

The k-turn is a widespread structural motif that introduces a tight kink into the helical axis of double-stranded RNA. The adenine bases of consecutive G•A pairs are directed toward the minor groove of the opposing helix, hydrogen bonding in a typical A-minor interaction. The authors show here that the available structures of k-turns divide into two classes, depending on whether N3 or N1 of the adenine at the 2b position accepts a hydrogen bond from the O2′ at the −1n position. There is a coordinated structural change involving a number of hydrogen bonds between the two classes.
Keywords: k-turn, RNA structure, RNA-protein interaction, SAM-I riboswitch
Abstract
The k-turn is a widespread structural motif that introduces a tight kink into the helical axis of double-stranded RNA. The adenine bases of consecutive G•A pairs are directed toward the minor groove of the opposing helix, hydrogen bonding in a typical A-minor interaction. We show here that the available structures of k-turns divide into two classes, depending on whether N3 or N1 of the adenine at the 2b position accepts a hydrogen bond from the O2′ at the −1n position. There is a coordinated structural change involving a number of hydrogen bonds between the two classes. We show here that Kt-7 can adopt either the N3 or N1 structures depending on environment. While it has the N1 structure in the ribosome, on engineering it into the SAM-I riboswitch, it changes to the N3 structure, resulting in a significant alteration in the trajectory of the helical arms.
INTRODUCTION
The kink turn (k-turn) is an important and abundant structural motif in structured RNA. The basic k-turn motif comprises a double-stranded region with a three-base bulge followed on the 3′ side by a G•A and then A•G pair and frequently another non-Watson-Crick base pair (Fig. 1). The helix containing the G•A pairs is termed NC, while that 5′ to the bulge is called the C helix. The k-turn introduces a sharp kink between the axes of the C and NC helices, with an included angle of ∼60°.
FIGURE 1.
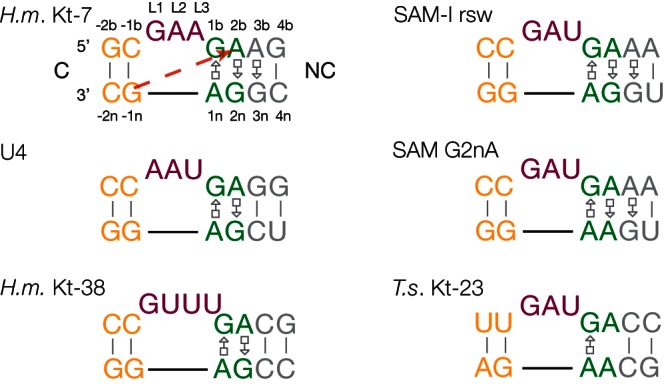
Sequence and secondary structures of the k-turn motifs analyzed in this work. The sequences of the k-turn structures are presented in our standard coloring, where the C helix (C) is shown yellow, the NC helix (NC) gray, the key G•A pairs green, and the loop magenta. The nomenclature of the nucleotide positions (Liu and Lilley 2007) are shown on the Haloarcula marismortui (H.m.) Kt-7 (Ban et al. 2000) sequence. The interaction between the O2′ at the −1n position and the nucleobase of A2b is indicated on this sequence by the broken red arrow. The other k-turns shown are those of U4 snRNA (Vidovic et al. 2000), H. marismortui Kt-38 (Ban et al. 2000), the SAM-I riboswitch (Schroeder et al. 2011) and its G2nA variant, and Thelohania solenopsae (T.s.) Kt-23 (Schroeder et al. 2012).
First found as a repeated motif in the large ribosomal subunit (Klein et al. 2001), k-turns are found in many functional RNA species and RNA-protein complexes, including both ribosomal subunits, in nucleolar box C/D and H/ACA guide RNA complexes (Hamma and Ferré-D’Amaré 2004; Moore et al. 2004), spliceosomal RNA U4 (Vidovic et al. 2000; Wozniak et al. 2005), and in untranslated regions of mRNA (Mao et al. 1999; White et al. 2004), including a number of riboswitches (Montange and Batey 2006; Blouin and Lafontaine 2007; Heppell and Lafontaine 2008; Smith et al. 2009). The sharp kink of the k-turn can create a functionally important change in the trajectory of a helix to facilitate tertiary contacts; a good example can be seen in the SAM-I riboswitch (Montange and Batey 2006). Many k-turns also serve as protein binding sites. We have created a web-based database of k-turn sequences and structures that is available at http://www.dundee.ac.uk/biocentre/nasg/kturn/ (Schroeder et al. 2010).
The k-turn can be induced to fold into the tightly kinked conformation in three different ways. First, k-turns with sequences that conform well to the consensus can fold in isolated duplexes upon addition of metal ions (Matsumura et al. 2003; Goody et al. 2004). Second, the binding of proteins such as members of the L7Ae family can induce the formation of the kinked conformation (Turner et al. 2005). Third, we have recently found that tertiary interactions within larger RNA species can stabilize k-turn structure in sequences that are unable to fold as an isolated duplex structure (Schroeder et al. 2011). In large RNA-protein structures such as the ribosome, it is likely that all three mechanisms will operate to stabilize k-turn structure in a coordinated way. Moreover, it is conceivable that the k-turn structure could be molded to some degree by its environment.
The folded structure of the k-turn is stabilized by a number of well-conserved hydrogen bonding interactions. The minor groove edges of the adenine nucleobases of the two G•A pairs are directed into the minor groove of the C helix, forming a standard A-minor interaction (Nissen et al. 2001). A series of hydrogen bonds involving 2′-hydroxyl groups lace up the structure (Liu and Lilley 2007). These include two donated by O2′ groups within the loop that are strongly conserved throughout the known k-turn structures; these are L1 O2′ to A1n N1 (by definition this position is adenine in the standard k-turn) and L3 O2′ to the proS nonbridging O of the L1/L2 phosphate. Some interactions are more variable, depending on the sequence of the individual k-turn. For example, where the second nucleotide of the bulge (L2) is adenine (e.g., Kt-7), an additional hydrogen bond between AL2 N6 and A2n O2′ is frequently observed.
Another group of hydrogen bonds connects the backbones of the C and NC helices. The O2′ of the nucleotide at the −1n position is normally hydrogen bonded to the nucleobase of the conserved adenine at position 2b, but the acceptor can be either N3 or N1, and in this work, we show that this difference divides the known k-turn structures into two groups. The alternative orientations of the 2b nucleobase between the two classes of structure have a consequence for the nature of the pairing with the 2n nucleobase. In the standard k-turn, 2b is guanine, in principle forming a trans Hoogsteen (A)-sugar edge (G) pair. However, we note that the distance between A2b N6 and G2n N3 is too long to be a stable hydrogen bond in k-turns of the N1 class.
This raises the question of what factors determine whether a given k-turn will adopt the N3 or N1 structure and whether a given sequence can exist in either conformation depending on its environment or protein binding. We have approached this question using the near-consensus k-turn Kt-7. In the 50S ribosomal subunit of Haloarcula marismortui, this k-turn adopts the N1 conformation, although we had previously noted that the folding of Kt-7 upon removal of selected 2′-hydroxyl groups was not completely explicable in terms of its structure in the ribosome (Liu and Lilley 2007). In this work, we have transferred the H. marismortui Kt-7 sequence into the SAM-I riboswitch, finding that, in the new context, it now adopts the N3 structure. This shows that k-turn structure can be plastic to some degree, switching between conformations depending on its environment. The change can have important longer-range structural consequences, leading to significant alteration in the relative disposition of the C and NC helices.
RESULTS
k-turns fall into two structural classes
The core of the k-turn structure forms when the minor grooves of the NC and C helices are juxtaposed, whereby the minor groove edges of the adenine nucleobases of the two G•A pairs are directed into the minor groove of the C helix. Within this region, the O2′ of the −1n ribose donates a hydrogen bond to the nucleobase of the conserved A2b in all k-turns. However, comparison of the crystal structures of a number of different k-turns shows that these interactions fall into two classes that differ in the hydrogen bond acceptor (Table 1). In one class, the acceptor in A2b is N3 (Fig. 2A,C); we term this the N3 class. This is exemplified by the k-turns of box C/D snoRNA (Moore et al. 2004), U4 snRNA (Vidovic et al. 2000), the c-diGMP riboswitch (Smith et al. 2009), and the SAM-I riboswitch, both as free RNA (Montange and Batey 2006) and protein-bound RNA (Baird et al. 2012). In this group, the mean O-N length is 2.90 ± 0.24 Å. In two members of the N3 class (box C/D and U4), −1n O2′ is also hydrogen bonded to A2b O2′. In the second class, the acceptor of the −1n O2′ proton is A2b N1 (Fig. 2B,D). These form the N1 class and include H. marismortui Kt-7 and Kt-38 (Ban et al. 2000). The mean O-N length in the N1 class is 2.75 ± 0.05 Å. In three members of the N1 class (H. marismortui Kt-7, Thermus thermophilus Kt-23 [Wimberly et al. 2000], and L30e-mRNA [Chao and Williamson 2004]), there is a corresponding O2′ to O2′ hydrogen bond between the backbones that is translated 1 bp further from the core on the C and NC helices, i.e., between the −2n and 3b riboses, but this interaction is not universal in all the N1 class k-turns.
TABLE 1.
Inter-atomic distances (Å) for potential hydrogen bonding interactions between the C and NC helices of k-turns
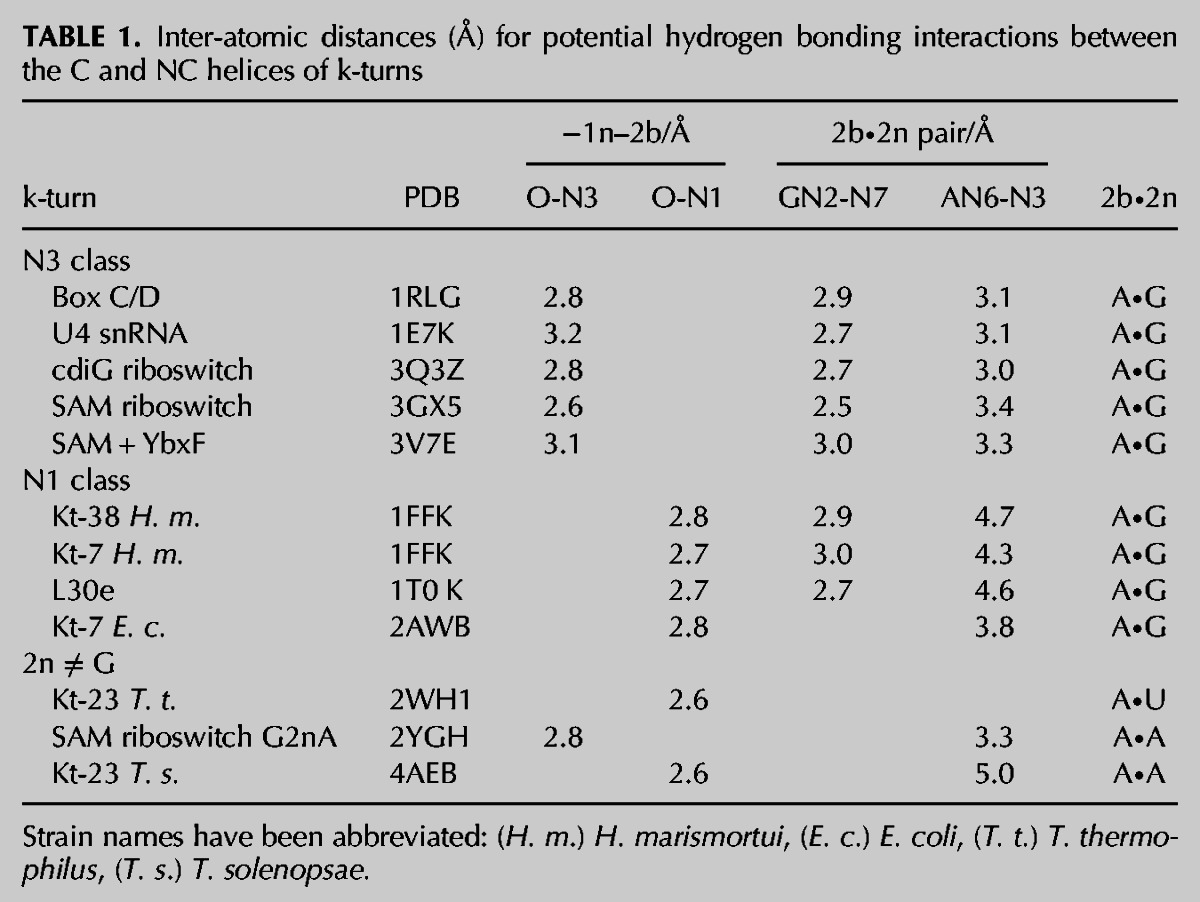
FIGURE 2.
Two structural classes of k-turn. (A,B) Parallel-eye stereo images of two representative k-turns viewed from the side of the nonbulged strand, with G2n, A2b, and A−1n highlighted using a wider bond radius. The U4 snRNA k-turn (Vidovic et al. 2000) (A) is a member of the N3 class of k-turns, while H. marismortui Kt-38 (Ban et al. 2000) (B) is a member of the N1 class. (C–G) Relative disposition and interactions of nucleotides 2n, 2b, and −1n taken from the k-turns indicated below. Hydrogen bonds are indicated by broken lines and the distances (in Å) indicated. Distances too long to be considered hydrogen bonded are indicated in red. (C,D) U4 and Kt-38 k-turn structures, respectively. (E) Kt-23 of T. thermophilus. This has a uridine at the 2n position. (F,G) Two k-turns in which adenine replaces the normal guanine at the 2n position. F is taken from the structure of the SAM-I riboswitch k-turn with a G2nA substitution (Schroeder et al. 2011). G is taken from the structure of the SAM-I riboswitch where the normal k-turn has been replaced by Kt-23 of T. solenopsae (Schroeder et al. 2012).
Hydrogen bonding to N3 or N1 obviously requires different orientations of the A2b nucleobase, and this has ramifications for the pairing with the nucleobase at the 2n position. There is a structural correlation between the hydrogen bonding observed in the 2b•2n pair and membership of the N3 or N1 class. Although the bond donated by GN2 to AN7 is always present, that from AN6 to GN3 is variable between k-turns. In the N3 class, this bond is present (mean N–N distance 3.18 ± 0.16 Å), but in the N1 class, the distance is too long to indicate a stable hydrogen bond (mean N–N distance 4.35 ± 0.16 Å) (Fig. 2C,D; Table 1). Rotation of the nucleobase to direct A2b N1 toward the −1n O2′ increases the AN6 to GN3 length. In contrast, hydrogen bonding to A2n N3 is compatible with the AN6 to GN3 hydrogen bond. Thus, there is a coordinated structural change involving a number of hydrogen bonds between the N3 and N1 classes of k-turn.
The classification extends to k-turns without guanine at the 2n position
This classification is not restricted to k-turns with A•G at the 2b•2n position. In some k-turns, well-exemplified by Kt-23 (Schroeder and Lilley 2009; Schroeder et al. 2012), the G normally found at the 2n position is often replaced by a different nucleotide. However, the adenine at the 2b position is usually retained, and it makes equivalent A-minor interactions with the C helix. T. thermophilus Kt-23 has a uridine at the 2n position, with the A2b oriented so that its N1 accepts a hydrogen bond from O2′ of G − 1n (Table 1); it can thus be classed as an N1 k-turn. A2b forms two hydrogen bonds to U2n, donating the proton of N6 to U2n O2, and accepting at N7 from U2n N3 (Fig. 2E). It is interesting to examine k-turns where the 2n position is adenine, creating a potential A•A pair, because this can be isosteric with the G•A pair (Lescoute et al. 2005). Table 1 contains two examples of such k-turns. We solved the crystal structure of a SAM-I riboswitch containing a G2nA substitution (Schroeder et al. 2011)—the adenine at the 2b position accepts a hydrogen bond at N3 from the O2′ of G−1n (N–O distance 2.8 Å), and it donates a single hydrogen bond from N6 to A2n N3 (N–N distance 3.3 Å) (Fig. 2F). This places the k-turn in the N3 class. We have recently presented the structure of Kt-23 from the small ribosomal subunit of Thelohania solenopsae (also engineered into the SAM-I riboswitch) (Schroeder et al. 2012) that naturally contains an adenine at the 2n position. In contrast to the previous structure, A2b accepts a hydrogen bond at N1 from O2′ of G − 1n (N–O distance 2.5 Å), and there is no hydrogen bond made with A2n (the A2b N6–A2n N3 distance 5 Å) (Fig. 2G). This identifies the T. solenopsae k-turn as a member of the N1 class. Thus, k-turns with an A•A pair at the 2b•2n position have been identified in both structural classes.
Kt-7 structure in the ribosome
The structure of Kt-7 found in the H. marismortui ribosome as determined by Steitz and coworkers (Ban et al. 2000) is shown in Figure 3. The structure is clearly of the N1 class; the G−1n O2′ proton is donated to N1 of A2b (O–N distance 2.7 Å), and the resulting position of the A2b nucleobase leads to a long A2b N6–G2n N3 distance (N–N distance 5 Å). In the ribosomal Kt-7 structure, we also observe a hydrogen bond between the C and NC helix backbones 1 nt further from the loop, between the O2′ groups of C−2n and A3b (O–O distance 2.8 Å). This interaction is not found in the N3 class k-turns; for example, the O–O distances in the U4 and box C/D k-turns are 8.5 and 7.2 Å, respectively, consistent with a significant difference in the relative disposition of the C and NC helices. The C−2n–A3b hydrogen bond found in Kt-7 has a standard length and acceptable tetrahedral angles at the two oxygen atoms, so it might be expected to contribute to the stabilization of the folded k-turn structure. However, we previously found that replacement of the 2′-hydroxyl group at C−2n by a hydrogen atom had no detectable effect on ion-induced folding of Kt-7 in free solution (Liu and Lilley 2007). We therefore suggested that the structure adopted by Kt-7 in isolation might be different from that formed in the context of the ribosome, where it is constrained by the tertiary structure and bound by protein. It is also informative to compare the structure of the ribosomal Kt-7 with that of the k-turn found in the SAM-I riboswitch. This k-turn is closely similar to Kt-7 in sequence (with the same A•G pairing at the 3b•3n position, for example), yet, in the riboswitch, it adopts the N3 structure (Montange and Batey 2006). This led us to ask if the Kt-7 sequence might be capable of adopting alternative conformations, and we therefore sought to determine its structure when replacing the k-turn of the SAM-I riboswitch.
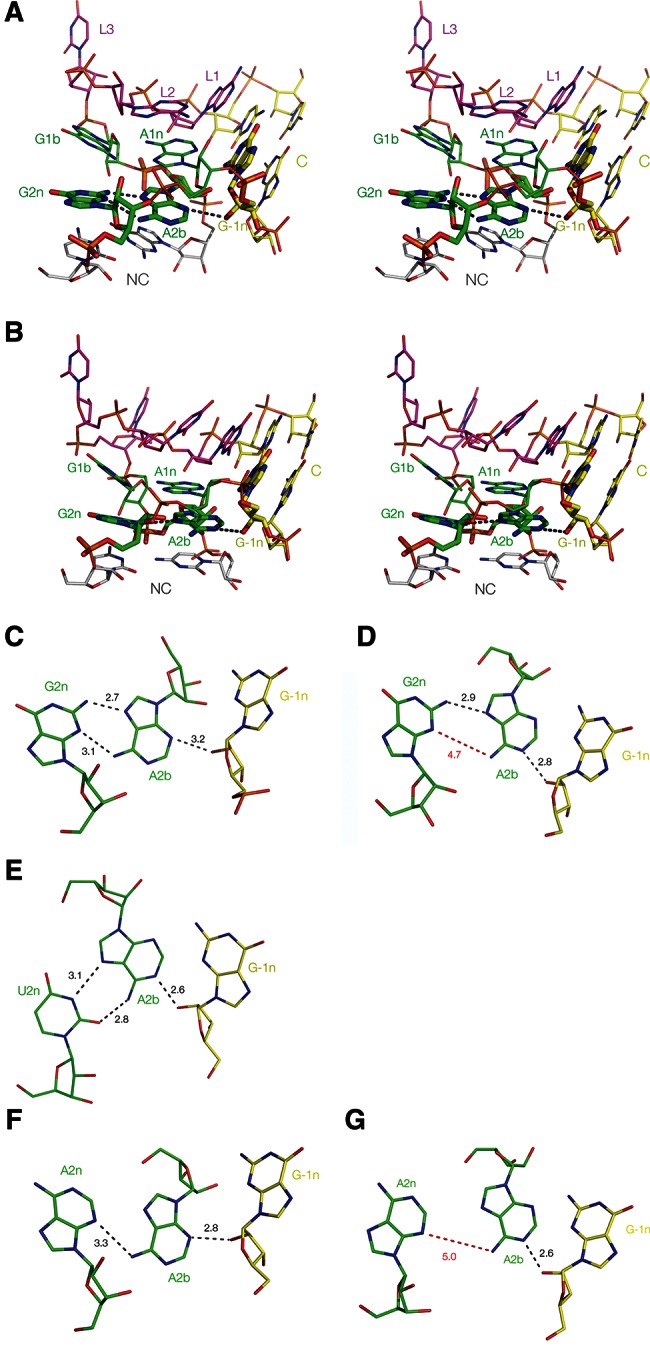
FIGURE 3.
Parallel-eye stereo images of Kt-7 in the H. marismortui 50S ribosomal subunit. (A) The whole k-turn viewed from the side of the nonbulged strand. Key hydrogen bonds are shown as gray broken lines. (B) Nucleotides participating in the key A-minor interactions in the core of the k-turn. Note that G−1n donates its O2′ proton to N1 of A2b, i.e., this is an N1-class k-turn. As a consequence, the distance between A2b N6 and G2n N3 is too long for hydrogen bonding, at 4.3 Å (shown in red). There is an additional hydrogen bond between the O2′ groups of C−2n and A3b that is never observed in the N3 structures.
Kt-7 structure in the SAM-I riboswitch
The RNA sequence of the SAM-I riboswitch was modified to replace its natural k-turn with that of Kt-7, as we have previously done for other k-turns such as Kt-23 of T. solenopsae (Schroeder et al. 2012). The modified species crystallized under very similar conditions to those used for the natural riboswitch, with retention of the P43212 space group and unit cell parameters (Montange et al. 2010). Diffraction data were collected and processed to a resolution of 2.95 Å (Table 2), and initial phases were obtained by molecular replacement using the structure of the natural riboswitch, PDB code 2GIS (Montange and Batey 2006). The overall structure of the modified SAM-I riboswitch was virtually identical to that of the natural species, with a root-mean-square deviation (RMSD) calculated over all P atoms of 0.97 Å (Supplemental Fig. S1).
TABLE 2.
Details of data collection and refinement statistics for the crystallographic data as deposited in the PDB

The structure of the k-turn is shown in Figure 4. It has a standard overall conformation that can be superimposed with the natural k-turn of the SAM-I riboswitch with an RMSD of 0.57 Å (Supplemental Fig. S2). The near-universal standard hydrogen bonds involving 2′-hydroxyl groups are present (Fig. 4A; Liu and Lilley 2007), including the L1 O2′ to A1n N1 and the L3 O2′ to PL1/PL2 proS O bonds. Interestingly, however, the structure differs from that of the same Kt-7 sequence observed in the ribosome when we examine the interactions at the critical A2b position (Fig. 4B,C). In contrast to the ribosomal structure, the nucleobase of A2b accepts a hydrogen bond from the G−1n O2′ at its N3, not N1. As a consequence, the A2b N6–G2n N3 distance has shortened to 3.2 Å and thus can be considered hydrogen bonded. A composite omit map was calculated (Supplemental Fig. S3) to exclude the possibility of model bias. All the nucleotides are very well resolved in the omit map, leading to the same model for the k-turn structure. Significantly, the A2b, G2n, and G−1n nucleotides are well-resolved (Supplemental Fig. S3C), supporting the pattern of hydrogen bonding seen in the 2Fo-Fc map (Fig. 4C).
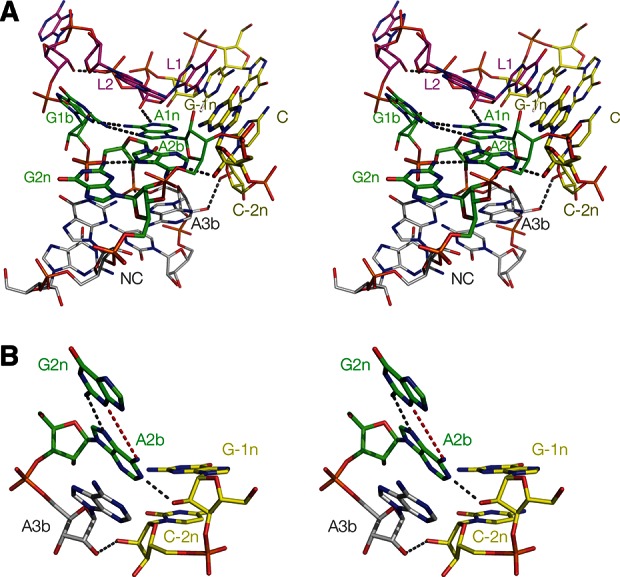
FIGURE 4.
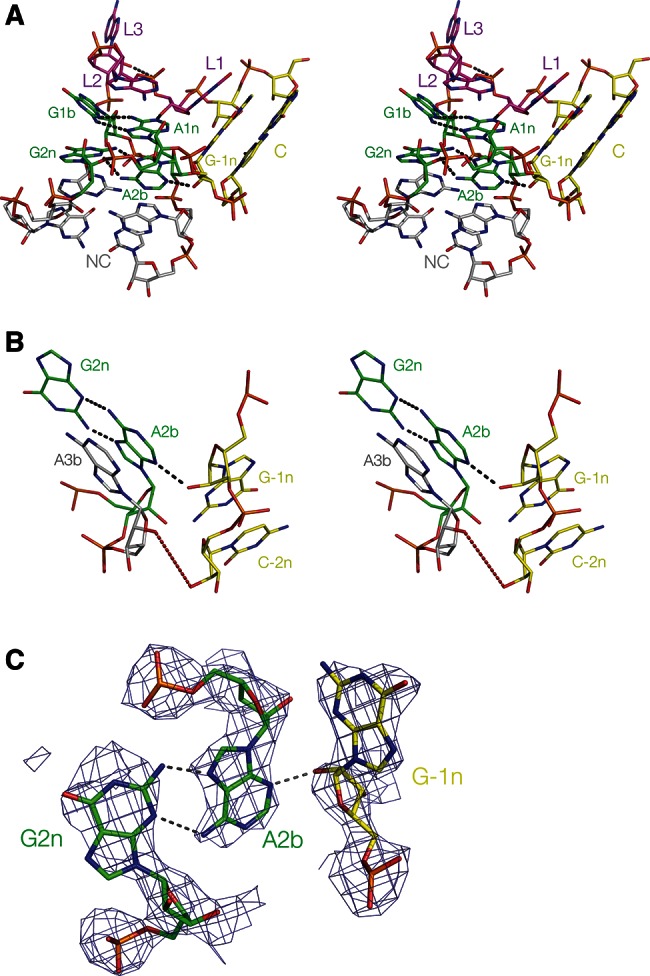
The structure of Kt-7 inserted into the SAM-I riboswitch, determined by X-ray crystallography in this study. (A) Parallel-eye stereo image of the whole k-turn viewed from the side of the nonbulged strand. Key hydrogen bonds are shown as gray broken lines. (B) Parallel-eye stereo image of the nucleotides participating in the key A-minor interactions in the core of the k-turn. G−1n donates its O2′ proton to N3 of A2b, i.e., this is an N3-class k-turn. The distance between A2b N6 and G2n N3 is now 3.2 Å. In contrast to the structure of Kt-7 observed in the H. marismortui ribosome, the distance between the O2′ groups of C−2n and A3b is now 6.9 Å (shown in red). (C) The G2n•A2b base pair and the A-minor interaction with the O2′ of G−1n, showing the 2Fo-Fc electron density map contoured at 1.2 σ.
Evidently transferring the Kt-7 sequence into the SAM-I riboswitch context has resulted in a change in conformation from the N1 to the N3 structure. Moving away from the core, we see that the O–O distance between the O2′ groups of C−2n and A3b is now 6.9 Å (compared to 2.8 Å in the ribosome). This shows that Kt-7 is conformationally plastic, able to alter its structure depending on context.
DISCUSSION
Analysis of the available atomic resolution structures for the k-turns has revealed that they fall into two distinct classes, depending on whether the O2′ proton of the −1n nucleotide is donated to N3 or N1 of the adenine nucleobase at the 2n position. This classification can be extended to k-turns that have either uridine or adenine at the 2b position and to some of the complex k-turns (such as Kt-7 of Escherichia coli), where the key nucleotides of the core do not map linearly onto the nucleotide sequence.
Kt-7 of the H. marismortui 50S ribosomal subunit has a sequence that is close to the consensus for k-turns and adopts the N1 conformation in the ribosome. When we placed the identical sequence into the SAM-I riboswitch in place of the natural k-turn, the riboswitch folded normally, but Kt-7 changed conformation into the N3 structure. This reveals a hitherto unsuspected structural plasticity of the k-turn. Kt-7 is subject to tertiary interactions both in the ribosome and in the riboswitch; in both cases, the terminal loop of the C helix makes tertiary contacts. The most obvious difference in environment is the binding of the L24 protein to the k-turn in the ribosome, whereas in the riboswitch, Kt-7 is completely free of protein. However, we note that binding the YbxF protein to the natural k-turn of the SAM-I riboswitch does not alter its conformation—it is N3 in both cases (Baird et al. 2012). If Kt-7 were found to adopt the N3 structure in free solution, as it does in the riboswitch, this would explain our earlier failure to observe a structural consequence of removal of the O2′ at the C−2n position (Liu and Lilley 2007).
We have seen that the nature of the hydrogen bonding between the −1n O2′ and the A2b nucleobase in turn affects the base-pairing between A2b and the nucleobase at the 2n position. There is a coordinated set of changes as the A2b reorients to direct either N3 or N1 toward the −1n O2′.
We have also seen that the distance between the O2′ atoms at the −2n and 3b positions can vary widely, differing by >4 Å between Kt-7 in the N3 and N1 conformations. This indicates that the relative disposition of the C and NC helices is affected by the change in conformation and, thus, the trajectory of the helices of the k-turn. We have analyzed this systematically in the available k-turn structures.
To achieve this, we fitted the C and NC helices to ideal A-form RNA helices and then calculated the transformation matrix required to superimpose them using PyMol (DeLano 2002). From this, we calculated three rotation angles describing the relative trajectory of the C helix with respect to the NC helix (Fig. 5A,B). The variation in these angles is plotted in Figure 5C. One (termed α) is the angle included between the two main helix axes, i.e., this describes the macroscopic bend angle. α clusters around 50°, as expected for k-turns, and does not exhibit an interesting variation between the N3 and N1 class structures. It will not be discussed further. The second angle (β) is the direction of the NC helix vector projected onto the x-y-plane which is orthogonal to the main C helix axis (Fig. 5B). β describes the direction of the bend and is the most variable of the three angles but does not differ significantly between the N1 and N3 classes. The third angle (γ) represents the rotation around the NC helix axis. In contrast to the other two angles, γ differs between the two classes, with an average of 47.3° ± 1.9° for the N3 class and 55.7° ± 5.1° for the N1 class. For all the k-turn structures analyzed, N3 structures have a value of γ < 50°, while for all the N1 classes, γ > 50°. The H. marismortui Kt-7 structure has the largest value, with γ = 63.3°, while the same sequence in the SAM-I riboswitch has a standard value for an N3 structure of γ = 48.5°. Thus, the global geometry of Kt-7 changes significantly between the environments of the ribosome and the riboswitch. This rotation around the NC helix axis will influence the relative distances between the backbone of the C and NC helix, leading to a very different pattern of interaction, explaining the different hydrogen bonding we have observed at the 3b, −2n position. Thus, the switch between the N3 and N1 structures has significant larger-scale conformational consequences.
FIGURE 5.
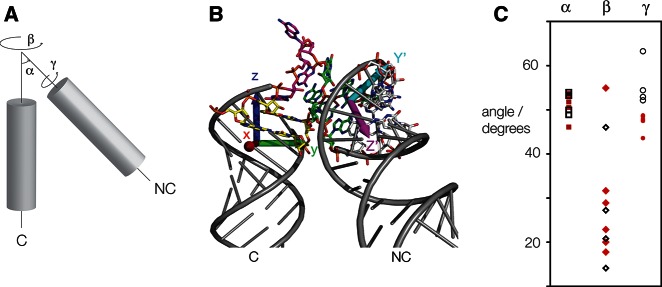
Conformational analysis of the global structure of k-turns. (A) Schematic showing the α, β, and γ angles of a k-turn. The angle α represents the angle included between the main helix axes of the C and NC helix. The angle β represents the direction of the NC helix vector projected onto a plane perpendicular to the C helix axis. Finally, γ represents the rotation around the main NC helix axis and is defined by the orientation of base pairs in the NC helix relative to the C helix axis. (B) H. marismortui Kt-7 (as determined in the SAM-I riboswitch [4B5R]) with fitted extended C and NC helices (shown in cartoon form, colored gray). The coloring scheme for the bases matches that from Figure 1. The x, y, and z axes of the atomic coordinates are shown in red, green, and blue, respectively, with the base of the arrow in the coordinate origin. The magenta arrow indicates the direction of the NC helix vector (Z′) and the cyan arrow the direction of the 4b•4n base pair of the NC helix (Y′). (C) Distribution of the calculated α, β, and γ angles of all the k-turns analyzed. k-turns of the N3 class are represented by filled red symbols and those of the N1 class by open black symbols. Note that, for the γ angle, the values for the two classes are separated into two distinct regions.
The k-turn is a key architectural element in functional RNA species that plays a critical role in mediating tertiary interactions. For example, mutating the SAM-I k-turn by an A1nC substitution disrupts the structure so that the riboswitch can no longer bind its S-adenonsyl methionine ligand (Schroeder et al. 2011). Such conformational influences are likely to be very important in the biogenesis of large RNA-protein assemblies such as the ribosome. It would, therefore, be anticipated that the possibility of switching between alternative conformations might add another dimension to the structural role of k-turns.
MATERIALS AND METHODS
Preparation of RNA
A plasmid containing a gene for the unmodified SAM-I riboswitch was changed by site-directed mutagenesis so that the k-turn region was converted to the sequence of Kt-7. The correct sequence of the resulting vector was verified by DNA sequencing. The template was amplified by PCR and transcribed in vitro using T7 RNA polymerase. The RNA was purified by denaturing polyacrylamide gel electrophoresis.
X-ray crystallography
The SAM-I riboswitch was crystallized using the hanging drop method. The crystal from which the data set used to solve the structure was obtained was grown by mixing 1 μL of 0.5 mM RNA in 40 mM Na-cacodylate (pH 7.0) plus 5 mM SAM with 1 μL of mother liquor. The mother liquor contained 40 mM Na-cacodylate (pH 7.0), 80 mM KCl, 30 mM BaCl2, 12 mM spermine-HCl, 8% (v/v) MPD. The crystal was then cryo-protected with mother liquor containing 30% (v/v) MPD and flash frozen in liquid nitrogen.
Diffraction data were collected on beamline ID14-1 at the European Synchrotron Radiation Facility in Grenoble, France. The data were indexed and integrated using MosFlm. The structure was solved by performing molecular replacement using MOLREP with PDB entry 2GIS (Montange and Batey 2006) as a preliminary model. The model was subsequently refined using Refmac5 (Vagin et al. 2004). Omit maps were calculated using PHENIX (Adams et al. 2010).
Calculation of global structural parameters
Ideal A-form helices of 3 bp (generated in COOT) were superimposed via the C1′, C4′, and O4′ atoms of all nucleotides of the C and NC helices of Kt-7 using PyMol. Conformational parameters were then calculated from the relative position of these helices using a PyMol plug-in written in Python. The main helix vectors were defined along the helix axis following the 5′ to 3′ direction of the bulge-containing strand, so that the helix vector of the C helix points toward the k-turn, while that of the NC helix points away from it. First, the C helix of the k-turn was fitted to an ideal 3-bp RNA helix, which was oriented such that the helix axis was parallel to the z-axis of the coordinate system, the −3b•−3n base pair is parallel to the y-axis, and the coordinate origin is in the center of the helix, with z = 0 at the height of the −3b•−3n base pair. Next, an ideal 3-bp helix was fitted to the NC helix. We then calculated the transformation matrix required to move the ideal C helix to the ideal NC helix. This matrix was then used to calculate the global structural parameters.
Angle α was calculated by α = 180-arccos(Z•Z′), where Z is the C helix vector (identical with the z-axis) and Z′ the NC helix vector. Z′ is the result of application of the transformation matrix to a vector parallel to Z of unit length starting in the coordinate origin, equivalent to the transformed z-axis. Next, β was calculated by projecting the NC helix vector onto the x-y-plane and calculating the angle between the resulting vector and the y-axis, leading to a value of 0° for the y-direction and 90° for the x-direction. The projected vector will be called G. Finally, γ was calculated from the orientation of the transformed y-axis vector (Y′), which is parallel to the direction of the 4n•4b base pair of the NC-helix. Y′ was projected onto the plane orthogonal to G, and the resulting vector was called J, and γ was calculated as the angle between J and Z. This defines γ as 0°, when Y′ is close to vertical and 90° when it contains no z-component.
DATA DEPOSITION
Coordinates have been deposited in the Protein Data Bank with accession code 4B5R.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Lin Huang for discussion and Cancer Research UK for financial support.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallog 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Zhang J, Hamma T, Ferré-D’Amaré AR 2012. YbxF and YlxQ are bacterial homologs of L7Ae and bind K-turns but not K-loops. RNA 18: 759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289: 905–920 [DOI] [PubMed] [Google Scholar]
- Blouin S, Lafontaine DA 2007. A loop–loop interaction and a K-turn motif located in the lysine aptamer domain are important for the riboswitch gene regulation control. RNA 13: 1256–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JA, Williamson JR 2004. Joint X-ray and NMR refinement of the yeast L30e-mRNA complex. Structure 12: 1165–1176 [DOI] [PubMed] [Google Scholar]
- DeLano WL 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- Goody TA, Melcher SE, Norman DG, Lilley DMJ 2004. The kink-turn motif in RNA is dimorphic, and metal ion-dependent. RNA 10: 254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamma T, Ferré-D’Amaré AR 2004. Structure of protein L7Ae bound to a K-turn derived from an archaeal box H/ACA sRNA at 1.8 Å resolution. Structure 12: 893–903 [DOI] [PubMed] [Google Scholar]
- Heppell B, Lafontaine DA 2008. Folding of the SAM aptamer is determined by the formation of a K-turn-dependent pseudoknot. Biochemistry 47: 1490–1499 [DOI] [PubMed] [Google Scholar]
- Klein DJ, Schmeing TM, Moore PB, Steitz TA 2001. The kink-turn: A new RNA secondary structure motif. EMBO J 20: 4214–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescoute A, Leontis NB, Massire C, Westhof E 2005. Recurrent structural RNA motifs, isostericity matrices and sequence alignments. Nucleic Acids Res 33: 2395–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lilley DMJ 2007. The role of specific 2′-hydroxyl groups in the stabilization of the folded conformation of kink-turn RNA. RNA 13: 200–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, White SA, Williamson JR 1999. A novel loop-loop recognition motif in the yeast ribosomal protein L30 autoregulatory RNA complex. Nat Struct Biol 6: 1139–1147 [DOI] [PubMed] [Google Scholar]
- Matsumura S, Ikawa Y, Inoue T 2003. Biochemical characterization of the kink-turn RNA motif. Nucleic Acids Res 31: 5544–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montange RK, Batey RT 2006. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature 441: 1172–1175 [DOI] [PubMed] [Google Scholar]
- Montange RK, Mondragon E, van Tyne D, Garst AD, Ceres P, Batey RT 2010. Discrimination between closely related cellular metabolites by the SAM-I riboswitch. J Mol Biol 396: 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Zhang Y, Fenley MO, Li H 2004. Molecular basis of box C/D RNA-protein interactions; cocrystal structure of archaeal L7Ae and a box C/D RNA. Structure 12: 807–818 [DOI] [PubMed] [Google Scholar]
- Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA 2001. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc Natl Acad Sci 98: 4899–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder KT, Lilley DMJ 2009. Ion-induced folding of a kink turn that departs from the conventional sequence. Nucleic Acids Res 37: 7281–7289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder KT, McPhee SA, Ouellet J, Lilley DMJ 2010. A structural database for k-turn motifs in RNA. RNA 16: 1463–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder KT, Daldrop P, Lilley DMJ 2011. RNA tertiary interactions in a riboswitch stabilize the structure of a kink turn. Structure 19: 1233–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder KT, Daldrop P, McPhee SA, Lilley DMJ 2012. Structure and folding of a rare, natural kink turn in RNA with an A•A pair at the 2b•2n position. RNA 18: 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA 2009. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat Struct Mol Biol 16: 1218–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B, Melcher SE, Wilson TJ, Norman DG, Lilley DMJ 2005. Induced fit of RNA on binding the L7Ae protein to the kink-turn motif. RNA 11: 1192–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN 2004. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr 60: 2184–2195 [DOI] [PubMed] [Google Scholar]
- Vidovic I, Nottrott S, Hartmuth K, Luhrmann R, Ficner R 2000. Crystal structure of the spliceosomal 15.5 kD protein bound to a U4 snRNA fragment. Mol Cell 6: 1331–1342 [DOI] [PubMed] [Google Scholar]
- White SA, Hoeger M, Schweppe JJ, Shillingford A, Shipilov V, Zarutskie J 2004. Internal loop mutations in the ribosomal protein L30 binding site of the yeast L30 RNA transcript. RNA 10: 369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339 [DOI] [PubMed] [Google Scholar]
- Wozniak AK, Nottrott S, Kuhn-Holsken E, Schroder GF, Grubmuller H, Luhrmann R, Seidel CA, Oesterhelt F 2005. Detecting protein-induced folding of the U4 snRNA kink-turn by single-molecule multiparameter FRET measurements. RNA 11: 1545–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.