exhibit favorable profiles with respect to human phase I and phase II isozymes, particularly those involving cytochrome P450 (CYP) and uridine 5'-diphospho-glucuronosyltransferase (UGT) (de Montellano, 2005; Tukey and Strassburg, 2000; Wienkers and Heath, 2005; Williams et al., 2004). These isozymes are pivotal determining factors in the occurrence of adverse drug-drug interactions.
HIV-1 integrase (Mr 32,000) is encoded at the 3′-end of the pol gene and is essential for the replication of HIV (Krishnan and Engleman, 2012). Integration of HIV DNA into the host cell genome requires metal ion cofactors and occurs through several steps including, site-specific endonuclease activity of the integrase-bound viral cDNA (3'-processing step), transport of the processed intasome complex through the nuclear envelope into the nucleus, integrase-catalyzed transfer of the processed viral cDNA ends into host chromosomal DNA (strand transfer step) and repair of the DNA at the integration sites (Esposito and Craigie, 1999; Frankel and Young, 1998; Hare et al., 2010; Haren et al., 1999). Research efforts on this crucial therapeutic target have resulted in two FDA-approved drugs, raltegravir and elvitegravir, for the treatment of HIV/AIDS (Shimura et al., 2008; Summa et al., 2008). Raltegravir is cleared primarily through glucuronidation involving the isozyme, UGT1A1, and to a lesser extent by UGT1A9 and UGT1A3 (Kassahun et al., 2007). Elvitegravir is a substrate for CYP3A4 and this compound and its metabolic products are also substrates for UGT1A1 and UGT1A3 (Mathias et al., 2009). The principal route for the metabolism of integrase inhibitor, S/GSK1349572 (Kobayashi et al., 2011), is also through UGT (Min et al., 2010).
To explore whether an authentic HIV-1 integrase inhibitor (Nair and Chi, 2007; Nair et al., 2006; Pommier et al., 2005; Taktakishvili et al., 2000) could be discovered, that would not only exhibit significant anti-HIV activity but also possess a favorable profile with respect to human CYP and UGT isozymes, we carried out structure-activity studies based on our earlier compounds with potent HIV-1 integrase inhibitory activities (Seo et al., 2011). These studies led to compound 1 (Figure 1) bearing a new integrase recognition motif. The compound, 4-(5-(2,6-difluorobenzyl)-1-(2-fluorobenzyl)-2-oxo-1,2-dihydropyridin-3-yl)-4-hydroxy-2-oxo-N-(2-oxopyrrolidin-1-yl)but-3-enamide, exhibited significant antiviral activity against a diverse set of HIV isolates and an excellent profile with respect to human cytochrome P450 and uridine 5'-diphospho-glucuronosyltransferase isozymes.
Fig. 1.
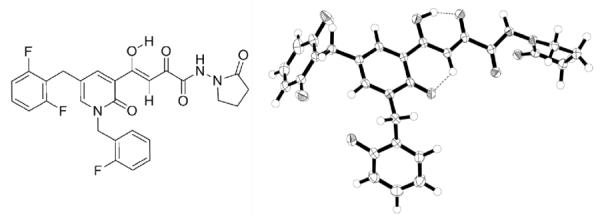
Integrase inhibitor 1 (left) and its single crystal X-ray structure (right) depicting the preferred tautomeric form and the conformation in the solid state.
2. Materials and Methods
2.1. Chemistry
2.1.1 General methods
NMR spectra were recorded on a Varian Inova 500 MHz spectrometer. HRMS data were obtained using Q-TOF Ion Mobility mass spectrometer. UV spectra were recorded on a Varian Cary Model 3 spectrophotometer. 5-Bromo-2-methoxy-pyridine, synthetic reagents and solvents were purchased from Aldrich, St. Louis, MO.
2.1.2. Synthesis
A concise methodology for the synthesis of compound 1 was developed that involved 8 steps and an overall yield of 25%. The key final step is described here. To a solution of 4-(5-(2,6-difluorobenzyl)-1-(2-fluorobenzyl)-2-oxo-1,2-dihydro-pyridin-3-yl)-2-hydroxy-4-oxobut-2-enoic acid (1.2 g, 2.71 mmol), prepared using modifications of methodologies previously described by us (Seo et al., 2011), in dimethylformamide (15 mL) was added 1-hydroxybenzotriazole (0.55 g, 4.07 mmol), followed by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (0.57g, 2.98 mmol) at 0oC. The resulting mixture was stirred at 0°C for 20 minutes and then 1-(amino)-2-pyrollidinone p-toluene sulfonate, (0.89 g, 3.25 mmol) and sodium bicarbonate (0.25 g, 2.98 mmol) were added. Stirring was continued for 2 h at 0–5°C. After completion of the reaction, the reaction mixture was quenched with water (50 mL). The resulting yellow solid was filtered and purified by trituration sequentially with methanol followed by chloroform: pentane (1:1 v/v) to afford compound 1 (1.11 g, 78% yield), m.p. 175–176 °C. UV (methanol) λ 401 nm (ε 9,139), 318 nm (ε 6,225). 1H-NMR (CDCl3, 500MHz): δ 15.2 (s,1H), 8.88 (s, 1H), 8.24 (s, 1H), 8.01 (s, 1H), 7.65 (s, 1H), 7.55 (t, 1H), 7.33-7.10 (m, 4H), 6.94 (t, 2H), 5.21 (s, 2H), 3.83 (s, 2H), 3.71 (t, 2H), 2.50 (t, 2H), 2.19 (m, 2H); 13C-NMR (CDCl3, 125MHz): δ 181.2, 179.3, 173.4, 162.2, 162.1, 162.1, 160.2, 160.2, 160.1, 159.5, 159.0, 144.0, 141.7, 132.1, 132.0, 130.4, 130.4, 128.9, 128.8, 124.7, 124.8, 122.5, 122.4, 122.3, 116.6, 115.6, 115.4, 115.0, 111.7, 111.6, 111.5, 111.4, 98.5, 47.8, 47.4, 28.4, 24.3, 16.8. HRMS: calcd for C27H23F3N3O5 [M+H]+ 526.1590, found 526.1589.
2.1.3. Compound Purity
Compound purity was 99.6% (from HPLC data, which was supported by high-field 1H and 13C NMR spectral data and quantitative UV data).
2.1.4. Molecular Modeling and Ligand Docking on Intasome
Molecular modeling of the crystal structure of prototype foamy virus (PFV) integrase intasome (PDB code 3OYA) with compound 1 docked within the catalytic site was achieved by using the Surflex-Dock package within Sybyl–X [Sybyl–X 1.3 (winnt_os5×) version] (Tripos, St. Louis, MO, 2011). Processing was done according to default conditions of Surflex-Dock and Biopolymer. The prepared ligand of compound 1 was docked to the intasome active site as guided by an appropriately generated protomol. The modeling was validated by screening a ligand set for compound 1 and a number of known anti-HIV integrase inhibitors and it was able to recognize all of the active compounds, including compound 1, as those with significantly high total scores.
2.3. Biology
2.3.1. Materials
All HIV-1 isolates (Gao et al., 1994; Gao et al., 1998; Jagodzinski et al., 2000; Michael et al., 1999; Vahey et al., 1999; Abimiku et al., 1994; Owen et al., 1998; Daniel et al., 1985), MT-4 cells, pNL4-3 plasmid DNA (Adachi et al., 1986), HeLa-CD4-LTR-βgal cells (Kimpton and Emerman, 1992), molecular clones for HIV-1 integrase mutations (Reuman et al., 2010), and Sup-T1 cells (Smith et al., 1984) were obtained from the NIH AIDS Research and Reference Reagent Program. Integrase-pBluescript was obtained from the HIV Drug Resistance Program, NCI, NIH. Other materials were purchased as follows: GeneTailor Site-Directed Mutagenesis System and High Fidelity Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA); PCR primers (Operon Biotechnologies, Germantown, MD), pBluescript SK(+) cloning vector and XL10-Gold Ultracompetent cells (Stratagene, La Jolla, CA); Plasmid Miniprep and Gel Extraction Kits (Qiagen, Valencia, CA); restriction enzymes AgeI and SalI (New England Biolabs, Ipswich, MA); Rapid DNA Ligation Kits (Roche Applied Science, Indianapolis, IN)
2.3.2. Antiviral assays in fresh human PBMCs
Fresh human peripheral blood mononuclear cells (PBMCs) were isolated and used in antiviral assays as previously described (Kortagere et al., 2012; Ptak et al., 2008). Inhibition of HIV-1 replication was measured based on the reduction of HIV-1 reverse transcriptase (RT) activity in the culture supernatants using a microtiter plate-based RT reaction (Buckheit and Swanstrom, 1991; Ptak et al., 2010). Cytotoxicity was determined using the tetrazolium-based dye, MTS (CellTiter®96, Promega).
2.3.3. Antiviral assays in MT-4 cells
Compound 1 was solubilized in DMSO to yield 80 mM stock solutions, which were stored at −20°C until the day of drug susceptibility assay setup and used to generate fresh working drug dilutions. The integrase inhibitors, raltegravir and elvitegravir, were included to study cross-resistance. AZT was a positive control compound. CPE inhibition assays were performed as described previously (Adachi et al., 1986). The wild-type parental virus used for this study was the HIV-1 molecular clone HIV-1 NL4-3. Stocks of the virus were prepared by transfection of pNL4-3 plasmid DNA into HeLa-CD4-LTR-βgal cells. Molecular clones for HIV-1 integrase mutations were prepared by transfection into 293T cells (see below) followed by expansion in Sup-T1 cells. Integrase mutations for these viruses were confirmed by sequencing following stock production. These virus stocks, as well as the site-directed mutant virus stocks produced in 293T cells (see below), were titrated in the MT-4 cells by serially diluting the virus stocks in tissue culture media and using the serial dilutions to infect MT-4 cultures. Samples were evaluated for antiviral efficacy in triplicate for EC50 and in duplicate for CC50 values.
2.3.4. Selection of drug resistant virus isolates
A standard dose escalation method (Buckheit and Swanstrom, 1991; Ptak et al., 2010) employing MT-4 cells infected with HIV-1 NL4-3 as the parental “wild-type” virus was used to select HIV-1 isolates that were resistant to compound 1. The virus was serially passaged, using the virus from the day of peak virus expression to generate a new acute infection of MT-4 cells and increasing the concentration of test compound with each passage until drug resistance was identified or compound cytotoxicity became a limiting factor. Elvitegravir was included in the passaging in order to provide comparative data. A no-drug control (NDC) culture was passaged in parallel with the drug-treated cultures. In order to monitor genotypic changes, the integrase coding region of the HIV-1 pol gene was sequenced for the viruses from each passage.
Acute infections were initiated by infecting 5 ×105 MT-4 cells with a 1:10 dilution of HIV-1 NL4-3 stock virus or peak virus. Cells and virus were incubated at 37°C for 2–4 h in a single well of a 96-well microtiter plate using a total volume of 200 μL. The cells and virus were then transferred to a T25 flask and the volume increased to 4 mL using media containing an appropriate concentration of compound 1, or elvitegravir. On day 2–3 post-infection, the volume was increased to 10 mL, maintaining the concentration of each test drug. On days post-infection where the supernatant RT activity was observed to increase to greater than 1,000 cpm, cells were collected by centrifugation, followed by re-suspension in 10 mL of fresh media containing each drug at the appropriate concentration. Supernatants removed from the pelleted cells on each of these days were collected and stored at −80°C. Virus collected on the peak day of virus production based on RT activity was used to initiate the next passage.
2.3.5 Sequencing of HIV-1 integrase coding region
Virion-associated RNA was extracted from the supernatant virus pools collected on the peak days of virus replication for each virus passage. The viral RNA was used as template RNA to amplify the entire HIV-1 integrase coding region. The DNA sequence of both strands of the PCR amplified region was determined by dsDNA sequencing (University of Alabama at Birmingham Center for AIDS Research Sequencing Facility). Comparison with the integrase coding region from wild-type HIV-1 NL4-3 NDC-culture was also performed.
2.3.6. Mutagenesis methodology
Site-directed mutagenesis of the integrase gene from HIV-1 NL4-3 was performed on a portion of pNL4-3, spanning from the AgeI to SalI restriction enzyme sites, that was sub-cloned into the pBluescript SK(+) cloning vector (“Integrase-pBluescript”) which was used to produce integrase site-directed mutants. Methylation and PCR amplification of the Integrase-pBluescript sub-clone using the mutagenesis primer sets were performed using the GeneTailor Site-Directed Mutagenesis System and Platinium Taq DNA Polymerase. The resulting plasmid PCR amplifications were verified on a 1% agarose gel and then transformed into DH5α −T1 E. coli cells. Transformed cells were spread on standard LB-agar plates containing ampicillin and incubated overnight (37°C) to allow for colony formation. Individual colonies were isolated, used to inoculate 5–10 mL of standard LB Broth containing ampicillin, and incubated overnight (37°C). Plasmids were extracted from cultures and sequenced to confirm the integrase coding region and presence of appropriate mutation. Mutated integrase genes were sub-cloned back into the pNL4-3 backbone. The final mutated NL4-3 plasmids were confirmed to be correctly constructed by restriction digest and sequence analysis. The mutated pNL4-3 clones were first quantified to determine DNA concentration, ethanol precipitated for sterility, and re-suspended in sterile water. Following transfection, the cells were incubated for an additional 48 hours at 37°C/5% CO2 and then the supernatant was collected and 1 mL aliquots were frozen at −80°C as stock virus. Each stock was subsequently analyzed for RT activity and then titrated in MT-4 cells. Sequence analysis of the virus stocks produced from transfection of the plasmids into 293T cells was performed to confirm that the resulting viruses maintained the point mutations associated with the site-directed mutagenesis.
2.4. Human Cytochrome P450 and Uridine 5'-diphospho-glucuronosyltransferase Isozyme Studies
2.4.1. Materials
Sources: human liver microsomes (mixed gender, 200 pooled, Xenotech LLC, Lenexa, KS) and human liver microsomes (mixed gender, 150 pooled) BD Biosciences, San Jose, CA; NADPH tetrasodium salt, UDPGA trisodium salt, G-6-P, G-6-P DH), alamethicin, D-saccharic acid 1,4-lactone, amodiaquine, dextromethorphan, testosterone, tolbutamide, triazolam, midazolam, omeprazole, 4-MU, 4-MU β-D-glucuronide and trifluoperazine (Sigma, St. Louis, MO); raltegravir potassium salt, elvitegravir (Selleck, LLC, Houston, TX). HPLC analyses were performed on a Beckman Coulter Gold 127 system using C18 columns.
2.4.2. Metabolism studies
Incubation mixture (final volume of 400 μL) contained human liver microsomes (protein, 0.5 mg/mL), compound 1 (50 μM in DMSO (<1% of final mixture), G-6-P-DH (0.5 U/mL), and G-6-P (5 mM) in potassium phosphate buffer (100 mM, pH 7.4) containing MgCl2 (5 mM). The reaction mixture was pre-incubated for 3 min at 37 °C before addition of NADPH (final, 2 mM) and then incubated further at 37 °C. An aliquot (60 μL) of the incubation mixture was taken for each sampling and was quenched with acetonitrile (60 μL). Proteins were removed by centrifugation at 5,000g. The supernatant was analyzed on a Beckman Coulter Gold 127 system using C18 analytical columns (UV 360 nm, retention times: compound 1 9.7 min, minor cleavage product (<5%) 13.2 min. The data were analyzed and the results are summarized in Figure 3.
Fig. 3.
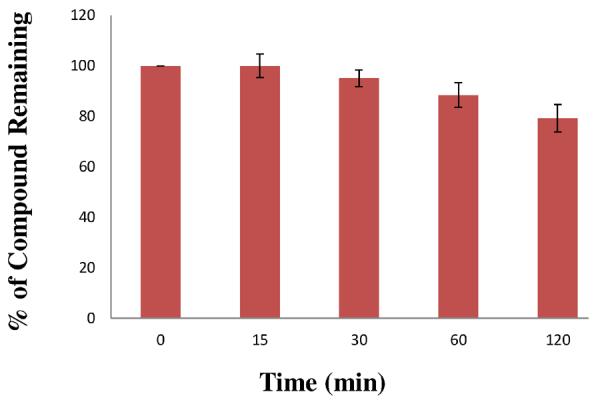
In vitro human liver microsome stability of compound 1. Each bar represents the average of three determinations with error bars showing standard deviations from the mean. The positive control compound for these studies was midazolam (Km for 1'-hydroxylation was 2.52 μM). The percent of midazolam (10 μM) remaining after 1 h under similar conditions was 61.5%.
2.4.3. Cytochrome P-450 isozyme inhibition assays
Incubation mixtures contained potassium phosphate buffer (100 mM, pH 7.4), MgCl2 (5 mM), requisite substrate dissolved in an appropriate solvent, compound 1 (in DMSO) and human liver microsomes. The reaction mixture (final volume 100 μL) was pre-incubated for 3 min at 37 °C prior to the addition of NADPH (final, 2 mM). Organic solvent was kept below 1.5%. Each reaction was terminated by acetonitrile (100 μL), centrifuged and the supernatant analyzed by HPLC-UV. A summary of the data and findings are presented (see Table 3).
Table 3.
Inhibition data for compound 1 against human cytochrome P450 isozymes
| Isozyme | Substrate (Metabolite) | Substrate Conc. (μM) | Protein Conc. (mg/mL) | Incubation Time (min) | IC50, μMa (% Inhibition)b |
|---|---|---|---|---|---|
| CYP 3A4 | Testosterone (6-Hydroxytestosterone) |
100 | 0.3 | 30 | >200 (28.0±4.9) |
| CYP 3A4 | Triazolam (4-Hydroxytriazolam) |
200 | 0.4 | 30 | >200 (33.4±4.5) |
| CYP 2D6 | Dextromethorphan (Dextrophan) |
200 | 2.0 | 60 | >200 (9.2±1.3) |
| CYP 2C8 | Amodiaquine (N-Desethylamodiaquine) |
200 | 0.4 | 30 | >200 (9.1±1.2) |
| CYP 2C9 | Tolbutamide (4-Hydroxytolbutamide) |
500 | 0.5 | 30 | >200 (15.5±2.1) |
| CYP2C19 | Omeprazole (5-Hydroxyomeprazole) |
200 | 0.5 | 30 | >200 (21.8±3.0) |
IC50 = concentration for 50% inhibition of the specified CYP isozyme from kinetic data.
The IC50s for isozymes listed above were not reached at 200 μM.
The IC50 for CYP3A4 (testosterone) was extrapolated from the 100 μM data in this study.
Percent inhibition of the isozymes by compound 1. The standard deviation is derived from the mean of three determinations.
2.4.4. UGT substrate activity in human liver microsomes or cDNA-expressed isozymes
Incubation mixtures (final volume of 500 μL) contained human liver microsomes (1.0 mg/mL proteins), compound 1 or raltegravir (50 μM in DMSO, <1% of final mixture), UDPGA (4 mM), alamethicin (0.024 mg/mg protein), D-saccharic acid 1,4-lactone (10 mM) in potassium phosphate buffer (100 mM, pH 7.4) containing MgCl2 (5 mM). Initially, the mixture of human liver microsomes and alamethicin in the buffer containing MgCl2 was kept in ice (0 °C) for 15 min. D-saccharic acid-1,4-lactone and the test compound were then added. This mixture was preincubated at 37 °C for 3 minutes and UDPGA (4 mM) was then added to initiate the reaction. An aliquot (60 μL) was removed each sampling time, quenched with acetonitrile (60 μL), centrifuged and the supernatant analyzed by HPLC and HRMS (see Table 4).
Table 4.
Summary of results for glucuronidation studies of compound 1 and raltegravir
| Compound | Microsome or UGT Isoform | Glucuronide | Reversed-phase HPLC retention time (min), UV detection (nm) | Substrate for UGT? Yes or No |
|---|---|---|---|---|
| 1 | - | - | 18.6 min, 350 nm | |
| 1 | pHLM (Xenotech) | Nonea | - | No |
| 1 | pHLM (BD Biosciences) | Nonea | - | No |
| 1 | UGT1A1 (BD Biosciences) | Nonea | - | No |
| 1 | UGT1A3 (BD Biosciences) | Nonea | - | No |
| Raltegravir | - | - | 30.3 min, 300 nm | |
| Raltegravir | pHLM (Xenotech) | Glucuronide | 16.4 min, 300 nm | Yes |
| Raltegravir | pHLM (BD Biosciences) | Glucuronide | 16.4 min, 300 nm | Yes |
| Raltegravir | UGT1A1 (BD Biosciences) | Glucuronide | 16.4 min, 300 nm | Yes |
| Estradiol | - | - | 27.9 min, 280 nm | |
| Estradiolb | pHLM (BD Biosciences) | Glucuronides (two) | 9.7 min, 11.2 min, 280 nm | Yes |
| Estradiolb | UGT1A1 (BD Biosciences) | Glucuronide | 9.6 min, 280 nm | Yes |
| Methylumbelliferone | - | - | 21.1 min, 318 nm | |
| Methylumbelliferoneb | pHLM (Xenotech) | Glucuronide | 6.1 min, 318 nm | Yes |
| Trifluoperazine | - | - | 19.2, 256 nm | |
| Trifluoperazineb | pHLM (Xenotech) | N-Glucuronide | 18.0 min, 256 nm | Yes |
The glucuronide of compound 1, if formed, would have been observed at a retention time of approximately 8 min. However, no such peak in the HPLC chromatograms using 254 nm, 300 nm or 350 nm detection wavelengths was observed.
Estradiol, 4-methylumbelliferone and trifluoperazine were used as positive control substrates.
2.4.5. UGT inhibition studies (UGT 1A1, 1A6, 1A9, 2B7)
Incubation mixture (total volume 200 μL) used human liver microsomes (0.2 mg/mL microsomal proteins), alamethicin (0.024 mg/mg protein), in potassium phosphate buffer (100 mM, pH 7.4) containing MgCl2 (5 mM), which was kept at 0° C for 15 min. A solution of compound 1 in DMSO (0 to 300 μM) and 4-methylumbelliferone (4-MU, 200 μM, dissolved in methanol) were added (Uchaipichat et al., 2004). The organic solvents in incubations were <1.6 %. The above mixture was preincubated at 37 °C for 3 min, UDPGA (final, 4 mM) was added and the mixture was incubated for 15 min. The reaction was terminated with acetonitrile (200 μL), centrifuged and the supernatant analyzed by HPLC-UV.
2.4.6. UGT inhibition studies (UGT1A4)
These studies were done in a similar manner to the above UGT inhibition study, but with trifluoperazine as substrate (Uchaipichat et al., 2006).
3. Results and Discussion
Compound 1 (Figure 1)was synthesized in8 steps and 25% overall yield from 5-bromo-2-methoxypyridine. Its structure was confirmed by single-crystal X-ray, UV, HRMS, 1H/ 13C NMR data, including gCOSY, HSQC and HMBC correlations. The purity of the compound used in these studies was 99.6%.
The in vitro anti-HIV activity of compound 1 in human PBMC cultures is shown in Table 1. The collective data indicate that this compound has significant activity against a broad and diverse set of HIV-1 subtypes of major group M, as well as against HIV-2 and SIV (mean EC50 35.0 nM). For key Group M subtypes A, B, C and F, the mean EC50 was 18.9 nM. Therapeutic indices varied from 1,119 to 13,962, with the mean being 4,618. Cytotoxicity data (CC50 96,200 nM ±18,600) gave strong evidence that the compound possessed low toxicity in human PBMC cultures. Major group M of HIV-1 and its subtypes are responsible for most HIV infections (Keele et al., 2006).
Table 1.
Anti-HIV activity of compound 1 against HIV-1, HIV-2 and SIV isolates in PBMC
| Isolates | Type, Subtype | EC50 (± SD)a nM | Therapeutic Indexb |
|---|---|---|---|
| HIV-1 (Group M) | |||
| 92RW016 | A | 20.8 (3.3) | 4,625 |
| 91US004 | B | 6.89 (13.4) | 13,962 |
| 98IN022 | C | 29.8 (31.8) | 3,228 |
| 93UG067 | D | 48.5 (23.5) | 1,984 |
| CMU08 | E | 52.4 (1.6) | 1,836 |
| 93BR029 | F | 18.3 (16.1) | 5,257 |
| G3 | G | 34.9 (25.0) | 2,756 |
| CDC310342 | CRF02-AG | 33.4(31.9) | 2,880 |
| HIV-2 | |||
| CDC310319 | HIV-2 | 86.0 (75.7) | 1,119 |
| SIV | |||
| MAC251 | SIV | 19.1(22.4) | 5,037 |
| All of the above isolates (HIV-1, HIV-2 and SIV) | 35.0 (31.4) | ||
| For HIV-1 Group M subtypes A, B, C, F | 18.9 (9.1) |
EC50 = concentration for inhibition of virus replication by 50%. Data are the mean of three determinations.
SD = standard deviation from the mean of three independent determinations.
CC50/EC50 = Therapeutic index.
Docking studies suggest that the mechanism of anti-HIV activity for compound 1 is through inhibition of HIV integrase (Figure 2). This is supported by the fact that compound 1 was discovered in our laboratory from structure-activity studies of closely related prototypes of compound 1 and also of their precursors, which showed IC50 data for integrase strand transfer inhibition at low nM levels (Seo et al., 2011). Further validation of integrase inhibition came from the observed mutation in the integrase coding region of the HIV-1 genome, as well as from the cross-resistance data (discussed below). In addition, the T66I mutation observed for compound 1 has also been observed in a resistant virus isolate of elvitegravir, a well-known integrase inhibitor (Goethals et al., 2008).
Fig. 2.
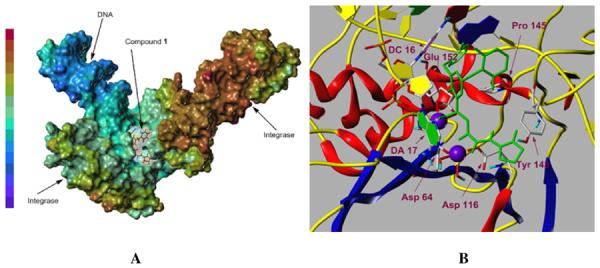
(A) Representation of compound 1 (black) docked within the active site of modeled HIV-1 integrase intasome complex (PDB code 3OYA). The inhibitor is shown inside a cleft in the modeled HIV-1 intasome electrostatic potential map. The red-brown color represents weak local electrostatic potential (most positive) while the blue-purple color symbolizes strong local electrostatic potential (most negative). (B) Docking picture of compound 1 (stick model in green) within the active site of modeled HIV-1 integrase intasome illustrating the interaction of compound 1 with the amino acid residues of integrase (indicated in purple). Stacking interaction of the mono-fluorophenyl ring (green) with cytosine of DC 16 can be easily discerned. The deoxyribose rings are shaded in yellow.
In dose escalation studies employing MT-4 cells infected with HIV-1 NL4-3, the identification of HIV-1 isolates resistant to compound 1 was investigated. The selection of a single amino acid mutation from threonine to isoleucine at amino acid 66 (T66I) of integrase, began to emerge following passage #4 with 600 nM of compound 1 and became a complete change following passage #9 (at 19.2 μM). Continued passaging with 20 μM of 1 (up to passage #15) did not result in the emergence of any additional mutations in integrase. The T66I mutation is in the catalytic core domain of the integrase coding region. In drug susceptibility studies in MT-4 cells, the fold change in the EC50 of compound 1 against resistant viruses with clinically-relevant integrase mutations were compared to raltegravir and elvitegravir. These integrase mutant viruses retained susceptibility to AZT, which was included as the positive control. The results are summarized in Table 2.
Table 2.
Fold change in susceptibility in MT-4 cells against resistant viruses with key clinical mutations in integrase.
| WT virus or IN substitutions of resistant viruses | EC50 fold change compared to wild-type virus |
|||
|---|---|---|---|---|
| Compound 1 | Raltegravir | Elvitegravir | AZT | |
| WT HIV-1 NL4-3 | 1.0 | 1.0 | 1.0 | 1.0 |
| Y143C | 1.87 | 826 | 46.7 | 1.18 |
| N155H | 8.20 | 88.8 | 200 | 3.29 |
| Q148R | 18.8 | 43.3 | 96.1 | NA |
| E92Q/N155H | 6.13 | 85.5 | 134 | 3.45 |
| T66I | 5.20 | 1.0 | 5.5 | 1.0 |
The EC50 against the Wild-Type HIV-1 NL4-3 were 63.5, 6.54, 0.82 and 1.84 nM for compound 1, raltegravir, elvitegravir and AZT, respectively.
A major focus of this investigation was determination of the profile of compound 1 towards key human CYP and UGT isozymes (Dye and Williams, 2010; Tukey and Strassburg, 2000; Wienkers and Heath, 2005; Williams et al., 2004, Miners et al., 2004). The cytochrome P450 (CYP) isozymes used in this study are known to be involved in the clearance mechanisms of about 90% of known therapeutic drugs. As illustrated in Figure 3, compound 1 was relatively stable in pooled human liver microsomes. Two key CYP-mediated metabolites of compound 1 were formed from monooxidation of the phenyl rings and their structures were confirmed by bioanalytical data, including HRMS. CYP isozyme kinetic data revealed that the IC50 for inhibition for compound 1 of CYP isozymes (3A4, 2D6, 2C8, 2C9, 2C19) were all >200 μM (Table 3). In addition, compound 1 was not an activator of these CYP isozymes.
UDP-glucuronosyltransferases (UGTs) are a superfamily of human phase II metabolizing isozymes, which are involved in the glucuronidation and subsequent clearance through bile or urine of a significant number of drugs, including raltegravir (Kassahun et al., 2007). Thus, in this investigation, the UGT profile of compound 1 was considered to be of high significance, both in terms of HIV co-infection therapeutics (Dye and Williams, 2010; Russell et al., 2010), and the issue of utilization of UGT-cleared integrase inhibitors for HIV/AIDS during fetal development and early infancy, given the low UGT activity during this phase (Strassburg et al., 2002).
Glucuronidation studies of compound 1 and, for comparison, raltegravir, were determined in pooled human liver microsomes verified to contain UGT 1A1, 1A4, 1A6, 1A9 and 2B7. Compound 1 was not a substrate for these key UGTs in human liver microsomes or for specific cDNA-expressed UGT isozymes, UGT1A1 and UGT1A3 (Table 4). Furthermore, in the kinetic studies in human liver microsomes, there was no indication of the activation of UGT isozymes. In contrast, raltegravir was a substrate for UGT (Figure 4), which is consistent with previously reported data (Kassahun et al., 2007). We also examined the possible competitive inhibition of UGTs by compound 1 using 4-methylumbelliferone (4-MU), a substrate for multiple isoforms of UGT. However, no evidence for significant competitive inhibition of the key UGT isozymes 1A1, 1A6, 1A9 and 2B7 was found (IC50 >300 μM). In addition, compound 1 was not an inhibitor of another key UGT isozyme, namely UGT1A4.
Fig. 4.
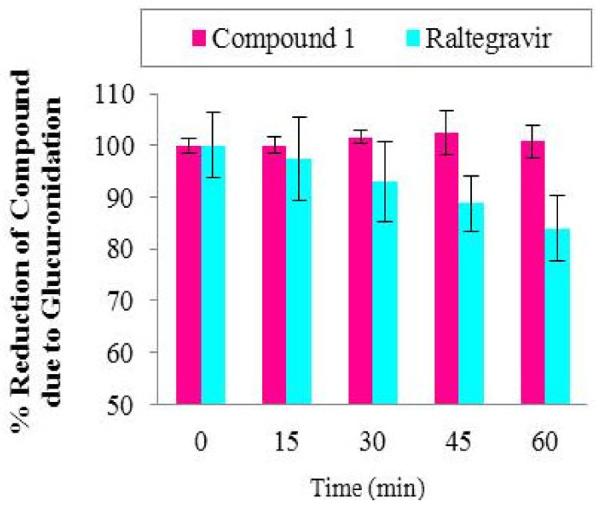
UGT-catalyzed glucuronidation of compound 1 and raltegravir. Reduction in percent of raltegravir is due to glucuronidation, and its glucuronide is easily identified (HPLC retention times: raltegravir, 30.6 min; raltegravir glucuronide, 16.7 min; monitored through UV detection at 300 nm). The glucuronide of 1 was not detected [HPLC retention times: compound 1, 14.1 min; its glucuronide expected at ~ 8 min (monitored at 350 nm, not seen)]. The control consisted of phosphate buffer (pH 7.4) containing MgCl2, alamethicin, D-saccharic acid 1,4-lactone, UDPGA and test compound. Error bars show standard deviations from the mean of 3 independent determinations.
In summary, we have discovered a new HIV integrase inhibitor (1), that exhibits significant antiviral activity against a diverse set of HIV-1 isolates, as well as against HIV-2 and SIV and that displays low in vitro cytotoxicity. It has a favorable resistance and related drug susceptibility profile. Compound 1 is not a substrate for key human UGT isoforms, which is of particular relevance, both in HIV co-infection therapeutics and in HIV treatments during fetal development and early infancy. Finally, the CYP isozyme profile of compound 1 suggests that it is not expected to interfere with normal human CYP-mediated metabolism.
Highlights
We have discovered an integrase inhibitor with in vitro anti-HIV activity.
The compound exhibited low cytotoxicity in peripheral blood mononuclear cells.
Resistance and related fold change in drug susceptibility are addressed.
Data on the CYP and UGT profile of the compound are presented.
Acknowledgements
Support of this research by the National Institutes of Health (R01 AI 43181 and NCRR S10-RR025444) is gratefully acknowledged. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. One of us (VN) also acknowledges support from the Terry Endowment (RR10211184) and from the Georgia Research Alliance Eminent Scholar Award (GN012726). The in vitro anti-HIV data were determined by Southern Research Institute, Frederick, MD, using federal funds from the Division of AIDS, NIAID, NIH, under contract HHSN272200700041C entitled “Confirmatory In Vitro Evaluations of HIV Therapeutics.” We acknowledge the help of Dr. Byung Seo and Dr. Pankaj Singh in the early structure-activity studies. We thank Dr. John Bacsa of Emory University for the X-ray crystal structure data.
4. Glossary
- 4-MU
4-methylumbelliferone
- NADPH
Nicotinamide adenine dinucleotide phosphate reduced form
- UDPGA
Uridine 5'-diphospho-glucuronosyltransferase
- G-6-P
Glucose 6-phosphate
- G-6-PDH
Glucose-6-phosphate dehydrogenase
- NMR
Nuclear Magnetic Resonance
- gCOSY
gradient Correlation Spectroscopy
- HSQC
Heteronuclear Single Quantum Coherence
- HMBC
Heteronuclear Multiple Bond Correlation
- HSQC
Heteronuclear Single Quantum Coherence
- HRMS
High Resolution Mass Spectrometry
- HPLC
High Performance Liquid Chromatography
- UV
Ultraviolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abimiku AG, Stern TL, Zwandor A, Markham PD, Calef C, Kyari S, Saxinger WC, Gallo RC, Robertguroff M, Reitz MS. Subgroup-G HIV type-1 isolates from Nigeria. AIDS Res. Hum. Retroviruses. 1994;10:1581–1583. doi: 10.1089/aid.1994.10.1581. [DOI] [PubMed] [Google Scholar]
- 2.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckheit RW, Swanstrom R. Characterization of an HIV-1 isolate displaying an apparent absence of virion-associated reverse-transcriptase activity. AIDS Res. Hum. Retroviruses. 1991;7:295–303. doi: 10.1089/aid.1991.7.295. [DOI] [PubMed] [Google Scholar]
- 4.Daniel MD, Letvin NL, King NW, Kannagi M, Sehgal PK, Hunt RD, Kanki PJ, Essex M, Desrosiers RC. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science. 1985;228:1201–1204. doi: 10.1126/science.3159089. [DOI] [PubMed] [Google Scholar]
- 5.de Montellano PR, editor. Cytochrome P450: structure, Mechanism, and Biochemistry. third ed. Kluwer Academic/Plenum; New York: 2005. [Google Scholar]
- 6.Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328:856–861. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 7.Frankel AD, Young JAT. HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Gao F, Robertson DL, Carruthers CD, Morrison SG, Jian BX, Chen YL, Barre-Sinoussi F, Girard M, Srinivasan A, Abimiku AG, Shaw GM, Sharp PM, Hahn BH. A comprehensive panel of near-full-length clones and reference sequences for non-subtype B isolates of human immunodeficiency virus type 1. J. Virol. 1998;72:5680–5698. doi: 10.1128/jvi.72.7.5680-5698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao F, Yue L, Craig S, Thornton CL, Robertson DL, Mccutchan FE, Bradac JA, Sharp PM, Hahn BH, Osmanov S, Belsey EM, Heyward W, Esparza J, Galvaocastro B, Vandeperre P, Karita E, Wasi C, Sempala S, Tugume B, Biryahwaho B, Rubsamenwaigmann H, Vonbriesen H, Esser R, Grez M, Holmes H, Newberry A, Ranjbar S, Tomlinson P, Bradac J, Mccutchan F, Louwagie J, Hegerich P, Lopez-Galindez C, Olivares I, Dopazo J, Mullins JI, Delwart EL, Bachmann HM, Goudsmit J, Dewolf F, Saragosti S, Schochetman G, Kalish M, Luo CC, George R, Pau CP, Weber J, Cheingsongpopov R, Kaleebu P, Nara P, Fenyo EM, Albert J, Myers G, Korber B. Genetic-variation of HIV type-1 in 4 world-health-organization-sponsored vaccine evaluation sites - generation of functional envelope (Glycoprotein-160) clones representative of sequence subtype-A, subtype-B, subtype-C, and subtype-E. AIDS Res. Hum. Retroviruses. 1994;10:1359–1368. doi: 10.1089/aid.1994.10.1359. [DOI] [PubMed] [Google Scholar]
- 10.Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, Dockx K, Strijbos R, Smits V, Vos A, Meersseman G, Jochmans D, Vermeire K, Schols D, Hallenberger S, Hertogs K. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 2008;82:10366–10374. doi: 10.1128/JVI.00470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haren L, Ton-Hoang B, Chandler M. Integrating DNA: transposases and retroviral integrases. Annu. Rev. Microbiol. 1999;53:245–281. doi: 10.1146/annurev.micro.53.1.245. [DOI] [PubMed] [Google Scholar]
- 13.Jagodzinski LL, Wiggins DL, McManis JL, Emery S, Overbaugh J, Robb M, Bodrug S, Michael NL. Use of calibrated viral load standards for group m subtypes of human immunodeficiency virus type 1 to assess the performance of viral RNA quantitation tests. J. Clin. Microbiol. 2000;38:1247–1249. doi: 10.1128/jcm.38.3.1247-1249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Josephson F. Drug-drug interactions in the treatment of HIV infection: focus on pharmacokinetic enhancement through CYP3A inhibition. J Intern Med. 2010;268:530–539. doi: 10.1111/j.1365-2796.2010.02301.x. [DOI] [PubMed] [Google Scholar]
- 15.Kassahun K, McIntosh I, Cui D, Hreniuk D, Merschman S, Lasseter K, Azrolan N, Iwamoto M, Wagner JA, Wenning LA. Metabolism and disposition in humans of raltegravir (MK-0518), an Anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 2007;35:1657–1663. doi: 10.1124/dmd.107.016196. [DOI] [PubMed] [Google Scholar]
- 16.Keele BF, Van Heuverswyn F, Li YY, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen YL, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JFY, Sharp PM, Shaw GM, Peeters M, Hahn BH. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiang TKL, Ensom MHH, Chang TKH. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Therapeut. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human-immunodeficiency-virus with a sensitive cell-line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. In vitro antiretroviral properties of S/GSK1349572, a next-generation hiv integrase inhibitor. Antimicrob. Agents Chemother. 2011;55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang XZ, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J. Virol. 2012;86:8472–8481. doi: 10.1128/JVI.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnan L, Engleman A. Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem. 2012;287:40858–40866. doi: 10.1074/jbc.R112.397760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathias AA, West S, Hui J, Kearney BP. Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin. Pharmacol. Ther. 2009;85:64–70. doi: 10.1038/clpt.2008.168. [DOI] [PubMed] [Google Scholar]
- 23.Michael NL, Herman SA, Kwok S, Dreyer K, Wang J, Christopherson C, Spadoro JP, Young KKY, Polonis V, McCutchan FE, Carr J, Mascola JR, Jagodzinsi LL, Robb ML. Development of calibrated viral load standards for group M subtypes of human immunodeficiency virus type 1 and performance of an improved AMPLICOR HIV-1 MONITOR test with isolates of diverse subtypes. J. Clin. Microbiol. 1999;37:2557–2563. doi: 10.1128/jcm.37.8.2557-2563.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min S, Song I, Borland J, Chen SG, Lou Y, Fujiwara T, Piscitelli SC. Pharmacokinetics and safety of S/GSK1349572, a next-generation hiv integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010;54:254–258. doi: 10.1128/AAC.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miners JO, Smith PA, Sorich MJ, McKinnon RA, Mackenzie PI. Predicting human drug glucuronidation parameters: application of in vitro and in silico modeling approaches. Annu. Rev. Pharmacol. 2004;44:1–25. doi: 10.1146/annurev.pharmtox.44.101802.121546. [DOI] [PubMed] [Google Scholar]
- 26.Nair V, Chi G, Ptak R, Neamati N. HIV integrase inhibitors with nucleobase scaffolds: discovery of a highly potent anti-HIV agent. J. Med. Chem. 2006;49:445–447. doi: 10.1021/jm0508890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nair V, Chi G. HIV integrase inhibitors as therapeutic agents in AIDS. Rev. Med. Virol. 2007;17:277–295. doi: 10.1002/rmv.539. [DOI] [PubMed] [Google Scholar]
- 28.Owen SM, Ellenberger D, Rayfield M, Wiktor S, Michel P, Grieco MH, Gao F, Hahn BH, Lal RB. Genetically divergent strains of human immunodeficiency virus type 2 use multiple coreceptors for viral entry. J. Virol. 1998;72:5425–5432. doi: 10.1128/jvi.72.7.5425-5432.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 30.Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, Bobardt MD, de Bethune MP, Neyts J, De Clercq E, Dumont JM, Scalfaro P, Besseghir K, Wenger RM, Rosenwirth B. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob. Agents Chemother. 2008;52:1302–1317. doi: 10.1128/AAC.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ptak RG, Gentry BG, Hartman TL, Watson KM, Osterling MC, Buckheit RW, Townsend LB, Drach JC. Inhibition of human immunodeficiency virus type 1 by triciribine involves the accessory protein nef. Antimicrob. Agents Chemother. 2010;54:1512–1519. doi: 10.1128/AAC.01443-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reuman EC, Bachmann MH, Varghese V, Fessel WJ, Shafer RW. Panel of prototypical raltegravir-resistant infectious molecular clones in a novel integrase-deleted cloning vector. Antimicrob. Agents Chemother. 2010;54:934–936. doi: 10.1128/AAC.01345-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell DG, Barry CE, Flynn JL. Tuberculosis: what we don't know can, and does, hurt us. Science. 2010;328:852–856. doi: 10.1126/science.1184784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo BI, Uchil VR, Okello M, Mishra S, Ma XH, Nishonov M, Shu QN, Chi GC, Nair V. Discovery of a potent hiv integrase inhibitor that leads to a prodrug with significant anti-HIV activity. ACS Med. Chem. Lett. 2011;2:877–881. doi: 10.1021/ml2001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. Broad Antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137) J. Virol. 2008;82:764–774. doi: 10.1128/JVI.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith SD, Shatsky M, Cohen PS, Warnke R, Link MP, Glader BE. Monoclonal-antibody and enzymatic profiles of human-malignant lymphoid-T cells and derived cell-lines. Cancer Res. 1984;44:5657–5660. [PubMed] [Google Scholar]
- 37.Strassburg CP, Strassburg A, Kneip S, Barut A, Tukey RH, Rodeck B, Manns MP. Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut. 2002;50:259–265. doi: 10.1136/gut.50.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Paz OG, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 39.Taktakishvili M, Neamati N, Pommier Y, Pal S, Nair V. Recognition and inhibition of HIV integrase by novel dinucleotides. J. Am. Chem. Soc. 2000;122:5671–5677. doi: 10.1016/s0960-894x(99)00677-0. [DOI] [PubMed] [Google Scholar]
- 40.Trono D, Van Lint C, Rouzioux C, Verdin E, Barre-Sinoussi F, Chun TW, Chomont N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science. 2010;329:174–180. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- 41.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 42.Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2006;34:449–456. doi: 10.1124/dmd.105.007369. [DOI] [PubMed] [Google Scholar]
- 43.Uchaipichat V, Mackenzie PI, Guo XH, Gardner-Stephen D, Galetin A, Houston JB, Miners JO. Human UDP-glucuronosyltransferases: isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab. Dispos. 2004;32:413–423. doi: 10.1124/dmd.32.4.413. [DOI] [PubMed] [Google Scholar]
- 44.Vahey M, Nau ME, Barrick S, Cooley JD, Sawyer R, Sleeker AA, Vickerman P, Bloor S, Larder B, Michael NL, Wegner SA. Performance of the Affymetrix GeneChip HIV PRT 440 platform for antiretroviral drug resistance genotyping of human immunodeficiency virus type 1 clades and viral isolates with length polymorphisms. J. Clin. Microbiol. 1999;37:2533–2537. doi: 10.1128/jcm.37.8.2533-2537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005;4:825–833. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 46.Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUC(i)/AUC) ratios. Drug Metab. Dispos. 2004;32:1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]