

This paper describes the development of a three-dimensional (3D) computational approach to enable prediction of miRNA–target duplexes and their interactions with the Argonaute protein. The authors did not develop novel modeling techniques; rather, they used available modeling tools. This is a pilot study that demonstrates that structure modeling can be a useful approach in the study of miRNA targeting.
Keywords: microRNA, miRNA tertiary structures, duplex binding free energy, entropy of duplex formation, miRNA–Argonaute docking
Abstract
Current computational analysis of microRNA interactions is based largely on primary and secondary structure analysis. Computationally efficient tertiary structure-based methods are needed to enable more realistic modeling of the molecular interactions underlying miRNA-mediated translational repression. We incorporate algorithms for predicting duplex RNA structures, ionic strength effects, duplex entropy and free energy, and docking of duplex–Argonaute protein complexes into a pipeline to model and predict miRNA–target duplex binding energies. To ensure modeling accuracy and computational efficiency, we use an all-atom description of RNA and a continuum description of ionic interactions using the Poisson–Boltzmann equation. Our method predicts the conformations of two constructs of Caenorhabditis elegans let-7 miRNA–target duplexes to an accuracy of ∼3.8 Å root mean square distance of their NMR structures. We also show that the computed duplex formation enthalpies, entropies, and free energies for eight miRNA–target duplexes agree with titration calorimetry data. Analysis of duplex–Argonaute docking shows that structural distortions arising from single-base-pair mismatches in the seed region influence the activity of the complex by destabilizing both duplex hybridization and its association with Argonaute. Collectively, these results demonstrate that tertiary structure-based modeling of miRNA interactions can reveal structural mechanisms not accessible with current secondary structure-based methods.
INTRODUCTION
Recent crystallographic studies of guide DNA–mRNA–Argonaute complexes have yielded new insights into the molecular mechanisms of mRNA targeting by small interfering RNAs (siRNAs) or microRNAs (miRNAs) (Wang et al. 2008a,b, 2009; Nakanishi et al. 2012). Analysis of X-ray structures supports the view that siRNA/miRNA binding to the target site involves a two-step process, in which initial binding of the target sequence by the seed sequence region (5′ region) of the guide RNA strand is followed by annealing of the 3′ region. These data also reveal the nature of contacts between the Argonaute protein and duplex RNA and specific structural requirements for mRNA cleavage. In complementary NMR studies, the structures of single-chain constructs of Caenorhabditis elegans let-7 miRNA binding to two complementary sites in the lin-41 3′ UTR have provided valuable information about the stability and conformational states of miRNA–target duplex structures, especially the conformations of internal loops and bulges (Cevec et al. 2008, 2010). Thus, tertiary structure analysis of miRNA–target interactions provides a more detailed understanding of siRNA/miRNA function that could open new avenues for developing more accurate algorithms for target prediction.
Current computational methods to identify mRNA targets of miRNAs use rules based on primary and secondary structure information. Target prediction algorithms primarily consider conservation of sequence complementarity to miRNA seeds and miRNA–target duplex hybridization energies (e.g., PicTar [Krek et al. 2005; Lall et al. 2006] and TargetScan [Friedman et al. 2009]) and may incorporate additional features of mRNAs such as target site accessibility (e.g., PITA [Kertesz et al. 2007], mirWIP [Hammell et al. 2008], and Vfold [Cao and Chen 2012]). The common element of these algorithms is the presence of base-pair matches in the seed region—a 7-nt stretch starting at the first or second position from the 5′ end of the miRNA (Ambros 2004)—with base pairs in the 3′ region playing only a minor role in target selection. This partitioning of the miRNA–target duplex is consistent with crystal structures of Argonaute–duplex complexes, which show that the 3′ base pairs are annealed only after the seed base pairs are formed (Wang et al. 2009). Importantly, by anchoring its backbone to the Argonaute protein, the miRNA adopts an ordered conformation that exposes the bases in the seed region to solvent (Wang et al. 2009), which lowers its entropy and thereby enhances its affinity for the target (measured by the dissociation constant) by up to ∼300-fold compared with the free seed sequence (Parker et al. 2009). Although current target prediction algorithms do not incorporate these protein–RNA interactions, their employment of secondary structure hybridization makes them efficient, allowing genome-scale surveys of 3′-UTR targets. Such surveys have revealed that miRNA families can potentially target a significant percentage of protein-coding transcripts (Farh et al. 2005; Grun et al. 2005; Krek et al. 2005; Lall et al. 2006; Chi et al. 2009; Friedman et al. 2009; Zisoulis et al. 2010).
The development of three-dimensional (3D)–based computational prediction of miRNA–target interactions could provide additional tools for improving current target prediction algorithms. Essential features such as conformational diversity of duplex RNAs generated by mutations, flexible internal loops, interactions with the Argonaute protein, and charge screening by ions can only be adequately assessed using 3D-based models. Interestingly, recent mapping of the spatial distribution of ions in C. elegans using tomography showed wide concentration variations throughout the organism, suggesting that local ionic conditions could significantly influence molecular interactions (McColl et al. 2012). These effects highlight some of the current challenges to the modeling of miRNA–target binding. However, modeling such effects is becoming increasingly feasible in view of recent advances in RNA structure prediction (Das and Baker 2008; Parisien and Major 2008; Das et al. 2010) and the availability of efficient algorithms for computing ion screening interactions (Baker et al. 2001; Rocchia et al. 2001).
Several molecular dynamics simulation studies and thermodynamic analyses have been performed to elucidate the interactions within ternary complexes of Argonaute bound to guide and target RNAs and to characterize the conformations of miRNA–target duplexes (Balasubramanian et al. 2010; Wang et al. 2010; Paciello et al. 2011). Dynamic simulations based on a crystal structure of the ternary complex have shown that correlated movements of the Argonaute’s PAZ, MID, and PIWI domains facilitate target binding and product release (Wang et al. 2010). All-atom dynamic simulations of C. elegans lin-4::lin-14 and let-7::lin-41 interactions have provided useful data on binding energies and have helped characterize the conformations generated by imperfect base-pairings in the structures (Balasubramanian et al. 2010; Paciello et al. 2011). While such dynamic studies have yielded mechanistic insights, their requirement for extensive simulations has so far limited their focus to detailed analysis of specific systems. For larger-scale structural analyses of miRNA–target interactions, more efficient computational methods need to be developed.
To overcome some of the limitations of current computational approaches, we have built a structure-based pipeline to enable the analysis of miRNA–target duplexes and their interactions with the Argonaute protein under varying ionic conditions. We use an (all-atom) RNA structure assembly method to generate structure ensembles from experimental or predicted secondary structures; we then use a hybrid all-atom and implicit-solvent force field for ions and water molecules to predict native-like RNA duplex structures in the ensembles. This approach avoids computationally expensive simulations of solvent molecules. Comparisons of predicted results with experimentally determined RNA structures and duplex binding energies show that our computational approach is fairly accurate. Moreover, this approach expands our understanding of post-transcriptional miRNA regulation by demonstrating that structural distortions induced by mutations in the seed duplex influence miRNA activity both by destabilizing the duplex itself and by weakening duplex–Argonaute interactions.
RESULTS
Development of computational pipeline
Our computational pipeline for predicting RNA structures and computing duplex binding energies, illustrated in Figure 1 and detailed in Materials and Methods, incorporates the following steps:
We first generate an ensemble of de novo all-atom RNA duplex structures for a given secondary structure by assembling a series of short fragments (≤4 nt) derived from experimentally determined structures using the MC-Sym algorithm (Parisien and Major 2008).
We predict RNA 3D structures based on the lowest energy criterion, where RNA interaction energies are computed at specific ionic conditions using a continuum electrostatic model.
We compute the duplex binding free energy by evaluating the enthalpy and entropy changes associated with duplex or protein–duplex formation.
FIGURE 1.
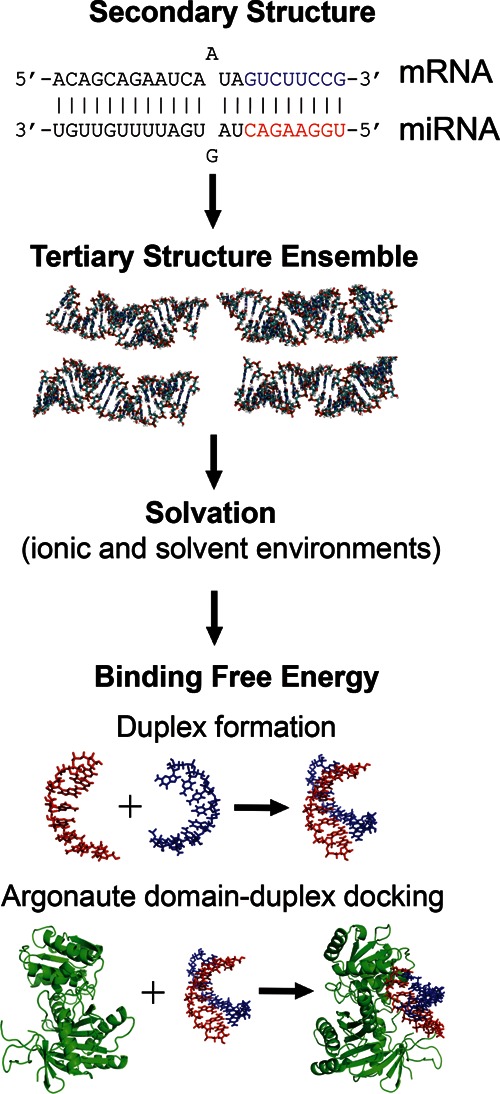
Computational pipeline for generating, solvating, and computing binding energies of 3D RNA structures, starting from a secondary duplex structure. The guide (red) and target (blue) strands in the seed region are highlighted. First, a conformational ensemble is generated using the MC-Sym algorithm. Second, the RNA interaction energies are computed at specific ionic conditions using a continuum electrostatic model. Third, the binding free energy is obtained by evaluating the enthalpy and entropy changes associated with either duplex formation (vs. free strands) or Argonaute–duplex formation (vs. free duplex), as illustrated here for docking of the PIWI/MID domain of Thermus thermophilus Argonaute to the given seed duplex.
In step (a), we implement a hierarchical structure assembly approach, in which 3D structures for longer duplexes are built by sequential addition of short fragments from known 3D structures (Parisien and Major 2008), guided by base-pairing of specific secondary structures (which are known for many miRNA–target duplexes) (Sethupathy et al. 2006). In step (b), we use an all-atom, physics-based force field rather than a knowledge-based force field (derived from atom or residue contact frequencies in database structures), as used in prior work (Parisien and Major 2008).
To assess the utility of this 3D modeling approach, we evaluated our ability to accurately model experimental results for RNA duplex structures and binding energies and compared the performance with a two-dimensional (2D) folding algorithm (Hofacker 2003). We also calculated the binding energies of the 3D models in different ionic conditions to gain insight into how environmental factors impact miRNA–target recognition. Finally, we explored how different structural features of duplexes influence the stability of Argonaute–RNA interactions by performing docking simulations.
3D-based algorithm reproduces NMR structures of let-7–target constructs
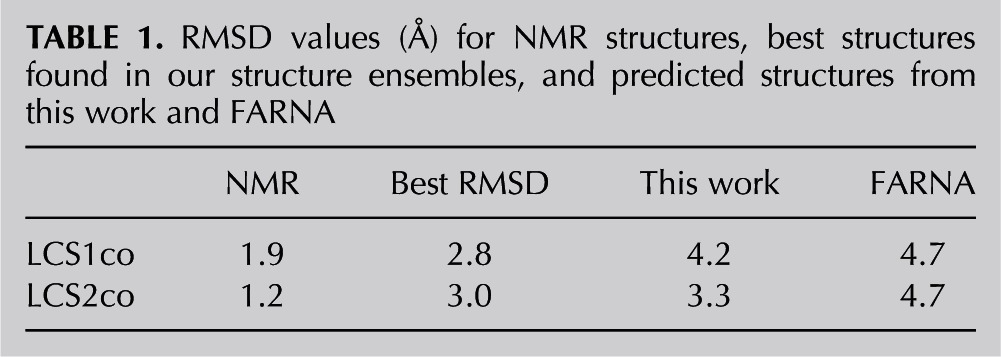
We first assessed performance by comparing our 3D predictions with solution structures of miRNA–target duplexes obtained by NMR spectroscopy. The C. elegans let-7 miRNA is known to bind two complementary sites in the lin-41 3′ UTR that are separated by 27 nt (Vella et al. 2004). Both let-7 complementary sites (LCSs) are required for down-regulation of lin-41, although the mechanism of their cooperative function is not yet understood (Vella et al. 2004). Recently, solution structures have been solved for let-7-LCS duplexes from both sites (LCS1co and LCS2co) using single-chain constructs with a closing loop and modified bases to enhance structural stability (Cevec et al. 2008, 2010; see also Materials and Methods). For reference, the 10 lowest-energy NMR solution structures for each construct are available in the Protein Data Bank (PDB); the root mean square deviation (RMSD) between these is 1.9 Å for LCS1co and 1.2 Å for LCS2co.
To test our procedures for constructing and assessing the energetics of RNA structures, we generated ensembles of 1000 3D structures for both LCS constructs using the MC-Sym algorithm, an RNA structure assembly method (Parisien and Major 2008). We then ranked the structures using the total energy, which includes contributions from bonded and nonbonded (van der Waals, electrostatic, and solvation) interactions, and we superimposed our predictions with the NMR solution structures. We define the average RMSD for each structure in an ensemble as the mean value derived from superimposing its structure with the 10 available corresponding NMR models (Materials and Methods; Fig. 2 shows representative examples; Table 1 summarizes all relevant RMSD values). Among the five top-ranking (lowest-energy) predicted structures for each construct, the lowest average RMSD values were 4.2 Å for LCS1co and 3.3 Å for LCS2co; the probabilities of obtaining these RMSD values in the structure ensembles are 0.085 and 0.007, respectively, indicating the greater significance of the predicted LCS2co structure. With a more stringent criterion combining the average RMSDs of all top five structures, the mean RMSD was 5.2 Å for LCS1co and 4.2 Å for LCS2co. The lowest average RMSD values in the ensembles were 2.8 Å and 3.0 Å, but these structures had slightly higher energies than the top-ranking structures because the model energy function used is imperfect. The better predictive performance we see (smaller RMSD) for LCS2co could reflect its higher stability, as observed experimentally (the melting temperature of LCS2co is 7 K higher than that of LCS1co) (Cevec et al. 2008, 2010). Overall, these results indicate that our structure assembly procedures sampled adequately small RMSD structures (e.g., the ensemble sizes required to attain the lowest RMSD values were well below the 1000: 281 for LCS1co and 21 for LCS2co). The RMSD values obtained here are comparable to those from previous studies of similarly sized RNAs; for example, the MC-Sym method reported an average RMSD of 2.7 Å for best structures of 30- to 47-nt RNAs (Parisien and Major 2008), and the FARNA approach yielded an average RMSD of 3.4 Å for 28- to 46-nt RNAs (Das and Baker 2007). This comparison of our best predictions with NMR structures shows that the structure-based approach we have developed, combining structure assembly and a physics-based force field, can reproduce the global conformational features of experimentally determined structures.
FIGURE 2.
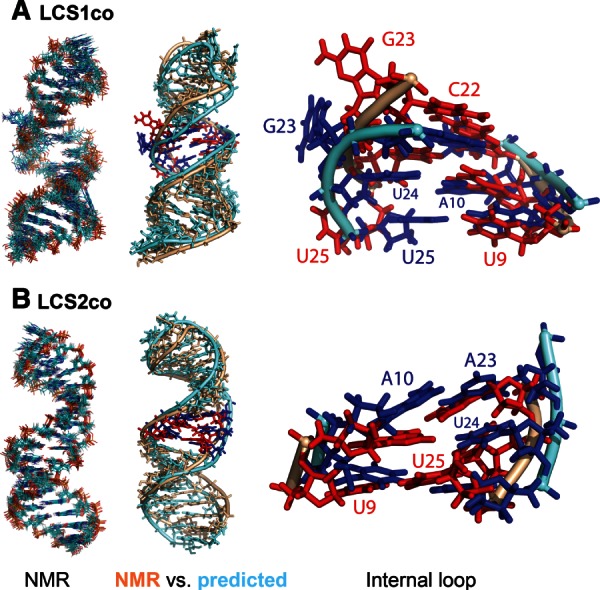
NMR models and predicted structures for single-stranded constructs of C. elegans let-7 miRNA–target site duplexes (A) LCS1co and (B) LCS2co. (Left) Superposition of 10 lowest-energy NMR structures for each construct. (Middle) Structure alignments of individual representative NMR (wheat color) and predicted (light blue) structures, highlighting bases in the internal loop (NMR, red; predicted, blue). (Right) Magnified view of internal loops from aligned structures, with bases labeled. For substructure alignments, structures were partitioned into internal loop, hairpin (upper), and stem (lower) regions. The predicted structures shown have average RMSD values of 4.2 Å for LCS1co and 3.3 Å for LCS2co (relative to the 10 lowest-energy NMR structures for the corresponding construct).
TABLE 1.
RMSD values (Å) for NMR structures, best structures found in our structure ensembles, and predicted structures from this work and FARNA
To evaluate the 3D modeling results in greater depth, we analyzed prediction accuracy by region upon partitioning each predicted duplex structure into three substructures: stem, internal loop, and hairpin loop (see Fig. 2). The respective RMSD values by region are 2.4, 3.1, and 4.1 Å for LCS1co and 1.4, 2.35, and 2.0 Å for LCS2co. These comparisons show that helical and internal loop regions are well reproduced and that, as expected, the stem region has the smallest RMSD among the three regions. The flexible internal loop of LCS1co has an RMSD value twice as large as that for LCS2co (4.1 Å vs. 2.0 Å). The predicted internal loops of both constructs show similarities, but also subtle differences, with their corresponding NMR structures. For LCS1co (Fig. 2A), we focus on the internal loop’s bases A10, U24, U25, and G23. The NMR structures exhibit a distorted base-pairing configuration between A10 and U24, and also show that the G23 base is mobile and flipped out of the helical axis. These features are also seen in the best predicted LCS1co structure. The main difference is that U25 is positioned near the helical axis in the predicted structure, but outside the helical axis in the NMR structure. For LCS2co (Fig. 2B), the NMR structure has an organized internal loop with noncanonical base pair A23:A10 and base triple U24:U25:U9, whereas the predicted LCS2co structure contained noncanonical base pairs A23:A10 and U25:U9 (without involving U24 in a base triple). Since base triples occur far less frequently than base pairs (Xin and Olson 2009), especially the base triple UUU (Abu Almakarem et al. 2012), a prediction algorithm that uses database fragments (like the one used here) is unlikely to generate such higher-order base interactions. Thus, our detailed structural comparisons reveal both strengths and limitations of our 3D-based algorithm to predict specific conformational features.
Total energy function discriminates native-like from nonnative structures
The quality of our physics-based energy function can be assessed by comparison with experimental structures and established computational methods. First, we plotted the total energy versus RMSD for all 1000 LCS1co and LSC2co ensemble structures aligned with the low-energy NMR structures (Fig. 3). The funnel-like shape of the energy–RMSD scatterplots indicates that low energies correlate with low RMSD values; the generated structures with the lowest average RMSD values (∼3 Å) have total energies that are only ∼2% higher than their reference NMR structures. Thus, the accuracy of predicted structures and the favorable RMSD–energy correlations indicate that our RNA energy function provides a satisfactory description of interactions in RNA molecules. Second, we evaluated the performance of our energy function in comparison with the FARNA (Das and Baker 2007) scores for the LCS1co and LCS2co structure ensembles (Supplemental Fig. S1). FARNA computes energy terms for a coarse-grained RNA model with a single interaction site at each base; while both methods use the fragment assembly approach, this simplified modeling permits more general RNA folding simulations when starting from a disordered fold than possible with the MC-Sym algorithm. Using the best of five top-ranked structures, the FARNA-predicted structures for LCS1co and LCS2co have the same RMSD value of 4.7 Å, compared with 4.2 Å and 3.3 Å, respectively, for results from our total energy function. For all five top-ranked FARNA structures combined, the mean RMSD values for LCS1co and LCS2co are 5.1 Å and 6.2 Å, respectively, versus 5.2 Å and 4.2 Å from our 3D method. These results show that our more detailed all-atom modeling of molecular interactions with continuum electrostatic forces performs favorably in comparison with established methods. A comprehensive assessment involving diverse RNA folds will be required to more rigorously test the performance of different existing energy functions (Parisien and Major 2008; Jonikas et al. 2009; Das et al. 2010).
FIGURE 3.
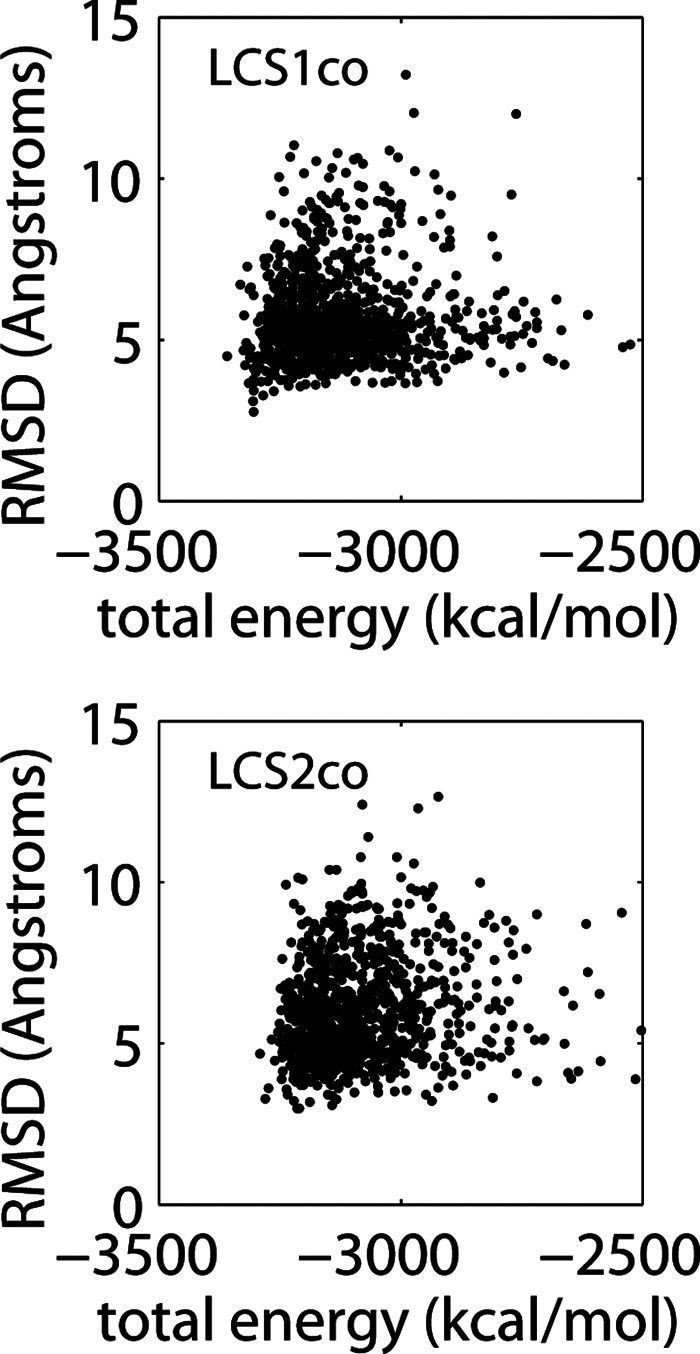
Total energy versus RMSD for LCS1co (top) and LCS2co (bottom). Plotted are individual data points for 1000 computed structures, each representing an individual structure’s total energy and its average RMSD value obtained from superposition with the 10 low-energy NMR structures.
3D models reproduce experimental binding enthalpies, entropies, and free energies
Current miRNA–target prediction programs rely on secondary structure models to compute the miRNA–target binding free energy. To test the performance of our 3D computational model, we compared computed enthalpic and entropic terms of the binding free energy with experimental results from titration calorimetry measurements (Parker et al. 2009) for eight duplexes with and without bound Argonaute under a constant solvent condition (150 mM KCl and 10 mM MgCl2 at 20°C) (Fig. 4; Supplemental Table S1). Overall, the binding energies for the free duplexes (Ago-free) computed using the 3D computational model showed reasonable agreement with experimental data. For enthalpic energies (ΔE), the agreement is generally better for duplexes containing 7 and 8 bp than for duplexes with 6 bp or with a mismatched base pair, reflecting the accuracy of MC-Sym’s procedures for assembling longer perfect duplex structures. The average computed enthalpy for the eight Ago-free duplexes is −40.5 kcal/mol, whereas the corresponding experimental data for Ago-free and Ago-bound duplexes are −47.5 and −36.6 kcal/mol, respectively. (Note that one duplex [duplex 2] has a large standard deviation [70 kcal/mol] due to the experimental limitations of finding optimal parameters [guide RNA concentration and dissociation constant] to accurately determine the duplex’s enthalpy [JS Parker, pers. comm.]). For the entropic term (−TΔS), the values from the 3D model are consistently lower (by ∼20% on average) than experimental data for the Ago-free duplexes, while for Ago-bound duplexes, agreement is within 15% except for two cases (one with 6 bp and another with a mismatch). The free energies (ΔG = ΔE − T ΔS) showed fair agreement with the experimental data: the average computed value is −9.6 kcal/mol versus experimental values of −7.8 and −9.1 kcal/mol for Ago-free and Ago-bound duplexes, respectively.
FIGURE 4.

Comparison of experimental and computed binding free energy terms for different RNA duplexes. (A) The eight structures analyzed, labeled 1–8. (B,C) Enthalpy (ΔE), entropy (−T ΔS), and free energy (ΔG) of free (left) or Argonaute-bound (right) duplexes, as determined from titration calorimetry experiments (crosses) versus tertiary (circles) and secondary (squares) structure computational methods (Supplemental Table S1 also summarizes all numerical values). Experiments and 3D calculations were performed under a constant solvent condition (150 mM KCl and 10 mM MgCl2 at 20°C); 2D calculations assumed 1 M NaCl. All entropies were computed at experimental guide miRNA concentrations. Error bars represent the standard deviation (SD) of experimental uncertainty in enthalpy.
For the Argonaute-free case, the less favorable agreement between computed and experimental duplex binding energies (ΔG = Gduplex − Gstrand1 − Gstrand2) stems from the approximations used for the conformations of free RNA strands: Since our method approximates these using the strands in the duplexes (which are more ordered than free strands), the computed entropy change is lower than the experimental value (average of 31.0 kcal/mol vs. 39.8 kcal/mol). In contrast, we find a better agreement between the computed binding energies for free duplexes and the experimental duplex binding energies where the guide strand is bound to the Argonaute (31 kcal/mol vs. 27 kcal/mol) because the strand conformation is stabilized by the protein. Since a free RNA strand has floppy conformations, its entropy cannot be computed accurately using the vibrational entropy method used here (which provides an efficient but rough estimate of duplex formation entropy).
Since the entropy of macromolecules is a difficult quantity to compute (Leach 1996), we have followed prior studies by estimating the entropy using the molecule’s normal mode frequencies, which assume harmonic interactions between the atoms (Tidor and Karplus 1994; Kollman et al. 2000). The semiquantitative agreement (deviations of 15%–20%) between our results and calorimetry data for duplex entropies reflects this approximation (Supplemental Table S1). The normal mode method is also limited to small systems (<1500 atoms or an ∼20-bp duplex with hydrogen atoms) because of the need to diagonalize a large interaction matrix of size 3N by 3N, where N is the number of atoms. Alternative methods for computing the entropy currently under development could improve the accuracy of the free energy and increase the molecular size that can be considered (Liu and Chen 2010; Xu et al. 2011).
The generality of our 3D approach is illustrated by comparison with how binding free energies are obtained from secondary structure algorithms. Current 2D folding programs typically assume a standard ionic condition (i.e., 1 M NaCl) (Xia et al. 1998) and do not allow specification of monovalent and divalent ionic concentrations. For short perfect duplexes and duplexes with a single GU wobble or mismatch, binding free energies can be predicted with reasonable accuracy: Both 2D and 3D structure calculations agree well with thermodynamic data (Fig. 4; Supplemental Table S1). For the eight Argonaute-free duplexes we considered, the average free energy predicted by 2D calculations is −8.6 kcal/mol (at the standard ionic condition of 1 M NaCl), compared with −9.6 kcal/mol and −7.8 kcal/mol for 3D-computed and experimental values, respectively (at 150 mM KCl and 10 mM MgCl2). Thus, although both 2D and 3D methods provide satisfactory agreement with experimental data, they involve difference input ionic conditions: Unlike our 3D method, which computes binding free energies at specified ionic conditions, 2D algorithms assume a (fixed) standard ion condition (1 M NaCl). As shown below, this approximation is only satisfactory above threshold ionic strengths (∼150 mM monovalent or ∼1 mM divalent ions), when the duplex binding free energy saturates. Thus, our 3D method aims to provide more consistent and general free energy estimates over a broad range of ionic strengths (e.g., 0–1 M for monovalent ions and 0–10 mM for divalent ions).
Duplex binding free energy saturates at threshold mono- and divalent ion concentrations
Different cell types can have widely varying concentrations of monovalent and multivalent ions. For example, the mammalian cell typically maintains ∼150 mM monovalent ions (e.g., K+, Na+), whereas the squid axon contains ∼500 mM monovalent ions and <1 mM divalent (e.g., Mg2+, Ca2+) ions (Lodish et al. 2000). In addition, recent tomography mapping of ion distributions in C. elegans showed that for intestinal cells the spatial distributions of ions are highly nonuniform (e.g., 0–1 mM for Ca2+, 0–14 µM for Mn2+, and 0–18 µM for Fe3+) (McColl et al. 2012). Because ion species can significantly alter nucleic acid interactions, it is important to quantify the effects of ionic conditions on miRNA–target binding free energy.
To investigate the effects of ions on miRNA binding properties, we calculated the binding free energy for a perfect duplex and another with a GU wobble across different mono- or divalent ion concentration regimes (Fig. 5). In each regime, the concentration of one ion species is varied across a range (0–300 mM for monovalent and 0–10 mM for divalent ions) while the other is held constant, either at zero (Fig. 5, crosses) or above saturation for the binding free energy (Fig. 5, circles). These concentration ranges explore the entire range of binding free energy behavior under saturating and nonsaturating conditions. Since ion species in cells can span wide concentration ranges (McColl et al. 2012), the concentration mixtures investigated are relevant for understanding miRNA activity in vivo. In particular, understanding the sensitivity of duplex binding free energy in environments with multiple ions could aid computation of duplex free energies. Moreover, comparing duplexes with and without a GU base pair probes the influence of noncanonical base pairs on the saturation behavior.
FIGURE 5.

Concentration dependence and saturation behavior of the binding free energy as a function of monovalent (top) and divalent (bottom) ions for a perfect duplex (left; duplex 1 in Fig. 4) and the same duplex but with a GU wobble (right; duplex 4 in Fig. 4). In each plot, the concentration of the other ion species is held constant either at 0 mM (crosses) or above its own saturation (circles). (Arrow) Concentration at which the binding free energy plateaus (solid line).
We find that at low ionic concentrations (monovalent ions <25 mM or divalent ions <1 mM), the duplex free energy is sensitive to concentration change. In contrast, when one ion species is already saturating, the binding free energy remains nearly constant; a saturation plateau is reached for both duplexes when the concentration exceeds ∼150 mM for monovalent ions (in the absence of divalent ions, cdv = 0), or ∼1 mM for divalent ions (in the absence of monovalent ions, cmv= 0). Both duplexes with and without a GU base pair have a similar concentration dependence behavior. In all the cases shown, the duplex binding free energies at saturation are about −10.3 kcal/mol and −8.3 kcal/mol for the perfect duplex and GU wobble-containing duplex, respectively. This suggests a 2 kcal/mol loss of affinity for the duplex with a GU wobble.
The predicted saturation regime of miRNA–target duplexes has several implications. First, there are spatial regions in C. elegans where divalent ion (e.g., Ca2+ and Mn2+) concentrations fall below the saturation concentration of ∼1 mM (McColl et al. 2012). It is not known whether this deficit is compensated by other unmapped ions. Second, the threshold concentrations can be exploited to guide both computational approaches and in vitro miRNA experiments. In particular, it implies that the solution structures for LCS1co and LCS2co at 30 mM monovalent ions (discussed above) were obtained below the threshold monovalent ion concentration. Third, the saturation behavior of duplex free energy implies that computations of miRNA–target duplexes using 2D algorithms, which assume standard ionic condition (1 M monovalent ions), are only valid in the saturation regime; below the saturation region, explicit treatment of ionic effects must be taken into consideration. A schematic of the free energy landscape using the computed enthalpy and entropy values illustrates the sensitivity of miRNA–target recognition to ion concentrations (Supplemental Fig. S2; Supplemental Material).
Characterization of energetic and conformational features of the Argonaute–duplex complex reveals the contributions of different interaction forces to its stability
Apart from Argonaute’s role in reducing the entropy of guide miRNA conformations, the nature of its interactions with the duplex RNA and the effects of RNA sequence variations have not been extensively explored. Specifically, distortions caused by mutations and naturally occurring bulges in the duplex can potentially alter Argonaute–duplex binding affinity and thus influence miRNA activity. We therefore assessed the effects of mutations in mRNA sequence on computed binding affinities of Argonaute–duplex complexes and correlated the binding energies with reported in vivo activities (Fig. 6; Brennecke et al. 2005). Since neither the C. elegans nor Drosophila melanogaster ternary structures have been determined, we performed simple dockings of RNA duplexes to the Argonaute protein using the available protein–DNA:RNA duplex ternary structure for Thermus thermophilus (Wang et al. 2009) (Materials and Methods).
FIGURE 6.

Analysis of interactions between the T. thermophilus Argonaute MID/PIWI domain and seed duplexes of D. melanogaster miR-7 with single point mutations in mRNA at base positions 1–8; 0 indicates the wild-type duplex with no mutation in panels B, C, and E. (A) Composition of wild-type duplex (duplex 0), with labeled base-pair positions 1–8. (B) Duplex binding free energy versus Argonaute–duplex binding energy for each duplex, indicating the specific substitutions in the mRNA at each position. (C) Interaction energy components (van der Waals, nonpolar solvation, and electrostatic) of Argonaute–duplex binding energy for all mutation positions. (D) r2 statistics versus the Q value (weight of the duplex energy term) derived from linear least squares fit between miRNA activity and an effective Argonaute–duplex binding free energy (Eq. 1). (E) 3D structure models of Argonaute–seed duplexes, illustrating wild-type duplex (0) and duplexes with point mutations (highlighted by red nucleotides) at each of the eight positions in the mRNA strand (1–8). Significant structural distortions occur in the mRNA strand (green) but only minor distortions in the miRNA strand (blue). These distortions depend on the mutation position, and they weaken the duplex’s binding affinity for Argonaute.
To identify the trends of binding affinities, we compared all complexes with and without single-nucleotide mismatches in the RNA duplex (Fig. 6A,B). Relative to the wild-type (perfect) duplex, we find that point mutations clearly destabilize both the duplex and Argonaute–duplex binding energies (Fig. 6B), which range from around −15 kcal/mol to −10 kcal/mol, and −140 kcal/mol to −90 kcal/mol, respectively (the latter energy range excludes the relatively constant Argonaute entropy contribution; see Materials and Methods). The distribution of binding energies for single base-pair mismatches has a funnel-like shape (Fig. 6B), revealing variable dispersion away from that of the wild-type duplex due to the complex interplay of the type and position of mismatches. Additional insights can be gained by examining the individual contributions of van der Waals, nonpolar solvation (or hydrophobic), and electrostatic forces involved in the Argonaute–duplex complex formation (Fig. 6C). The van der Waals energies (around −115 kcal/mol) are the dominant stabilizing force but remain nearly constant for all mutations, as do the hydrophobic (nonpolar solvation) energies (−49 kcal/mol). Thus, these two energy terms provide nonspecific stabilizing forces. In contrast, the electrostatic energies (10–70 kcal/mol with a mean of 36 kcal/mol) show significant variations across mutations. For example, duplexes 0–3 (numbered by the position of the single point mutation) show increasingly unfavorable electrostatic energies; a similar trend is seen for duplexes 4–8. These results indicate a possible positional dependence of electrostatic interactions arising from the mutations and the periodic nature of the RNA helix.
Models of Argonaute–duplex complexes (Fig. 6E) show that single base-pair mismatches distort the RNA helix and also alter its interactions with the Argonaute protein. Mutations in the mRNA sequence distort both its backbone and base conformations, as well as its interactions with the guide miRNA strand, which in contrast displays relatively small backbone distortions due to its contacts with the protein (see, for example, the bulges formed by the mutations at positions 3, 4, and 5 in Fig. 6E). These structural distortions are local, however, since superimposing the conformations of docked native and mutated duplexes yields small RMSD values between 1.5 Å and 2 Å. The consequence of the distortions is a less stable association of the Argonaute-bound guide strand with the mRNA target sequence (Fig. 6B), leading to reduced recognition efficiency and consequently lower miRNA activity.
miRNA activity is influenced by both duplex and Argonaute–duplex binding affinities
Current analysis of miRNA function is primarily based on RNA–RNA interactions exclusively (Bartel 2009; Cao and Chen 2012). The contribution of the above-characterized RNA–protein interactions to miRNA activity is not known. To quantify the relative importance of duplex and Argonaute–duplex binding energies, we propose a simple model for an effective binding free energy function for a given “query” duplex:
 |
(1) |
where ΔGdup is the duplex binding free energy; ΔEAgo–dup, ΔE0Ago–dup are the Argonaute–duplex binding energies for the query and wild-type duplexes, respectively (including electrostatic, van der Waals, and hydrophobic contributions); and Q describes the relative contributions of the duplex and duplex–protein complex binding energies. The binding energy of the complex is defined relative to that of the wild-type (perfect) duplex, so the wild-type duplex activity is determined solely by its duplex binding free energy. Thus, the second term of Equation (1) measures the effects of mutation-induced structural distortions on duplex–protein interactions. We then performed linear regression for ΔGAgo-dup and experimental miRNA activities associated with the mutations (excluding the mutation at position 1 because the X-ray structure shows that the first base pair is disrupted in the complex) (Wang et al. 2009). The r2 versus Q plot shows a peak at Q = 0.96 (Fig. 6D), meaning the duplex binding free energy ΔGdup accounts for most (96%) of the variation, with duplex–protein binding contributing only 4%; Supplemental Figure S3 illustrates the linear dependence of miRNA activity on ΔGAgo-dup for fixed Q = 0.96. RNA–RNA binding affinity dominates in these examples because the single base-pair mismatches induced only minor structural changes. Larger structural deviations from the perfect seed duplex, which are present in imperfect duplexes with bulges or multiple base-pair mismatches, could magnify the role of the duplex–protein interaction term (i.e., larger 1 − Q value). Examples of such imperfect seed duplexes include C. elegans let-7::lin-41, let-7::daf-12, and lsy-6::cog-1 (Sethupathy et al. 2006), as well as various G-bulge sites regulated by mouse miR-124 (Chi et al. 2012).
This analysis indicates that the influence of Argonaute on miRNA function depends on the conformations of the duplex and its interactions with the protein, features that cannot be captured using 2D analysis. Future work to understand the general miRNA activity–energy relation will require a more comprehensive analysis of many duplexes with different types of mutations and bulges in the seed regions.
DISCUSSION
Tertiary structure-based computational methods are required for more detailed analysis of the structural mechanisms of miRNA target recognition and post-transcriptional regulation. We have integrated a set of computational methods to enable analysis of structural and energetic contributions to the activity of Argonaute–miRNA–target complexes. Specifically, we have exploited advances in RNA folding (MC-Sym) (Parisien and Major 2008), efficient implementation of the Poisson–Boltzmann equation solver (APBS) (Baker et al. 2001) to treat ionic strength effects, hybrid all-atom and continuum interactions (Srinivasan et al. 1998; Kollman et al. 2000), and various molecular modeling tools (minimization algorithms, vibrational frequency solver). These analysis tools have allowed us to predict with good accuracy the NMR structures of let-7–target constructs (Cevec et al. 2008, 2010), compute duplex entropy and binding free energy necessary for comparing with calorimetric data (Parker et al. 2009), determine the threshold monovalent and divalent ion concentrations for seed miRNA–target duplexes, and analyze the interactions in the Argonaute–duplex complex that correlate with miRNA activity (Brennecke et al. 2005).
Unlike molecular dynamic simulations of miRNA systems (Balasubramanian et al. 2010; Wang et al. 2010; Paciello et al. 2011), our use of the combined all-atom and continuum approach to miRNA interactions leads to greater computational efficiency, as shown by the feasibility of analyzing a number of duplex-only structures and duplex–Argonaute complexes. On a modern processor, to minimize a duplex structure and compute its energy components typically takes ∼0.5 h of CPU time; we computed the electrostatic component using the APBS software on multiprocessors (up to 16). The computation of the entropy change associated with the formation of an 8-bp duplex takes ∼1 h. Even with modest computing resources, we estimated that our computational scheme can analyze 102–103 structures. This advantage over dynamic simulation methods could prove useful for assessing candidate miRNA targets from current prediction algorithms and high-throughput experiments.
Computing the electrostatic term of the binding energy using the Poisson–Boltzmann equation provided a realistic modeling of ionic screening of RNA duplexes and RNA–protein complexes. We found that only the electrostatic contribution to the duplex–Argonaute binding showed positional dependence reflecting mutations in the target mRNA sequence, unlike the hydrophobic and van der Waals energy contributions. In addition, our approach allowed us to compute the threshold concentrations for monovalent and divalent ions beyond which the seed duplex binding energy saturates. These studies highlight the advantages of using continuum electrostatics in structure prediction and assessment.
We have used NMR let-7-target structures to test the structure assembly approach and the hybrid force field. The attained accuracy of ∼3.8 Å RMSD for two ∼35-nt RNAs indicates that the predicted miRNA–target conformations are sufficiently reliable for evaluating binding energies and for performing docking studies. Since the full miRNA–target duplexes contain ∼40 bases, the availability of experimental data for slightly larger systems would be preferable; the available crystal structure of a full duplex is a guide DNA–RNA hybrid (Wang et al. 2009). The accuracy of our structure prediction method can be improved by using larger structure ensembles (for 1000 structure ensembles used the lowest RMSD achieved is ∼3 Å), although at a linearly increased cost of evaluating the interaction energies. The structure sampling protocol could also be improved by using a recent physics-based algorithm for RNA structure prediction (Cao and Chen 2011).
Our analysis of duplex–Argonaute complexes with single base-pair mismatches in the seed region shows that assessment of the functional potential of candidate miRNA–target systems must incorporate the effects of both duplex and duplex–Argonaute binding affinities. Support for this scenario is implicit in existing experimental and computational works. For example, in vivo experimental studies of various miRNA–target constructs have demonstrated the shortcomings of duplex-only considerations to account for miRNA activities (Brennecke et al. 2005; Didiano and Hobert 2008). Moreover, a recent computational study suggests that the functional status of some imperfect seed duplexes with a bulge larger than 1 base cannot be correctly assessed based on secondary structure calculations (Cao and Chen 2012). It implies the existence of structural “selection rules” that depend on detailed aspects of base-pair mismatches or bulges (e.g., their positions in the seed duplex) and possibly their interactions with the Argonaute protein. Indeed, we observed in our analysis of Argonaute–duplex complexes the positional dependence of duplex interactions arising from electrostatic forces. This kind of analysis could be applied to derive energetic selection rules for various duplex systems (Brennecke et al. 2005; Kertesz et al. 2007) and thus improve assessment of miRNA function.
Our 3D structure-based approach provides complementary tools to current computational methods toward the development of a comprehensive algorithm that can more accurately identify miRNA target sites. Although current target-finding algorithms based on primary and secondary structure considerations can identify many known and candidate targets of various miRNA families, ∼30% of functional miRNA–target duplexes (Kertesz et al. 2007) may still be incorrectly assigned (Cao and Chen 2012). This is likely due to altered interactions between Argonaute proteins and imperfect duplexes with bulges and base-pair mismatches that naturally occur in miRNA–target systems and are more accurately modeled in a structural context. An integration of secondary and tertiary structure-based methods (like those we present) is thus needed to achieve greater accuracy in miRNA–target prediction. More broadly, improvements in computational tools are needed to meet the challenges of interpreting genome-scale data to probe post-translational regulatory mechanisms in different cell types and animal developmental stages (Hammell et al. 2008; Chi et al. 2009; Zhang et al. 2009; Zisoulis et al. 2010).
MATERIALS AND METHODS
Generation and refinement of 3D structure ensemble
We used the MC-Sym algorithm to build single- and double-stranded RNA structures using input secondary structures (Parisien and Major 2008). MC-Sym builds RNA structures from single-stranded RNA fragments and stacked base pairs from double-stranded nucleotide cyclic motifs (NCMs) or fragments found in solved structures; this approach is especially suited for building structured RNAs. As elaborated below, we used a physics-based force field rather than a knowledge-based potential used in previous structure prediction studies using the same algorithm (Parisien and Major 2008). For each 2D structure, we generated a maximum of 1000 3D structures, which is ample for the small, structured RNAs (typically <20 nt) considered here. The speed and number of 3D structures generated are determined by the availability of candidate RNA fragments in the fragment database; we used structure diversity parameter values (smallest RMSDs allowed among fragments) between 1 Å and 3 Å. For single-stranded RNA folds (LCS1co and LCS2co) with large internal loops, we specified single-stranded template fragments of 2–4 nt in structure generation to improve conformational sampling. For much more intensive ionic concentration dependence calculations involving seed duplexes (∼7 bp), smaller samples of 200 structures were used since the binding energy typically converges within ∼15% of samples of size 1000.
The assembled RNA structures may contain slight misalignments of consecutive backbone atoms from adjacent fragments. These misalignments of database RNA fragments were corrected by performing a minimization of the phosphate backbone atoms while fixing the sugar and base atoms. We performed the constrained minimization using the TINKER package’s routines (“minimize” and “Newton”) (Pappu et al. 1998) in two steps: The steepest-descent method was used to reduce the root-mean-square gradient to 0.1 kcal/mol per angstrom, and then the Newton minimization method was used to reduce the gradient to 0.01 kcal/mol per angstrom. The minimization was performed with a positive monovalent ion placed at 3 Å (in the direction rOP1 + rOP2) away from each phosphate group. We removed the dangling phosphate groups at the 5′ ends to avoid structural distortions upon minimization.
Binding free energy of double-stranded RNAs
Double-stranded RNAs are stabilized by van der Waals, electrostatic, and solvation energies. We use continuum or implicit solvent approaches to describe the effects of ions and water molecules. The electrostatic energy includes interactions between RNA strands and ions. The total duplex free binding energy is decomposed into electrostatic, non-electrostatic, and entropic components (Roux and Simonson 1999; Dong et al. 2008):
where ΔGentropic originates from the loss of translational, rotational, and vibrational motions upon duplex formation (Tidor and Karplus 1994) (elaborated below). The non-electrostatic component consists of the van der Waals and nonpolar solvation energies:
which are assumed to be additive (Dill 1997). The nonpolar solvation term accounts for the energy needed to create the cavity occupied by the solute or RNA molecule (Fennell et al. 2011). The electrostatic component is the excess energy of duplex formation when fixed, isolated conformations of strands 1 and 2 are brought to the final duplex configuration:
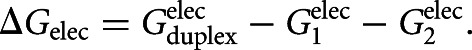 |
This electrostatic contribution was computed using the Adaptive Poisson–Boltzmann Solver (APBS) (Baker et al. 2001) version 1.3. We used APBS’s standard parameter values (e.g., RNA and water dielectric constants of 2.0 and 78.54, respectively) and monovalent salt concentration of 150 mM to approximate physiological conditions (unless explicitly stated otherwise). We set the grid spacing to ∼0.4 Å in electrostatic calculations. Similarly, the interstrand van der Waals energy is defined as:
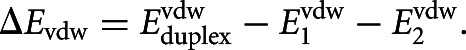 |
We computed this term using the TINKER routine “analyze” with the AMBER99 force field (Cornell et al. 1995). The nonpolar solvation energy ΔGsolvnonpolar was computed using the HCT model (a generalized Born solvation approach) as implemented in TINKER. This term is usually small compared with other energy terms. The total binding energy (sum of electrostatics, van der Waals, and nonpolar solvation) was computed as a Boltzmann-weighted average of the structures in the ensemble at the given temperature. This binding energy reflects conformational fluctuations of duplexes in solution.
When two molecules bind, there is a change in the translational, rotational, and vibrational degrees of freedom, which is entropic in origin. The free energy change associated with these degrees of freedom is given by Tidor and Karplus (1994):
where these free energy components are written in the form ΔGtrans = ΔEtrans − TΔStrans, etc.:
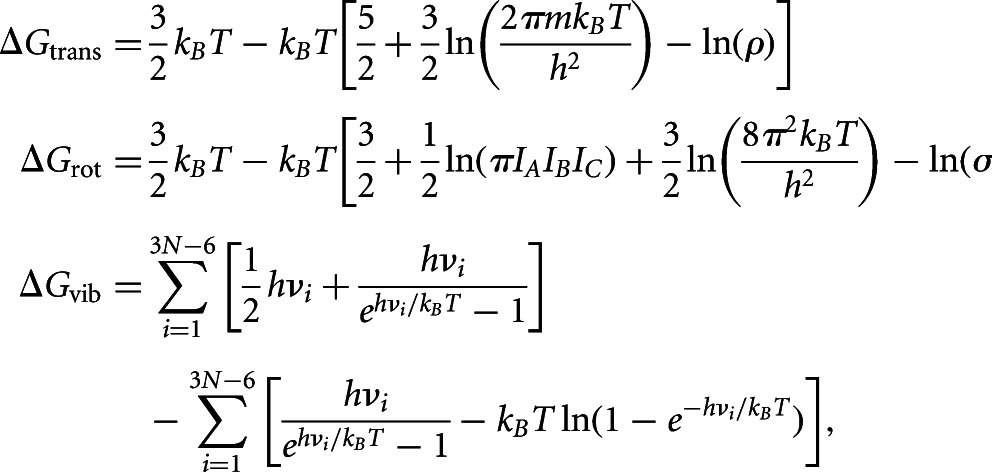 |
where IAIBIC is the product of the three principal moments of inertia; ρ is the solute number density; σ is the symmetry factor (1 for nonsymmetric molecules); m is the mass; h is the Planck’s constant; νi are the vibrational frequencies of the macromolecule computed using normal mode analysis; and N is the number of atoms. We computed the vibrational frequencies νi using TINKER’s “vibrate” routine. For duplex formation, the free energy change is ΔGentropic = ΔGtrans + ΔGrot + ΔGvib, where ΔGα = Gαduplex − Gαstrand1 − Gαstrand2. We estimated the average change using the 25 lowest-energy structures representing the native-like states in the ensemble. These low-energy structures were further subject to a stringent minimization (root-mean-square convergence of 10−5 kcal/mol per angstrom) with GBSA (generalized Born Surface Area) to obtain nonnegative vibrational frequencies.
Total energy of single-stranded RNAs
The total energy is used to predict energetically favorable conformations of a single chain. It is a sum of bonded, nonbonded, electrostatic, and solvation terms:
 |
where Ebond-stretching is the bond-stretching energy, Ebond-angle is the bond-angle energy, Etorsion and Eimproper-torsion are the torsion and improper torsion energies, Evdw is the van der Waals energy, Gelec is the electrostatic energy, and Gsolvnonpolar is the nonpolar solvation energy. We computed the non-electrostatic and nonpolar solvation energy terms using the TINKER routine “analyze” as described above. The total electrostatic energy was computed using the formula (Dong et al. 2008):
where
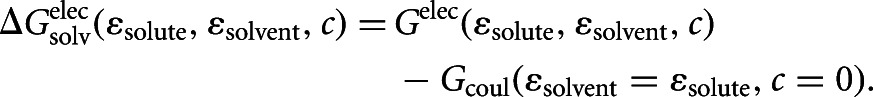 |
Here, ε’s are the dielectric constants and c is the ion concentration. The electrostatic energy term is expressed as the difference between energy terms with heterogeneous and homogeneous dielectric media to reduce errors in quantities computed on a 3D grid. The electrostatic solvation term was computed using APBS and the Coulomb term using TINKER.
RNA secondary structure calculations
We performed all secondary structure calculations using the Vienna RNA package (Hofacker 2003). For free energy calculations using the module RNAeval, we used the default setting, which allows interactions in dangling end bases.
Comparison of calculated and NMR solution structures
The LCS1co and LCS2co miRNA–target constructs analyzed by NMR are 34 and 33 nt long, respectively, and contain several modifications of the natural duplexes (Cevec et al. 2008, 2010): the length of the duplex is shortened from 18 to 12 bp; the shortened duplexes are closed with a GAAA hairpin loop; and the closing base pair for the tetraloop and the 5′,3′-end base pairs are changed from A:U to G:C to enhance structural stability. We constructed duplex structure ensembles for these using MC-Sym software (Parisien and Major 2008) as described above. We note that for both constructs, in an ensemble of 1000 structures, the lowest-energy structure is attained when the subensemble size is <600, and the improvement in energy is <3% from the lowest energy value at sample size of 200. To match experimental conditions, for all electrostatic calculations, we used a monovalent ion concentration of 30 mM at 278 K for LCS1co and 298K for LCS2co. We consider a “predicted” structure to be within the top five scoring structures based on the total energy described above. For comparison with NMR models, we computed the RMSD for each predicted structure with all the 10 available (low-energy) NMR models in the PDB file.
Argonaute–duplex docking simulations
The predicted docking configuration of a duplex to Argonaute was obtained by aligning the structure of the guide RNA strand to its corresponding guide DNA structure in the solved X-ray structure for T. thermophilus Argonaute bound to a DNA:RNA hybrid duplex (Wang et al. 2009); a similar approach was used to set up the ternary structure for MD simulations (Balasubramanian et al. 2010). (Since the recent X-ray structure of a eukaryotic Argonaute PIWI/MID domain lacks the bound duplex [Boland et al. 2011], the T. thermophilus structure was the best approximation available for the docking simulation. Most recently, the structure of budding yeast Argonaute with bound guide RNA has been solved [Nakanishi et al. 2012].) The resulting ternary complex was energy-minimized with the Amber99 and GBSA force fields (Bashford and Case 2000). Since conformational relaxation of the ternary complex was not performed using molecular dynamics or Monte Carlo, we confined docking to the small seed duplex structure (nucleotide positions 1–8), which is exposed to the solvent environment (Wang et al. 2009); the seed duplex with the lowest energy conformation in the structure ensemble was chosen for docking. We defined the energy of complex formation as the total energy of the complex subtracted from the independent duplex and Argonaute energies.
The binding energy ΔEAgo-dup (sum of electrostatic, van der Waals, and nonpolar solvation) between the duplex RNA and Argonaute protein was then computed at 150 mM monovalent ions using the same procedures as described above for isolated RNA duplexes: ΔEAgo-dup = EAgo-dup − EAgo − Edup, where EAgo-dup, EAgo, and Edup are the total energies of the complex, Argonaute and duplex, respectively. This Argonaute–duplex binding energy does not include the entropy contributions because the matrix storage requirement for computing the vibrational entropy term scales as (3N)2, where N, the number of atoms, approaches 104. The lack of entropy information is not critical because entropy contribution roughly shifts the free energy by a constant amount, as we found in our analysis of duplexes (−TΔS∼ 30 kcal/mol).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Sean West and Richard Bonneau for useful comments on the manuscript, Ashish Agarwal and Douglas Renfrew for technical assistance, and NYU Information Technology Services for providing high-performance computing resources. We also thank Jay Ponder for comments on usage of TINKER routines to calculate vibrational frequencies, and James Parker for clarifying experimental measurements of duplex enthalpy. This work was supported by NIH grants U01-HG004276, U01-HG004276-S, and RC2-HG005639.
REFERENCES
- Abu Almakarem AS, Petrov AI, Stombaugh J, Zirbel CL, Leontis NB 2012. Comprehensive survey and geometric classification of base triples in RNA structures. Nucleic Acids Res 40: 1407–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V 2004. The functions of animal microRNAs. Nature 431: 350–355 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian C, Ojha RP, Maiti S, Desideri A 2010. Sampling the structure of the noncanonical lin-4:lin-14 microRNA:mRNA complex by molecular dynamics simulations. J Phys Chem B 114: 16443–16449 [DOI] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashford D, Case DA 2000. Generalized Born models of macromolecular solvation effects. Annu Rev Phys Chem 51: 129–152 [DOI] [PubMed] [Google Scholar]
- Boland A, Huntzinger E, Schmidt S, Izaurralde E, Weichenrieder O 2011. Crystal structure of the MID-PIWI lobe of a eukaryotic Argonaute protein. Proc Natl Acad Sci 108: 10466–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM 2005. Principles of microRNA–target recognition. PLoS Biol 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen SJ 2011. Physics-based de novo prediction of RNA 3D structures. J Phys Chem B 115: 4216–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen SJ 2012. Predicting kissing interactions in microRNA–target complex and assessment of microRNA activity. Nucleic Acids Res 40: 4681–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevec M, Thibaudeau C, Plavec J 2008. Solution structure of a let-7 miRNA:lin-41 mRNA complex from C. elegans. Nucleic Acids Res 36: 2330–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevec M, Thibaudeau C, Plavec J 2010. NMR structure of the let-7 miRNA interacting with the site LCS1 of lin-41 mRNA from Caenorhabditis elegans. Nucleic Acids Res 38: 7814–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB 2009. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 460: 479–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB 2012. An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19: 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Das R, Baker D 2007. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci 104: 14664–14669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Baker D 2008. Macromolecular modeling with rosetta. Annu Rev Biochem 77: 363–382 [DOI] [PubMed] [Google Scholar]
- Das R, Karanicolas J, Baker D 2010. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 7: 291–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didiano D, Hobert O 2008. Molecular architecture of a miRNA-regulated 3′ UTR. RNA 14: 1297–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill KA 1997. Additivity principles in biochemistry. J Biol Chem 272: 701–704 [DOI] [PubMed] [Google Scholar]
- Dong F, Olsen B, Baker NA 2008. Computational methods for biomolecular electrostatics. Methods Cell Biol 84: 843–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP 2005. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310: 1817–1821 [DOI] [PubMed] [Google Scholar]
- Fennell CJ, Kehoe CW, Dill KA 2011. Modeling aqueous solvation with semi-explicit assembly. Proc Natl Acad Sci 108: 3234–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun D, Wang YL, Langenberger D, Gunsalus KC, Rajewsky N 2005. microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput Biol 1: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell M, Long D, Zhang L, Lee A, Carmack CS, Han M, Ding Y, Ambros V 2008. mirWIP: microRNA target prediction based on microRNA-containing ribonucleoprotein-enriched transcripts. Nat Methods 5: 813–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL 2003. Vienna RNA secondary structure server. Nucleic Acids Res 31: 3429–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB 2009. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA 15: 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E 2007. The role of site accessibility in microRNA target recognition. Nat Genet 39: 1278–1284 [DOI] [PubMed] [Google Scholar]
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. 2000. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem Res 33: 889–897 [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, Macmenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500 [DOI] [PubMed] [Google Scholar]
- Lall S, Grun D, Krek A, Chen K, Wang YL, Dewey CN, Sood P, Colombo T, Bray N, Macmenamin P, et al. 2006. A genome-wide map of conserved microRNA targets in C. elegans. Curr Biol 16: 460–471 [DOI] [PubMed] [Google Scholar]
- Leach AR 1996. Molecular modeling: Principles and applications. Addison Wesley Longman, Singapore [Google Scholar]
- Liu L, Chen SJ 2010. Computing the conformational entropy for RNA folds. J Chem Phys 132: 235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Beck A, Zipursky S, Matsudaira P, Baltimore D, Darnell J 2000. Molecular cell biology. Freeman, New York [Google Scholar]
- McColl G, James SA, Mayo S, Howard DL, Ryan CG, Kirkham R, Moorhead GF, Paterson D, de Jonge MD, Bush AI 2012. Caenorhabditis elegans maintains highly compartmentalized cellular distribution of metals and steep concentration gradients of manganese. PLoS ONE 7: e32685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Weinberg DE, Bartel DP, Patel DJ 2012. Structure of yeast Argonaute with guide RNA. Nature 486: 368–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciello G, Acquaviva A, Ficarra E, Deriu MA, Macii E 2011. A molecular dynamics study of a miRNA:mRNA interaction. J Mol Model 17: 2895–2906 [DOI] [PubMed] [Google Scholar]
- Pappu RV, Hart RK, Ponder JW 1998. Analysis and application of potential energy smoothing and search methods for global optimization. J Phys Chem B 102: 9725–9742 [Google Scholar]
- Parisien M, Major F 2008. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452: 51–55 [DOI] [PubMed] [Google Scholar]
- Parker JS, Parizotto EA, Wang M, Roe SM, Barford D 2009. Enhancement of the seed-target recognition step in RNA silencing by a PIWI/MID domain protein. Mol Cell 33: 204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchia W, Alexov E, Honig B 2001. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. J Phys Chem B 105: 6507–6514 [Google Scholar]
- Roux B, Simonson T 1999. Implicit solvent models. Biophys Chem 78: 1–20 [DOI] [PubMed] [Google Scholar]
- Sethupathy P, Corda B, Hatzigeorgiou AG 2006. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 12: 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J, Miller J, Kollman PA, Case DA 1998. Continuum solvent studies of the stability of RNA hairpin loops and helices. J Biomol Struct Dyn 16: 671–682 [DOI] [PubMed] [Google Scholar]
- Tidor B, Karplus M 1994. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J Mol Biol 238: 405–414 [DOI] [PubMed] [Google Scholar]
- Vella MC, Reinert K, Slack FJ 2004. Architecture of a validated microRNA::target interaction. Chem Biol 11: 1619–1623 [DOI] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ 2008a. Structure of an Argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456: 921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sheng G, Juranek S, Tuschl T, Patel DJ 2008b. Structure of the guide-strand-containing Argonaute silencing complex. Nature 456: 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Wardle GS, Tuschl T, Patel DJ 2009. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 461: 754–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Ma Z, Yang W, Ai C 2010. Mechanism of microRNA–target interaction: Molecular dynamics simulations and thermodynamics analysis. PLoS Comput Biol 6: e1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, SantaLucia J Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37: 14719–14735 [DOI] [PubMed] [Google Scholar]
- Xin Y, Olson WK 2009. BPS: A database of RNA base-pair structures. Nucleic Acids Res 37: D83–D88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Shen H, Zhu X, Li G 2011. Fast and accurate computation schemes for evaluating vibrational entropy of proteins. J Comput Chem 32: 3188–3193 [DOI] [PubMed] [Google Scholar]
- Zhang L, Hammell M, Kudlow BA, Ambros V, Han M 2009. Systematic analysis of dynamic miRNA–target interactions during C. elegans development. Development 136: 3043–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, Pasquinelli AE, Yeo GW 2010. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol 17: 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.