

The authors study the impacts of human gene annotations in diverse databases and report on a systematic comparison of RefSeq, Ensembl, and AceView on related studies, especially genomic and transcriptomic comparisons. The paper provides solid data highlighting the problems in current gene model databases and is a warning call to the community.
Keywords: RNA-seq, short-read mapping, gene expression quantification, differential expression calling, genetics
Abstract
Human gene annotation is crucial for conducting transcriptomic and genetic studies; however, the impacts of human gene annotations in diverse databases on related studies have been less evaluated. To enable full use of various human annotation resources and better understand the human transcriptome, here we systematically compare the human annotations present in RefSeq, Ensembl (GENCODE), and AceView on diverse transcriptomic and genetic analyses. We found that the human gene annotations in the three databases are far from complete. Although Ensembl and AceView annotated more genes than RefSeq, more than 15,800 genes from Ensembl (or AceView) are within the intergenic and intronic regions of AceView (or Ensembl) annotation. The human transcriptome annotations in RefSeq, Ensembl, and AceView had distinct effects on short-read mapping, gene and isoform expression profiling, and differential expression calling. Furthermore, our findings indicate that the integrated annotation of these databases can obtain a more complete gene set and significantly enhance those transcriptomic analyses. We also observed that many more known SNPs were located within genes annotated in Ensembl and AceView than in RefSeq. In particular, 1033 of 3041 trait/disease-associated SNPs involved in about 200 human traits/diseases that were previously reported to be in RefSeq intergenic regions could be relocated within Ensembl and AceView genes. Our findings illustrate that a more complete transcriptome generated by incorporating human gene annotations in diverse databases can strikingly improve the overall results of transcriptomic and genetic studies.
INTRODUCTION
The human transcriptome bridges the human genome to gene functions and is much more intricate than we initially thought. The number of human genes has been estimated from 20,000 to 120,000 (Aparicio 2000; Ewing and Green 2000; Liang et al. 2000). However, to date, the exact number of genes in the human genome and their encoded transcripts is still unknown. Because there are lots of repetitive and homologous sequences interspersed throughout the human genome, these sequences are very intricate and make it difficult to identify bona fide genes within them. In addition, the assembled human reference genome is still incomplete, and some genomic regions may be misassembled (Eichler et al. 2004; Stein 2004). The missing genomic sequences of the human reference genome could contain novel genes that have unknown biological functions (Kidd et al. 2010; Li et al. 2010; Chen et al. 2011a). This is further complicated by the fact that diverse computational approaches/pipelines with the distinct algorithms can result in different predictions for identifying the genes harbored in a genome (Pennisi 2003; Stanke and Waack 2003; Shi et al. 2006; Guttman et al. 2010; Trapnell et al. 2010; Garber et al. 2011).
Several public databases annotate genes and isoforms in the human genome, such as RefSeq (Pruitt et al. 2012), Ensembl (Flicek et al. 2011), GENCODE (Harrow et al. 2012), and AceView (Thierry-Mieg and Thierry-Mieg 2006). RefSeq human gene models are well supported and broadly used in various researches. Ensembl gene predictions contain both automated genome annotation and manual curation, while the gene set of GENCODE corresponds to the Ensembl annotation since GENCODE version 3c (equivalent to Ensembl 56). AceView provides a comprehensive nonredundant curated representation of available human cDNA sequences. On account of the differences in source data and pipelines used for gene annotation among these databases, the annotated human genes in each database have distinct properties and vary in terms of annotation completeness, quality, gene structure, and so on. In general, the human gene annotations in these databases are valuable resources for conducting biological studies and promoting our deeper appreciation of the human genome. Nevertheless, no study has yet systematically compared the impacts of human transcriptome annotations in these databases on related studies. To make full use of these annotation resources, it is necessary to carry out comprehensive analysis and better understand the coverage and limitations for each database.
Human gene annotation is fundamental for carrying out various transcriptomic studies. Nowadays, RNA-seq technologies are widely used to investigate the diverse aspects of the transcriptome and have greatly facilitated our understanding of the intricate transcriptomes of diverse organisms (Mortazavi et al. 2008; Sultan et al. 2008; Wang et al. 2009; Marguerat and Bahler 2010; Chen et al. 2011b; Ozsolak and Milos 2011). To comprehensively conduct transcriptomic studies, such as identifying alternative splicing, quantifying the expression of genes and isoforms, and calling differential expression, the related applied bioinformatics algorithms generally need the gene annotation of the studied genome to guide the analyses. Usually the RNA-seq reads are mapped onto the reference genome or transcriptome to investigate the expression profiles and/or conduct other analyses of annotated genes. Therefore, the more complete the gene annotation to be used, the more comprehensive the read mapping information of genes will be obtained, which can enrich the results of subsequent analyses.
During genetic studies, gene annotation is also important for inferring the connections between genetic variations and genes. Genetic variations may lead to changes in expression of genes or protein structures, and the mechanisms behind these changes are vital for both medical and evolutionary genetics (Strausberg et al. 2004; Williams et al. 2007; Pickrell et al. 2010). Single nucleotide polymorphisms (SNPs) are the most common form of human DNA variation, and their associations with human disorders are often interrogated in genome-wide association studies (GWAS). Currently, most of the potentially functional SNPs have been found falling outside the genic regions (Stranger et al. 2007; Barreiro et al. 2008; Veyrieras et al. 2008), which made it difficult to accurately determine their affected genes and elucidate their functions. However, it is important to keep in mind that a large number of human genes and isoforms are still unannotated in current public databases, and therefore, many non-protein-coding and protein-coding genes in the intergenic and intronic regions remain to be identified (Wilusz et al. 2009; Guttman et al. 2010; Cabili et al. 2011). Full annotation of the genes and isoforms in the human genome should enable us to more precisely determine the relationships between genetic variations and genes and gain new insights into the mechanisms underlying their effects on human traits/diseases.
Our study aims to systematically compare the influences of human transcriptome annotations in RefSeq, Ensembl (GENCODE), and AceView on diverse transcriptomic and genetic studies and help researchers gain insights into human genome annotations and take full advantage of various annotation databases. First, we compared the annotated human transcriptomes in these databases and generated two new transcriptomes based on Ensembl and AceView to more comprehensively conduct the subsequent study. Then we investigated the impacts and differences of the five transcriptomes on aligning RNA-seq reads, profiling the expression of genes and isoforms, and calling differential expression. Moreover, we inspected the influences of different transcriptome annotations on genetic variation researches. Our results highlight that combing the human gene annotations of diverse databases can obtain a more complete human transcriptome, which can significantly enhance the relevant analyses of transcriptomics and genetics.
RESULTS
Comparison and integration of different human transcriptome annotations
To gain insights into the characteristics of the human gene annotations in RefSeq, Ensembl, and AceView, we first compared their annotated gene models. Considering that both the Ensembl and AceView transcriptome annotations use partial evidence of RefSeq and contain more genes and transcripts than RefSeq, we sought to integrate the annotations in Ensembl and AceView to generate a more complete human transcriptome. To avoid controversy and confusion resulting from overlapped gene structures between Ensembl and AceView, we generated two new transcriptomes by separately using each database as template and adding the genes from another database lying in its intergenic and intronic regions relative to each other. One was “EnsAce,” which was made up of the entire Ensembl human transcriptome plus the AceView genes located in the Ensembl intergenic and intronic regions; the other was the reverse one “AceEns,” which consisted of the whole AceView human transcriptome plus the Ensembl genes lying in the AceView intergenic and intronic regions.
The number of genes and transcripts in RefSeq, Ensembl, AceView, EnsAce, and AceEns varied greatly (Fig. 1A). RefSeq had the fewest genes and transcripts, while AceView annotated more transcripts but fewer genes than Ensembl. Moreover, the average isoform rate for AceView genes (4.92) was greater than that of Ensembl (3.41) or RefSeq (1.68). EnsAce included all of Ensembl human genes and 15,847 additional AceView genes (5574 in Ensembl intergenic regions and 10,273 in Ensembl intronic regions; these genes were annotated 18,111 transcripts) (Fig. 1B; Supplemental Data Set 1). According to the AceView annotation, 8162 of these added transcripts are potentially protein-coding and 9949 are non-protein-coding. In addition, the putative proteins encoded by these protein-coding transcripts are expected to localize in various places including nucleus, cytoplasm, membrane, and extracellular space, and a portion of them contain known functional protein domains. AceEns, on the other hand, contained all of the AceView human genes and 17,742 additional Ensembl genes (9268 in AceView intergenic regions and 8474 in AceView intronic regions; these genes were annotated 18,083 transcripts) (Fig. 1C). The added Ensembl transcripts can be classified into multiple categories and are mainly from the pseudogenes, small RNAs, long intergenic non-protein-coding RNAs (lincRNAs), protein-coding genes, and others (Fig. 1D). We found that the majority of those additional genes from Ensembl and AceView are single exons and possess shorter lengths than other multi-exon genes in general. Accordingly, the intergenic and intronic regions of both Ensembl and AceView had a large number of genes that were not annotated, and it should be much more for RefSeq. Furthermore, the numbers of unannotated genes per chromosome appeared to be positively correlated with chromosome size. Taken together, our initial results showed that the human transcriptome annotation in RefSeq, Ensembl, and AceView is far from complete, and integrating these databases can generate a more complete set of genes and transcripts.
FIGURE 1.

Human transcriptomes in RefSeq, Ensembl, AceView, EnsAce, and AceEns. (A) The number of human genes and transcripts in each transcriptome. The counts for RefSeq are its unique genes and transcripts in UCSC for which the duplicated genes were removed. The Ensembl human transcriptome is the sum of its protein-coding and non-protein-coding transcripts (release 67 of GRCh37, corresponding to GENCODE 12). AceView transcripts that contained unknown bases of “N” were not taken into account. (B) Chromosome distribution of AceView human genes located in the intergenic and intronic regions of the Ensembl annotation. (C) Chromosome distribution of Ensembl human genes within the intergenic and intronic regions of AceView annotation. (D) Categories of the 18,083 Ensembl transcripts located in the AceView intergenic and intronic regions.
Mapping rate of RNA-seq data on different human transcriptomes
To investigate discrepancies in RNA-seq read alignment among the transcriptomes of RefSeq, Ensembl, AceView, EnsAce, and AceEns, we compared the short-read mapping rate among the five transcriptomes with RNA-seq data. Short-read mapping is a basic step in RNA-seq data analyses, and to a certain extent, the percent of reads mapped onto a given transcriptome can reflect the completeness of its annotated genes and transcripts. To make the comparison more comprehensive and convincing, we used 58 other public RNA-seq data sets obtained from more than 20 human tissues and cell lines (Supplemental Table 1) (Beane et al. 2011; Cabili et al. 2011; Chen et al. 2011a,b; Shapiro et al. 2011; Toung et al. 2011; Gertz et al. 2012; Peng et al. 2012). The read lengths of these data sets are diverse (range 35–100 bp), and some samples are paired end while others are single end.
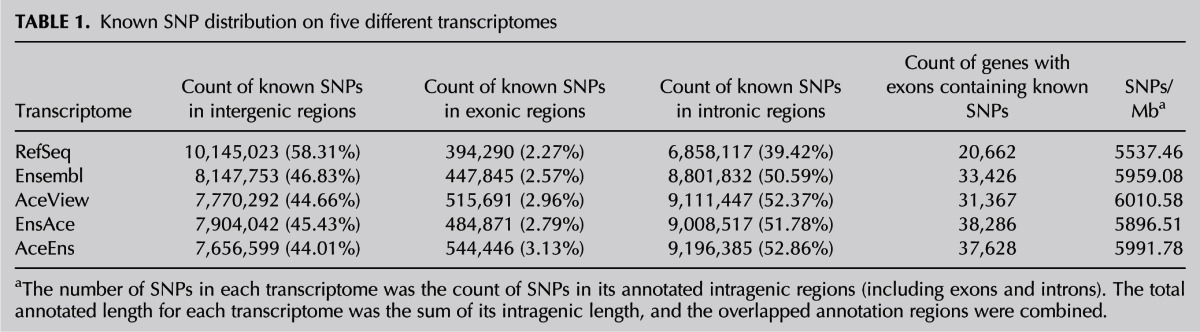
TABLE 1.
Known SNP distribution on five different transcriptomes
We separately mapped each RNA-seq data set onto the transcriptomes of RefSeq, Ensembl, AceView, EnsAce, and AceEns using Bowtie (Langmead et al. 2009), respectively. To guarantee the reads mapped onto the transcriptome as correctly as possible and to take multimapped reads into account, we required that Bowtie search multiple optimal alignments for each read (see Materials and Methods). Across all 58 data sets, we observed a consistent pattern in which the RNA-seq read mapping rate for the five human transcriptomes was in descending order: AceEns, AceView, EnsAce, Ensembl, and RefSeq (Fig. 2A). AceView and Ensembl can achieve conspicuously higher mapping rates than RefSeq, indicating that they annotated a portion of specific genes/transcripts that are unannotated in RefSeq. Although EnsAce includes 15,847 AceView genes that are not in Ensembl and AceEns has 17,742 Ensembl genes that are not included in AceView, the ratio of reads that mapped onto EnsAce and AceEns was not much greater than that of Ensembl (∼1% fewer than EnsAce) and AceView (∼0.06% fewer than AceEns), respectively. Since we used the same parameter setting for the five transcriptomes, the mapping results preliminarily suggest that the added genes from AceView in EnsAce and the Ensembl genes in AceEns may not be actively expressed in diverse tissues and cell lines.
FIGURE 2.

RNA-seq read mapping and expression profile comparison among the five transcriptomes. (A) Short-read mapping rate for RefSeq, Ensembl, AceView, EnsAce, and AceEns using 58 RNA-seq data sets from more than 20 human tissues and cell lines (Supplemental Table 1). (B) Number of expressed genes detected in the five transcriptomes across all 58 data sets. (C) Number of expressed transcripts examined in each data set among the five transcriptomes.
For all 58 RNA-seq data sets, the short-read mapping rates for the five human transcriptomes were lower than 100%, and all had a fraction of unmapped reads. These unmapped reads could occur for a variety of reasons. One possible reason is that the genes/transcripts in each transcriptome set are incomplete, and thus the reads from the unannotated genes/transcripts could not be aligned. On the other hand, some of the unmapped reads could have sequencing errors or post-transcriptional modifications (e.g., RNA editing) or even arise from contamination, and therefore these reads would fail to align to the transcriptome. Of course, limitations of the Bowtie aligner could also result in some reads being unable to be mapped onto the transcriptome.
Expression profiles of different transcriptomes in diverse tissues and cell lines
To assess the differences among the five human transcriptomes in profiling gene and isoform expression, we separately quantified the gene and isoform expression of the five transcriptomes using all 58 data sets with the MMSEQ pipeline (Turro et al. 2011). Because some human genes and transcripts might be expressed at very low level and the mapping ambiguities could introduce false positives, we used a threshold of 0.1 FPKM (fragments per kilobase of transcript per million mapped reads or read pairs) for all samples.
For a given sample, the numbers of expressed genes and transcripts detected among the five transcriptomes varied considerably, and variation was in the thousands for genes and even larger for transcripts (Fig. 2B,C). At both the gene and isoform levels, the number of detected expressed genes and transcripts in RefSeq was the lowest. Despite the fact that the Ensembl human transcriptome contained more genes than AceView, more expressed genes were detected in AceView than Ensembl for each data set. This could result from a large number of genes annotated in Ensembl being small RNA genes that were not annotated in AceView, whereas almost all of the small RNAs were selected out during RNA-seq library construction due to their short length. In addition, AceView annotated more transcripts than Ensembl, and more expressed transcripts were observed in AceView for each sample, indicating that the number of transcripts annotated in Ensembl is far lower than the actual number of transcripts. Interestingly, we also observed that many pseudogenes annotated in Ensembl were widely expressed in human tissues and cell lines, suggesting that they may execute important roles in human cells (Hirotsune et al. 2003; Kalyana-Sundaram et al. 2012). Strikingly, using EnsAce and AceEns could detect both more expressed genes and transcripts in a given sample than Ensembl and AceView, respectively. Using the RefSeq human transcriptome, 56.29%–81.97% of genes and 40.18%–71.15% of transcripts were detected across different data sets. Relatively lower expression ratios were found using Ensembl, AceView, EnsAce, and AceEns for the same sample. These results indicate that the five transcriptomes have large discrepancies in profiling the expression of genes and transcripts, and integrating these databases can significantly improve the overall results in detecting the gene and transcript expression more than only using an individual database, which helps us to study human gene expression profiles more comprehensively.
Expression distribution of genes and isoforms among different transcriptomes
Distinct variances in the distribution of gene and transcript expression were observed among the five transcriptomes. For the same sample, we found that the median gene expression level for RefSeq was the highest, followed by Ensembl, while AceView, EnsAce, and AceEns had similarly low levels (Fig. 3A, using testis tissue as an example). The median transcript expression level in RefSeq was also the highest, followed by Ensembl and EnsAce with their similar level, and the lowest is for AceView and AceEns (Fig. 3B). Furthermore, the expressed genes detected in AceView, EnsAce, and AceEns exhibited similar expression density distributions, which differed greatly from those of Ensembl and RefSeq (Fig. 3C). This difference should result from the fact that AceView, EnsAce, and AceEns had a significantly larger proportion of genes with low expression levels compared with RefSeq and Ensembl. That is, the variation could be mainly caused by the annotation completeness of genes and transcripts in each database. Accordingly, EnsAce, AceView, and AceEns also exhibited lower median gene expression levels than RefSeq and Ensembl. Although analogous density curves of transcript expression were observed between AceView and AceEns and between Ensembl and EnsAce, they differed from that of RefSeq (Fig. 3D). In fact, because most of the transcripts in EnsAce came from Ensembl, and similarly for AceEns from AceView, analogous median transcript expression levels and expression density curves were observed between each derived transcriptome and the original one.
FIGURE 3.

Expression distribution of genes and transcripts in the five transcriptomes. (A) Boxplot comparison of gene expression in our sequenced testis tissue for each transcriptome. (B) Boxplot comparison of transcript expression in testis for each transcriptome. (C) Density curves of gene expression level in testis among five transcriptomes. (D) Density curves of transcript expression level in testis. (E) Expression-level histogram of the AceView genes located in the intergenic and intronic regions of Ensembl annotation in testis. (F) Expression-level histogram of Ensembl genes located in the intergenic and intronic regions of AceView human annotation in testis.
To understand the expression properties of the intergenic and intronic genes relative to Ensembl and AceView in EnsAce and AceEns, we further inspected the expression profiles of these genes. The majority of these genes in EnsAce and AceEns showed lower or no expression levels, except for a small portion, which had relatively higher expression levels in each sample (Fig. 3E,F, using testis tissue as an example; Supplemental Data Set 2). Most of these genes are also not actively expressed in our analyzed human tissues and cell lines. Furthermore, this portion of AceView genes was also annotated as expressed at low level by the AceView database. Consequently, a possible reason that these genes were not annotated into Ensembl or AceView is due to their low and/or rare expression, and they were not detected in the source data used for human gene annotations in Ensembl and AceView. But it does not exclude the possibility that some of these genes are the annotation noise of Ensembl/AceView databases. The results indicate that it is necessary to conduct deep sequencing to capture genes and transcripts with low expression to study comprehensively the expression profiles of the human transcriptome.
Comparison of differential expression calling
To gain insights into the impacts of the five transcriptomes on the differential expression studies between differing conditions, we conducted differential expression calling between brain and UHR (universal human reference) samples (Chen et al. 2011a) used as the reference RNA samples by the Microarray Quality Control project (Shi et al. 2006). RNA-seq technologies provide opportunities to call differential expression at both the gene and isoform levels, and knowledge of the isoform level provides a more comprehensive understanding of the detailed gene expression changes between two experimental conditions. To this end, we called the differential expression of genes and isoforms between brain and UHR samples using NOISeq (Tarazona et al. 2011) (see Materials and Methods).
Comparing the differentially expressed tags (q = 0.95) among the five transcriptomes at both the gene and isoform levels, between brain and UHR, we detected 90 genes and 518 transcripts that were differentially expressed with the RefSeq human transcriptome, while relatively more differentially expressed genes and transcripts were observed when we used the Ensembl, AceView, EnsAce, and AceEns transcriptomes (Fig. 4). In theory, the differentially expressed features identified with the Ensembl and AceView transcriptomes should be entirely contained within the results of EnsAce and AceEns, respectively. However, we observed that a small fraction of differentially expressed genes and transcripts detected in Ensembl and AceView was not found in EnsAce and AceEns (Fig. 4). This is likely because the added genes from Ensembl and AceView in AceEns and EnsAce led to subtle changes in the expression distribution for the genes and transcripts. These changes then resulted in NOISeq failing to identify some of the genes and transcripts as being significantly differentially expressed. However, 31 and 98 more differentially expressed genes and transcripts were found in EnsAce than in Ensembl, while two more genes and 84 more transcripts were found to be differentially expressed in AceEns than in AceView (Fig. 4). These results indicate that the five transcriptomes have certain differences in differential expression calling and a more complete transcriptome may help us to capture more differentially expressed features.
FIGURE 4.

Comparison of differential expression calling among different transcriptomes using brain versus UHR samples. (A) Comparison of detected differentially expressed genes between Ensembl and EnsAce. (B) Comparison of detected differentially expressed genes between AceView and AceEns. (C) Comparison of detected differentially expressed transcripts between Ensembl and EnsAce. (D) Comparison of detected differentially expressed transcripts between AceView and AceEns.
Distribution of genetic variation in the human transcriptomes
To determine the distribution of genetic variations in the different transcriptomes, we first compared the genomic distribution of 17,397,430 SNPs from the Single Nucleotide Polymorphism database (dbSNP) among the five transcriptomes (see Materials and Methods). We observed that >50% of these SNPs lay within the intragenic (exonic and intronic) regions of Ensembl, AceView, EnsAce, and AceEns but not RefSeq (Table 1). AceEns had the largest number of SNPs in its intragenic regions, and the number for RefSeq is lower than both Ensembl and AceView. However, only a small portion (2.27%–3.13%) of SNPs was located in the exonic regions for all five transcriptomes. EnsAce had the most genes that contained SNPs in its exonic regions, followed by AceEns, Ensembl, AceView, and RefSeq (Table 1). Thus, the results show that different transcriptomes have distinct abilities to reveal genetic variations within their annotated genes and the integrated transcriptomes can more comprehensively ascertain the real locations between genetic variations and human genes.
To investigate whether genes present in Ensembl and AceView can further promote analyses to determine genome-wide association loci for human traits/disorders, we restudied the location relationships of 3041 trait/disease-associated SNPs previously reported to be in the intergenic regions of RefSeq (Hindorff et al. 2009). Since these trait/disease-associated SNPs were found in the RefSeq intergenic regions, it is difficult to determine their associations with human genes. Surprisingly, we found that 724 (34 in exonic areas and 690 in intronic regions) of these trait/disease-associated SNPs could be mapped in the intragenic regions of 532 Ensembl genes and 879 (42 in exonic regions and others in intronic areas) within 676 AceView genes (Supplemental Tables 2, 3). The distribution of the trait/disease-associated SNPs on each human chromosome is shown in Figure 5 with a Circos graph (Krzywinski et al. 2009). Moreover, some of these SNPs within the Ensembl and/or AceView genes were associated with two or more distinct traits/diseases, suggesting that they might be involved in different traits/diseases with the similar mechanisms. In total, 1033 trait/disease-associated SNPs involved in about 200 human traits/diseases that had previously been reported to be present in the RefSeq intergenic regions could be relocated within Ensembl and AceView genes. Our findings demonstrate that a large fraction of previously reported trait/disease-associated SNPs was misassigned into the intergenic regions, likely because of the incomplete gene annotations of RefSeq.
FIGURE 5.
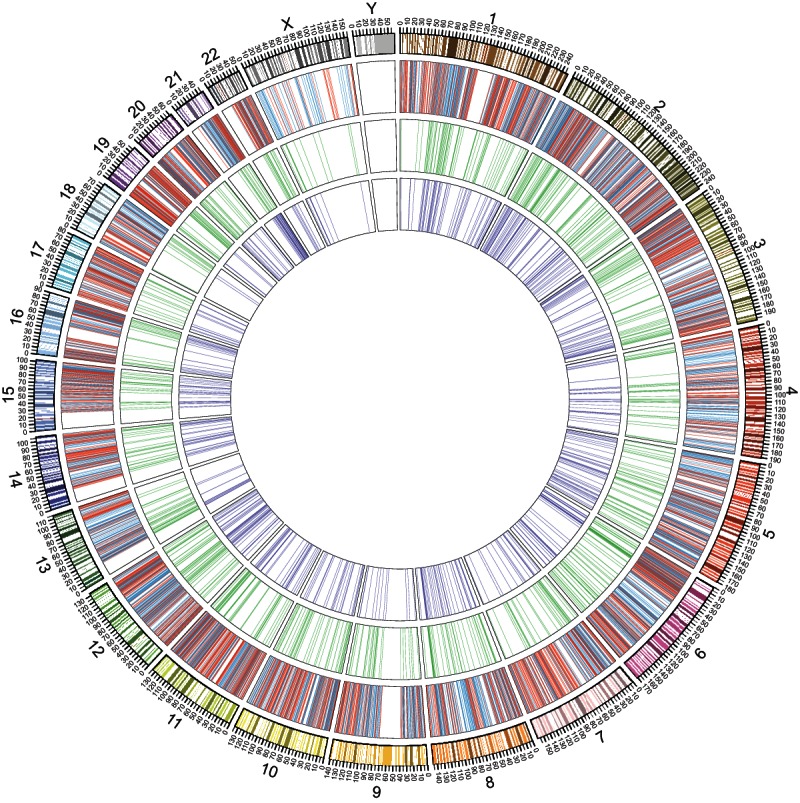
Distribution of reported trait/disease-associated SNPs on human chromosomes. From outside to inside, the first circle represents the human chromosomes. The 6522 previously reported trait/disease-associated SNPs are distributed on the second circle (one line for one SNP); 3481 of these SNPs were previously reported within RefSeq intragenic regions (red lines), while the other 3041 were reported in intergenic regions of RefSeq (blue lines). Among these 3041 trait/disease-associated SNPs, 879 could be remapped within 676 AceView genes (green lines in the third circle), and 724 could be relocated in 532 Ensembl genes (purple lines in the fourth circle).
DISCUSSION
We systematically compared the human transcriptome annotations of RefSeq, Ensembl, and AceView from diverse perspectives of transcriptomic and genetic studies. The human gene annotations in these databases are still far from complete. RefSeq annotated fewer genes and transcripts than both Ensembl and AceView, but 15,847 AceView (17,742 Ensembl) genes still could be located within the intergenic and intronic regions of the Ensembl (AceView) annotation. Intriguingly, most of these genes are expressed at low level in diverse human tissues and cell lines, suggesting that a large number of human genes that have low expression remain to be identified by ultra-deep sequencing. Moreover, considering that the human reference genome is still incomplete and lacks many novel genes (Li et al. 2010; Chen et al. 2011a), the real number of human genes and isoforms is certainly higher than that suggested by the integration of these databases. With the innovations of both sequencing technologies and bioinformatics strategies, we believe that the human genes and transcripts will be annotated more and more completely.
Our results indicate that the integration of different annotation databases can obtain a more complete transcriptome and strikingly improve the overall results of relevant transcriptomic analyses. The short-read mapping rate on a given transcriptome is largely determined by its completeness. Furthermore, profiling gene and isoform expression and calling differential expression are also closely associated with the completeness of the transcriptome we used. If we only use the annotation of a certain database, genes and isoforms that are not annotated in this database will be ignored in corresponding research, which will lead to incomplete results. For each RNA-seq data set, we found that using AceView, the human transcriptome could obtain a higher RNA-seq read mapping rate and detect a larger number of expressed genes and transcripts than both RefSeq and Ensembl. Moreover, a larger ratio of expressed genes and transcripts was examined in RefSeq than in Ensembl and AceView, suggesting that RefSeq contained a larger proportion of genes and isoforms with relatively higher activity. Remarkably, both of the integrated transcriptomes (EnsAce and AceEns) could achieve better performance than solely using either the original database alone in RNA-seq read mapping, gene and isoform expression quantification, and differential expression calling.
Our findings show that the location relationships between known genetic variations and human genes also vary differently among different transcriptomes. More identified SNPs were found within the AceView and Ensembl genes than the RefSeq genes. Furthermore, more existent SNPs were observed within human genes of the integrated transcriptomes of EnsAce and AceEns than of Ensembl and AceView, respectively. In addition, 1033 trait/disease-associated SNPs that were previously reported to be located in the RefSeq intergenic regions (Hindorff et al. 2009) and were involved in about 200 human traits/diseases could be relocated into the intragenic regions of Ensembl and AceView annotations. These potentially trait/disease-associated SNPs were originally thought to lie in intergenic regions, making it difficult to define their associations with human genes. Mapping them within the Ensembl and/or AceView genes could help us better infer their influences on human genes and human traits/diseases. Moreover, an increasing number of human traits/diseases-related SNPs will be identified in the future with the advancements in technologies and correlated genotype calling approaches, which are crucial for the realization of personalized medicine (Rosenfeld et al. 2012). Accordingly, combing various human gene databases will definitely facilitate determining the real location relationships between genetic variations and human genes, as well as improve result interpretation and functional study of those genetic variations.
Collectively, our study reveals the incompleteness and complementarity of the human gene annotations in different databases, which may help researchers make better use of diverse valuable public annotation resources. Furthermore, integrating gene annotations of diverse databases to generate a more complete transcriptome is greatly conducive to improving the overall results of relevant transcriptomic and genetic studies and providing new insights into corresponding research.
MATERIALS AND METHODS
RNA-seq data generation and other public data used
To carry out this study comprehensively, we used 58 RNA-seq data sets generated from more than 20 human tissues and cell lines (Beane et al. 2011; Cabili et al. 2011; Chen et al. 2011a,b; Shapiro et al. 2011; Toung et al. 2011; Gertz et al. 2012; Peng et al. 2012). These data sets include paired-end and single-end reads; moreover, the read lengths range from 35 to 100 bp. All the related information of these RNA-seq data sets can be found in Supplemental Table 1. We downloaded the transcriptome annotations of human reference genome GRCh37/hg19 of RefSeq, Ensembl, and AceView from UCSC (http://genome.ucsc.edu/), Ensembl (release 67 of GRCh37, corresponding to GENCODE 12, http://asia.ensembl.org/index.html), and AceView (version 2010-V3, http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/), respectively. The AceView transcripts that contain unknown bases of “N” were removed in the analyses. We also excluded the duplicated RefSeq human genes and transcripts to generate a unique transcriptome. The known SNPs of human reference genome hg19 were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/ARCHIVE/BUILD.37.1/mapview/). The previously reported trait/disease-associated SNPs (Hindorff et al. 2009) were downloaded from http://www.genome.gov/gwastudies/.
To better conduct this study, we generated two new transcriptomes based on the Ensembl and AceView human gene annotations. Specifically, the genes in one database whose loci overlapped with the exons in another database were not incorporated into the latter database. One new transcriptome was “EnsAce,” which contained the whole Ensembl human transcriptome plus the AceView genes in the intergenic and intronic regions of Ensembl annotation. Another was “AceEns,” and it included the entire AceView human transcriptome plus the Ensembl genes in the intergenic and intronic regions of AceView annotation. Then, the five human transcriptomes of RefSeq, Ensembl, AceView, EnsAce, and AceEns were used to conduct subsequent analyses.
Short-read mapping and expression quantification of genes and isoforms
The 58 RNA-seq data sets were separately aligned onto the human transcriptomes of RefSeq, Ensembl, AceView, EnsAce, and AceEns using Bowtie (version 0.12.8) (Langmead et al. 2009). To enable each mapped read to find its optimal alignments and also to take the multimapped reads into account, we set the parameters for Bowtie with “-a –best –strata -S -m 100 -X 500 –chunkmbs 256.” Bowtie permits two mismatches in the seed by default. Then we followed the MMSEQ (version 0.11.2) pipeline (Turro et al. 2011) (http://bgx.org.uk/software/mmseq.html) to quantify the gene and isoform expression in each sample using the transcriptomes of RefSeq, Ensembl, AceView, EnsAce, and AceEns. The MMSEQ pipeline uses multimapping RNA-seq reads to calculate the expression levels of genes and isoforms, and the expression estimates are in FPKM units (fragments per kilobase of transcript per million mapped reads or read pairs).
Calling differential expression
We separately carried out differential expression calling between brain and UHR (universal human reference RNAs) samples (Chen et al. 2011a) using the human transcriptomes (RefSeq, Ensembl, AceView, EnsAce, and AceEns) at both the gene and isoform levels. We first used the R script (readmmseq.R) in the MMSEQ pipeline to obtain the normalized mapped read counts (by their median deviation from the mean) of each gene and transcript. Then we called the differential expression of genes and isoforms between brain and UHR by using NOISeq (Tarazona et al. 2011) on the five human transcriptomes. Only the genes and transcripts that have the probability of differential expression calculated with NOISeq ≥ 0.95 (q = 0.95) were considered as differentially expressed features.
Calculation of genetic variation distribution
We first calculated the distribution of 17,397,430 known SNPs within the human transcriptome annotations of RefSeq, Ensembl, AceView, EnsAce, and AceEns. The counts of SNPs in the intergenic, exonic, and intronic regions of each transcriptome were determined according to the genomic coordinate of each SNP. We also relocated the genomic distribution of 3041 previously reported trait/disease-associated SNPs that were found in the intergenic regions of RefSeq by using the GWAS assay (Hindorff et al. 2009) on the human transcriptome annotations of Ensembl and AceView. The genomic coordinates of these trait/disease-associated SNPs were compared with the Ensembl and AceView gene annotations of GRCh37/hg19 to determine whether the trait/disease-associated SNPs could be remapped within Ensembl and/or AceView genes.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pengzhan Hu for her assistance in drawing some figures. We are also grateful to Dr. Keely Walker at City of Hope for her help in reviewing the manuscript. This work was supported by the National 973 Key Basic Research Program (Grant Nos. 2010CB945401 and 2012CB910400), the National Natural Science Foundation of China (Grant No. 31240038, 31071162, and 31000590), the Science and Technology Commission of Shanghai Municipality (11DZ2260300), and the Graduate School of East China Normal University.
REFERENCES
- Aparicio SAJR 2000. How to count … human genes. Nat Genet 25: 129–130 [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L 2008. Natural selection has driven population differentiation in modern humans. Nat Genet 40: 340–345 [DOI] [PubMed] [Google Scholar]
- Beane J, Vick J, Schembri F, Anderlind C, Gower A, Campbell J, Luo L, Zhang XH, Xiao J, Alekseyev YO, et al. 2011. Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev Res (Phila) 4: 803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Li R, Shi L, Qi J, Hu P, Luo J, Liu M, Shi T 2011a. Revealing the missing expressed genes beyond the human reference genome by RNA-Seq. BMC Genomics 12: 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Yin K, Shi L, Fang Y, Qi Y, Li P, Luo J, He B, Liu M, Shi T 2011b. Comparative analysis of human protein-coding and noncoding RNAs between brain and 10 mixed cell lines by RNA-Seq. PLoS ONE 6: e28318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Clark RA, She X 2004. An assessment of the sequence gaps: Unfinished business in a finished human genome. Nat Rev Genet 5: 345–354 [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P 2000. Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet 25: 232–234 [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. 2011. Ensembl 2011. Nucleic Acids Res 39: D800–D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Grabherr MG, Guttman M, Trapnell C 2011. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods 8: 469–477 [DOI] [PubMed] [Google Scholar]
- Gertz J, Varley KE, Davis NS, Baas BJ, Goryshin IY, Vaidyanathan R, Kuersten S, Myers RM 2012. Transposase mediated construction of RNA-seq libraries. Genome Res 22: 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: The reference human genome annotation for the ENCODE Project. Genome Res 22: 1760–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 106: 9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S, Yoshida N, Chen A, Garrett L, Sugiyama F, Takahashi S, Yagami K, Wynshaw-Boris A, Yoshiki A 2003. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature 423: 91–96 [DOI] [PubMed] [Google Scholar]
- Kalyana-Sundaram S, Kumar-Sinha C, Shankar S, Robinson DR, Wu YM, Cao X, Asangani IA, Kothari V, Prensner JR, Lonigro RJ, et al. 2012. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell 149: 1622–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Sampas N, Antonacci F, Graves T, Fulton R, Hayden HS, Alkan C, Malig M, Ventura M, Giannuzzi G, et al. 2010. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods 7: 365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA 2009. Circos: An information aesthetic for comparative genomics. Genome Res 19: 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Zheng H, Luo R, Zhu H, Li Q, Qian W, Ren Y, Tian G, Li J, et al. 2010. Building the sequence map of the human pan-genome. Nat Biotechnol 28: 57–63 [DOI] [PubMed] [Google Scholar]
- Liang F, Holt I, Pertea G, Karamycheva S, Salzberg SL, Quackenbush J 2000. Gene Index analysis of the human genome estimates approximately 120,000 genes. Nat Genet 25: 239–240 [DOI] [PubMed] [Google Scholar]
- Marguerat S, Bahler J 2010. RNA-seq: From technology to biology. Cell Mol Life Sci 67: 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM 2011. RNA sequencing: Advances, challenges and opportunities. Nat Rev Genet 12: 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, et al. 2012. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30: 253–260 [DOI] [PubMed] [Google Scholar]
- Pennisi E 2003. Bioinformatics. Gene counters struggle to get the right answer. Science 301: 1040–1041 [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR 2012. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res 40: D130–D135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Mason CE, Smith TM 2012. Limitations of the human reference genome for personalized genomics. PLoS ONE 7: e40294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB 2011. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 7: e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, et al. 2006. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24: 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 (Suppl 2): i215–ii225 [DOI] [PubMed] [Google Scholar]
- Stein LD 2004. Human genome: End of the beginning. Nature 431: 915–916 [DOI] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. 2007. Population genomics of human gene expression. Nat Genet 39: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausberg RL, Simpson AJ, Old LJ, Riggins GJ 2004. Oncogenomics and the development of new cancer therapies. Nature 429: 469–474 [DOI] [PubMed] [Google Scholar]
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. 2008. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960 [DOI] [PubMed] [Google Scholar]
- Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A 2011. Differential expression in RNA-seq: A matter of depth. Genome Res 21: 2213–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry-Mieg D, Thierry-Mieg J 2006. AceView: A comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 7 (Suppl 1): S12.1–S12.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toung JM, Morley M, Li M, Cheung VG 2011. RNA-sequence analysis of human B-cells. Genome Res 21: 991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro E, Su SY, Goncalves A, Coin LJ, Richardson S, Lewin A 2011. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome Biol 12: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK 2008. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4: e1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RB, Chan EK, Cowley MJ, Little PF 2007. The influence of genetic variation on gene expression. Genome Res 17: 1707–1716 [DOI] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, Spector DL 2009. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev 23: 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.