

Abstract
This study explores the possible use of reactive oxygen-activated DNA modifying agents against acute myeloid leukemia (AML). A key amine on the lead agent was investigated via cytotoxicity assays and was found necessary for potency. The two best compounds were screened via the NCI-60 cell panel. These two compounds had potency between 200 and 800 nM against many of the leukemia cancer cell types. Subsequent experiments explored activity against a transformed AML model that mimics the molecular signatures identified in primary AML patient samples. A lead compound had an IC50 of 760 nM against this AML cell line as well as a therapeutic index of 7.7 ± 3 between the transformed AML model cell line and non-cancerous human CD34+ blood stem/progenitor cells (UCB). The selectivity was much greater than the mainstays of AML treatment: doxorubicin and cytarabine. This manuscript demonstrates that this novel type of agent may be useful against AML.
Keywords: DNA-modifying agent, acute myeloid leukemia, 2′-deoxyguanosine, Reactive Oxygen Species, Structure-Activity
Anticancer agents that exert their mechanism of action through DNA modification form a backbone of many anticancer therapies. Leukemia is often treated with nucleoside analogs and/or DNA intercalating agents.1,2 For example, acute myeloid leukemia (AML) is treated with combination chemotherapy involving cytarabine, a nucleoside analog, and doxorubicin, a DNA intercalator.1 Among the various types of leukemia, AML has the poorest prognosis, with a 5-year survival rate of 5% to 60% depending on prognostic factors.3 Thus, there is a need for new, efficacious therapeutic agents with high selectivity for AML. Previously, we described novel agents that are activated by reactive oxygen species (ROS) to become DNA-modifying agents.4,5 Given the use of DNA-modifying agents in leukemia, we sought to determine if these novel agents could be useful against a leukemia with high levels of ROS.6
Elevated levels of ROS are thought to play a vital role in AML. Excess ROS benefits malignant cells by upregulating redox-regulated growth and survival factors.7–9 Importantly, examples of genetic changes that confer an elevated ROS phenotype are plentiful within AML. The internal tandem duplication of the FLT3 gene, a common AML genotype, leads to increased ROS.10,11 In addition, permanent activation of RAS oncogenes causes the overexpression of proteins that promote cancer cell proliferation as well as lower the concentration of cellular antioxidant enzymes, thereby increasing ROS.12–14 Because of the strong relationship between ROS and AML, we hypothesized that ROS-activated agents would be useful as treatment against AML.
In this manuscript we utilize a recently developed cellular model of AML whereby we express the leukemia-associated oncogene MLL-AF9 in human blood stem/progenitor cells and use these cells in vitro and in animal models to study the mechanisms associated with transformation.15 The molecular signatures associated with these AML cells closely mimic those identified in primary AML patient samples.15 Thus, potency experiments between these novel models and blood stem cells accurately reflect relative specificity of our ROS-activated DNA modifying agents.
Chemists have begun to recognize elevated ROS in cancer cells as a therapeutic design strategy.16,17 We should note that ROS-associated agents have been used for many years, as both arsenic trioxide and doxorubicin generate ROS as part of their mechanisms of action.18–20 Initial strategies to utilize elevated ROS in cancer cells focused on the inactivation of glutathione.21 Newer ROS-associated approaches use pro-drugs that possess a hydrogen peroxide sensitive boronic ester or agents that release toxic metabolites.22–24 In the approach described here, the agents were designed to have a unique activation mechanism, which requires ROS (Figure 1). These agents induce a large and bulky phenol lesion which requires DNA repair for cellular survival as part of their mechanism of action.5
Figure 1.

The general structure of the molecules synthesized and examined. In the presence of ROS, a potent electrophile is generated. These compounds transfer a phenol (grey) to DNA to form a cytotoxic hydroxy-N2,3-benzetheno-2′-deoxyguanosine adduct.
Here, we describe the evaluation of several ROS-activated DNA-modifying agents. The agents were initially screened against HeLa cells, which revealed the importance of a tethered aniline. Next, we investigated the potency of two lead compounds against a panel of 60 cell lines through the National Cancer Institute as well as on human AML cells. We found that one of the ROS-activated DNA-modifying agents had an IC50 of 760 nM against AML cells compared with an eight-fold lower potency against normal CD34+ blood stem/progenitor cells. The selectivity of this agent was greater than the DNA alkylating agents cisplatin and chlorambucil as well as doxorubicin and cytarabine (ara-C), two agents which are mainstays of AML treatment.
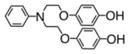
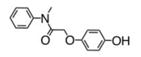
We previously published the structures of two novel ROS-activated DNA-modifying agents, compound 3 and 5.4,5 We systematically modified the agents in order to assess the role of the amine (Table 1). Synthesis of an aniline derivative which lacked an N-alkyl group, compound 1, was accomplished. Compared to previously published 3, in which the amine was an N-ethylaniline, 1 had a modest reduction in cytotoxicity, down to 30 ± 6 μM from 23 ± 5 μM. Potency was restored to 18 ± 3 μM when the agent possessed an N-methylaniline, as seen in compound 2. Replacement of the methyl with a much more bulky, isopropyl group at the aniline nitrogen, as in 4, resulted in a 3-fold decrease in potency relative to compound 2. The IC50 of 4 was 53 ± 7 μM. When a second, reactive hydroquinone moiety was added to the other alkyl position, as in 5, potency doubled relative to compound 3. We further investigated the effect of the carbon chain between the amine and the hydroquinone ether, based on both 2 and 3. In 6, a compound containing an N-ethylaniline and a three-carbon chain, the IC50 value was 14 ± 3 μM. The N-methylaniline with a three-carbon chain, 7, led to a change in IC50 to 9 ± 2 μM. Both 6 and 7 indicated that increasing the carbon chain length was favorable. The chain was further lengthened to four carbons for 8 and five carbons for 9, and IC50 values changed to 12 ± 3 and 13 ± 2 μM, respectively. Our results indicated a linker of three to five carbons yielded potent compounds. When we investigated the aniline ring system, we found that conversion to a benzylamine, 10, decreased potency to 33 ± 3 μM from 18 ± 3 μM. Furthermore, when the aniline amine was changed to an amide, as in compound 11, anticancer cell activity was eliminated. When the aniline was completely removed, 12, we found no potency, indicating that the presence of the amine is required. Therefore, potency was strongly altered by direct inactivation of the amine. Additional negative controls were synthesized. In compound 13, exchanging the hydroquinone ether to a phenol ether led to the complete loss of potency. Compound 13 is not oxidizeable, which infers that anticancer activity requires oxidation. Finally, compound 14, which lacked a hydroquinone moiety entirely, demonstrated loss of potency. Compound 14 is a traditional type of DNA alkylating agent, which infers that ROS-activated agents utilize a new and innovative mechanism of action, which requires an amine for potency.
Table 1.
Structures of Novel Agents and IC50 Values in HeLa Cells.
| Compound | Name | IC50 |
|---|---|---|

|
1 | 30 +/− 6 |

|
2 | 18 +/− 3 |

|
3 | 23 +/− 5 |

|
4 | 53 +/− 7 |
|
5 | 11 +/− 2 |

|
6 | 14 +/− 3 |

|
7 | 9 +/− 2 |

|
8 | 12 +/− 3 |

|
9 | 13 +/− 3 |

|
10 | 33 +/− 3 |
|
11 | >125 |
|
|
12 | >125 |

|
13 | >125 |

|
14 | >125 |
IC50 values, in μM, are the mean of six replicates from two independent experiments. All values normalized to 3.
In our previous work, a limited number of cell lines were assessed via MTT cell viability assay.4 The relationship was complex with some cell lines being affected much more than others. We, therefore, decided to assess compound toxicity on a larger panel of cells. Compounds 5 and 7 were evaluated for effects on the viability of the cell lines in the NCI-60 cell panel (Table 2). The NCI panel screen has a few changes compared to an MTT assay. First, viability is monitored using a Sulforhodamine B assay. The Sulforhodamine B assay is used to measures total protein content, and therefore, is a measure of cell number rather than metabolic activity.25 We found little difference in the measured values via the two methods. The second difference is that the data is reported as GI50 rather than IC50 to reflect the change in cell counts relative to the starting number of cells.
Table 2.
Potency of Compounds 5 and 7 Against the NCI-60 panel. [*] Indicates line not tested.

|
Compounds 5 and 7 showed selective potency against particular cell lines in the sixty-cell panel. The median GI50 for 5 was 3.9 μM and 3.0 μM for 7 (Table 2). Importantly, the distribution of GI50 values was large, with a range of 55-fold for 5 and 53-fold for 7. This data indicates that both 5 and 7 demonstrate high potency against several cell types, while some cancer cells were less targeted. We further analyzed the growth inhibition pattern induced by 5 and 7 based on the type of cancer. Table 2 shows all GI50 values among the various cancer types relative to the median. The majority of leukemia and renal cancer cell lines were above the median, indicating enhanced sensitivity to ROS-activating compounds. In previous work, we had identified renal carcinoma as a potential target, and the current results further substantiate this finding.5 We analyzed the leukemia cancer cell lines to determine if leukemia cells were attractive targets for ROS-activated DNA-modifying agents. Leukemia cells demonstrated the highest median potency, with all cell lines examined having GI50 values below the median for 5 and five of the six below the median for 7. Compound 5 had a median GI50 of 2.1 μM in all the leukemia cell lines tested, whereas compound 7 had a median GI50 value of 1.9 μM. A model for acute lymphoblastic leukemia (ALL), SR-91, had GI50 values of 470 nM for compound 5, and 2.5 μM for compound 7. Another ALL model line, CCRF-CEM, had GI50 values of 263 nM for 7 and 2.8 μM for 5. The final ALL model line tested, MOLT-4, had GI50 values of 2.0 μM for compound 7 and 1.3 μM for compound 5. In RPMI-8226, a plasmacytoma and myeloma cell line, 7 had a GI50 value of 1.2 μM and 5 gave a GI50 value of 3.8 μM. HL-60, a model of acute promyelocytic leukemia, had a GI50 value of 900 nM for compound 7, but was not tested for compound 5. Finally, K-562, a model for myelogenous leukemia, had GI50 values corresponding to 4.3 μM and 2.1 μM for compounds 7 and 5, respectively.
We then focused on AML since it is the leukemia with the poorest prognosis. To fully assess the anticancer activity against AML, we were interested in comparing transformed cells to closely related, yet non-transformed counterparts. In the case of AML, the generally accepted non-transformed cell of origin is the human CD34+ blood stem/progenitor cell.26 Assessing potency against normal blood stem cells is important since, without proper selectivity, a cytotoxic agent will yield reduced efficacy and therapeutic potential. One major limitation to current treatment is that many anticancer agents do not selectively inhibit growth or cause cytotoxicity to cancer cells relative to healthy cells. Therefore, a comparison of current agents and our ROS-activated chemotherapeutics was necessary. Accordingly, we measured cytotoxicity of our compounds on CD34+ blood stem/progenitor cells and on the MA9.3 AML cell line, which was derived from the transformation of human CD34+ cells by introduction of the leukemia oncogene MLL-AF9.15
Therapeutic indices for ROS-activated DNA-modifying agents and known anticancer agents were determined (Figure 2). The foundations of AML treatment are the anticancer agents doxorubicin and ara-C. Doxorubicin and ara-C showed strong potency against MA9.3 AML cells with IC50 values of 86 ± 5 nM and 0.92 ± 0.03 μM respectively; these agents had IC50 values against CD34+ blood stem/progenitor cells of 76 ± 18 nM and 0.59 ± 0.09 μM. These agents had very limited selectivity. The therapeutic index for doxorubicin was 0.9 ± 0.2 and the index for ara-C was 0.6 ± 0.1. The negative index for ara-C was statistically significant but small. Thus, these two agents showed low selectivity. We then determined if other commercially available DNA-modifying agents were selective, as a base of comparison for our ROS-activated DNA-modifying agents. We chose cisplatin for its high therapeutic use and chlorambucil for its utilization in chronic lymphocytic leukemia. The IC50 value for cisplatin was 14 ± 0.7 μM and for chlorambucil was 8.7 ± 0.3 μM against MA9.3 AML cells. Against CD34+ blood stem/progenitor cells, the former had an IC50 value of 15 ± 2 μM, while the latter had an IC50 value of 11 ± 1 μM. The therapeutic index for cisplatin was 1.0 ± 0.2, while the index for chlorambucil was 1.3 ± 0.1. Chlorambucil did, therefore, show some selectivity, although slight. Our ROS-activated DNA-modifying agents were investigated; Compound 5 had an IC50 value of 3.0 ± 0.2 μM and 7 had an IC50 value of 0.7 ± 0.2 μM against MA9.3 cells. The IC50 values against normal blood stem cells were 16 ± 1 μM and 5.4 ± 0.9 μM for 5 and 7, correspondingly. The therapeutic indices were 5.3 ± 0.5 and 7.7 ± 3 for 5 and 7, respectively. Consequently, ROS-activated DNA-modifying agents have favorable therapeutic indices and statistically significant selectivity against AML cancer cells.
Figure 2.

Evaluation of Therapeutic Indices. Cytotoxicity of two ROS-activated agents and several common anticancer agents were investigated in MA9.3 AML cancer cells (black lines) and human CD34+ stem/progenitor cell (grey lines). (A) Evaluation of agents 5 and 7. (B) Evaluation of doxorubicin, cytosine arabinoside (ara-C), cisplatin, and chlorambucil (CLB).
The systematic syntheses in this manuscript have helped to uncover important factors for potency of our ROS-activated DNA-modifying agents. We have found that a tethered amine is required. First, extreme changes like removal of the nucleophile, conversion to an amide, or steric blocking severely reduced potency. This manuscript identified a new lead compound, 7, which has many favorable anticancer agent properties such as a high therapeutic index and potency in the nM range against several leukemia cell lines.
The ROS-activated DNA-modifying agents we investigated were selective for AML over normal CD34+ blood stem/progenitor cells. We should note than when testing these agents on normal blood CD34+ cells, we found that 5 and 7 had a potency of 16 ± 1 μM and 5.4 ± 0.9 μM, respectively. Thus, many cancer cell lines were selectively targeted to some extent. The most potent compound, 7, had a therapeutic index of 7.7 ± 3 for the MA9.3 AML cell line. The NCI panel allows for analysis by COMPARE, which is an algorithm to correlate the cytotoxic profile with that of other agents.27 Interestingly, no significant correlation to known anti-cancer agents is observed for both 5 and 7. Therefore, ROS-activated DNA-modifying agents are a novel class of chemotherapeutics with distinct cellular mechanisms. In addition, this manuscript highlights AML with its poor prognosis, are natural targets for ROS-activated DNA-modifying agents as well as for the several other medicinal chemistry teams that are pursuing ROS-based agents.16–23.
Supplementary Material
Acknowledgments
Acknowledgments should be inserted at the end of the paper, before the references, not as a footnote to the title. Use the unnumbered Acknowledgements Head style for the Acknowledgments heading.
Footnotes
Synthesis and materials and methods. This material is available via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, Mulloy JC. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chabner BA, Roberts TG. Nat Rev Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, Cho H, Scoppa S, Hachey M, Kirch R, Jemal A, Ward ECA. Cancer J Clin. 2012;62:220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 4.Li GR, Bell T, Merino EJ. ChemMedChem. 2011;6:869–875. doi: 10.1002/cmdc.201100014. [DOI] [PubMed] [Google Scholar]
- 5.Jones AR, Bell-Horwath TR, Li G, Rollmann SR, Merino EJ. Chem Res Toxicol. 2012;25:2542–2552. doi: 10.1021/tx300337j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trachootham D, Alexandre J, Huang P. Nat Rev Drug Discovery. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 7.Hole PS, Darley RL, Tonks A. Blood. 2011;117:5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 8.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Solivio MJ, Nemera DB, Sallans L, Merino EJ. Chem Res Toxicol. 2012;25:326–336. doi: 10.1021/tx200376e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woolley JF, Naughton R, Stanicka J, Gough DR, Bhatt L, Dickinson BC, Chang CJ, Cotter TG. Plos One. 2012;7:34050. doi: 10.1371/journal.pone.0034050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sano H, Shimada A, Taki T, Murata C, Park MJ, Sotomatsu M, Tabuchi K, Tawa A, Kobayashi R, Horibe K, Tsuchida M, Hanada R, Tsukimoto I, Hayashi Y. Int J Hematol. 2012;95:509–515. doi: 10.1007/s12185-012-1033-x. [DOI] [PubMed] [Google Scholar]
- 13.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GRS, Chandel NS. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, Chiao PJ, Keating MJ, Garcia-Manero G, Huang P. Cell Res. 2012;22:399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, Zheng Y, Cancelas JA, Gu Y, Jansen M, DiMartino JF, Mulloy JC. Can Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng X, Gandhi V. Ther Deliv. 2012;3:823–33. doi: 10.4155/tde.12.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montero AJ, Jassem J. Drugs. 2011;71:1385–96. doi: 10.2165/11592590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Muller I, Niethammer D, Bruchelt G. Int J Mol Med. 1998;1:491–494. doi: 10.3892/ijmm.1.2.491. [DOI] [PubMed] [Google Scholar]
- 19.Kitchin KT, Conolly R. Chem Res Toxicol. 2010;23:327–335. doi: 10.1021/tx900343d. [DOI] [PubMed] [Google Scholar]
- 20.Alp O, Zhang YF, Merino EJ, Caruso JA. Metallomics. 2011;3:482–490. doi: 10.1039/c0mt00110d. [DOI] [PubMed] [Google Scholar]
- 21.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu JS, Huang P. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Major Jourden JL, Cohen SM. Angew Chem Int Ed Eng. 2010;49:6795–7. doi: 10.1002/anie.201003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuang Y, Baakrishnan K, Gandhi V, Peng X. J Amer Chem Soc. 2011;133:19278–19281. doi: 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagen H, Marzenell P, Jentzsch E, Wenz F, Veldwijk MR, Mokhir A. Aminoferrocene-Based Prodrugs Activated by Reactive Oxygen Species. J Med Chem. 2012;55:924–934. doi: 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- 25.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A, et al. J Natl Cancer Inst. 1991;83:757–66. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 26.Becker MW, Jordan CT. Blood Rev. 2011;25:75–81. doi: 10.1016/j.blre.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Hodes L, Paull K, Koutsoukos A, Rubinstein L. J Biopharm Stat. 1992;2:31–48. doi: 10.1080/10543409208835029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.