

It is well established that alternative splicing contributes to muscle development. This study shows that the KH-domain RNA-binding protein, Quaking, is a global regulator of alternative splicing and many of its targets overlap with targets of PTB.
Keywords: alternative splicing, myogenesis, splicing silencer, splicing enhancer
Abstract
Alternative splicing contributes to muscle development, but a complete set of muscle-splicing factors and their combinatorial interactions are unknown. Previous work identified ACUAA (“STAR” motif) as an enriched intron sequence near muscle-specific alternative exons such as Capzb exon 9. Mass spectrometry of myoblast proteins selected by the Capzb exon 9 intron via RNA affinity chromatography identifies Quaking (QK), a protein known to regulate mRNA function through ACUAA motifs in 3′ UTRs. We find that QK promotes inclusion of Capzb exon 9 in opposition to repression by polypyrimidine tract-binding protein (PTB). QK depletion alters inclusion of 406 cassette exons whose adjacent intron sequences are also enriched in ACUAA motifs. During differentiation of myoblasts to myotubes, QK levels increase two- to threefold, suggesting a mechanism for QK-responsive exon regulation. Combined analysis of the PTB- and QK-splicing regulatory networks during myogenesis suggests that 39% of regulated exons are under the control of one or both of these splicing factors. This work provides the first evidence that QK is a global regulator of splicing during muscle development in vertebrates and shows how overlapping splicing regulatory networks contribute to gene expression programs during differentiation.
INTRODUCTION
Alternative pre-mRNA splicing is regulated to control both the quantity and the coding potential of mRNAs from a gene. Quantity of mRNA can be controlled by regulating splicing that creates mRNA forms with premature stop codons, turning genes off by the nonsense-mediated decay pathway (Lewis et al. 2003; Lejeune and Maquat 2005; Ni et al. 2007). Coding potential can be controlled by regulating alternative exons to produce mRNAs for distinct proteins from the same gene, expanding the proteome in metazoans (Black 2003; Nilsen and Graveley 2010). Understanding the potential for alternative splicing to regulate important processes is only the first step toward an integrated view of how it is coordinated in complex systems (Kalsotra and Cooper 2011). There is a long but incomplete list of splicing factors, each with a complex set of cooperative and antagonistic relationships with other splicing factors that seem distinct at different regulatory sites and in different tissues. Several splicing factors act at other steps in gene expression, for example, binding to mRNA-untranslated regions to affect nuclear export, localization, stability, and translation (e.g., Sanford et al. 2005). Conversely, many RNA-binding proteins not characterized as splicing factors might yet be shown to regulate splicing.
Muscle cells have evolved alternative splicing for making specialized forms of cytoskeletal and mitochondrial proteins necessary for building, energizing, and controlling the contractile apparatus, such as the α-actinin SM exon, α-tropomyosin exon 2, and cardiac troponin T exon 5 (Gooding et al. 1998; Southby et al. 1999; Charlet-B et al. 2002). Inclusion of α-tropomyosin exon 2 is regulated in part by the antagonistic action of SFRS7 with hnRNPs H and F (Crawford and Patton 2006). Similarly, antagonism between PTB, CUG-BP1, MBNL1, and the CELF proteins regulates α-actinin splicing (Gromak et al. 2003). Cardiac troponin T is regulated by the opposing actions of CUG-BP2 and MBNL1 (Goo and Cooper 2009; Warf et al. 2009). A variety of other alternative splicing factors are expressed in muscle cells (e.g., MBNL2, RBFOX1, RBFOX2). Thus far, none of the proteins known to function as splicing factors in vertebrate muscle recognize the ACUAA motif that we identified near mouse heart and skeletal muscle-specific exons (Sugnet et al. 2006).
A role in splicing regulation is suspected for sequences related to the ACUAA motif since they appear enriched downstream from muscle exons (Sugnet et al. 2006; Das et al. 2007), downstream from alternatively spliced exons in general (Voelker and Berglund 2007), and upstream of exons that are spliced in hESCs but not other ES cell types (Yeo et al. 2007), as well as near exons activated during differentiation of C2C12 myoblasts to myotubes in vitro (Bland et al. 2010). This sequence has been called the “STAR” motif because it is contained within a more general binding site consensus for members of the mammalian STAR (Signal Transduction and Activation of RNA) family of proteins (Vernet and Artzt 1997). Despite its bioinformatic identification, direct evidence for its function and the identity of protein factors that might act through it are not available.
We investigated the role of the ACUAA motif in C2C12 myoblasts by searching for proteins that bind a natural intron RNA region containing it. This effort identified the RNA-binding protein QK as a muscle-splicing factor acting directly through ACUAA motifs near the exons that it controls. Unlike in oligodendrocytes, where QK affects splicing by controlling the expression of the direct splicing regulator hnRNPA1 through its mRNA 3′ UTR (Zhao et al. 2010; Zearfoss et al. 2011), QK acts both positively and negatively as a direct splicing regulator through intronic splicing enhancers and silencers in muscle precursor cells. The changes in alternative splicing at QK-regulated exons during myogenesis are likely due to an increase in QK expression. Levels of the splicing factor PTB are reduced during myogenesis (Boutz et al. 2007a), and its regulatory network overlaps that of QK, such that the net regulation of a subset of QK-dependent exons is further refined by relief of PTB repression during myogenesis.
RESULTS
Capzb exon 9 is a muscle-activated exon with conserved ACUAA motifs downstream
To study the role of ACUAA motifs in splicing, we chose exon 9 from the Capzb gene as a model. Capzb encodes an actin filament capping protein; its C terminus is altered by alternative inclusion of exon 9 in heart and skeletal muscle (Schafer et al. 1994). Capzb exon 9 was identified as a muscle- and heart-specific exon with adjacent ACUAA elements in our study on splicing in adult tissues (Sugnet et al. 2006). We confirmed the activation of Capzb exon 9 during differentiation of C2C12 myoblasts (Fig. 1A; Bland et al. 2010) by RT-PCR (Fig. 1B). Alignment of sequences downstream from the Capzb exon from mouse and other vertebrates reveals conservation of multiple copies of the ACUAA element (Fig. 1C).
FIGURE 1.
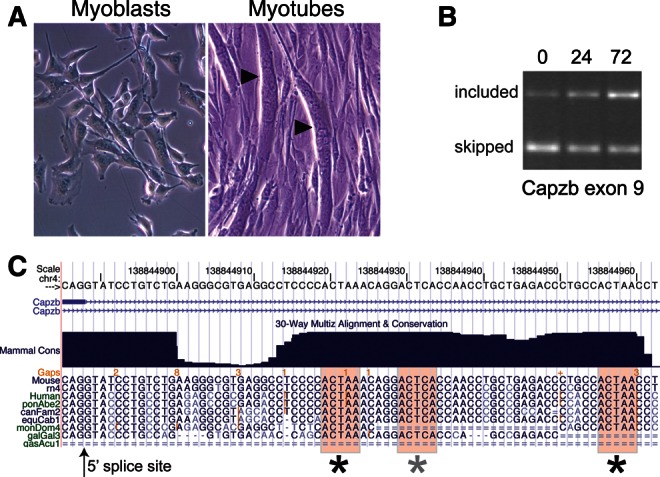
Capzb exon 9 is activated during myogenesis and has intronic ACUAA elements. (A) Phase contrast photographs of proliferating C2C12 or cells treated with low serum for 72 h to induce myogenesis. Multinucleate myotubes are indicated by arrowheads. (B) RT-PCR measurement of changes in Capzb exon 9 inclusion during differentiation using primers in exons 8 and 10 of the endogenous Capzb gene. Exon 9 inclusion increases during differentiation. (C) Alignment of intronic sequences downstream from the 5′ splice site bordering Capzb exon 9 from several vertebrates. The ACUAA elements are shaded and marked with asterisks.
ACUAA motifs are required for exon inclusion
To determine whether the ACUAA elements downstream from Capzb exon 9 contribute to proper splicing of this exon, we used a β-globin reporter system (Dominski and Kole 1991), inserting Capzb exon 9 along with adjacent intron sequences ∼200 nucleotides upstream and ∼250 nucleotides downstream, between human β-globin exons (Fig. 2A). A series of mutations focusing on the conserved region from 32 to 72 nucleotides downstream from Capzb exon 9 containing the ACUAA motifs was tested by transfection into proliferating C2C12 myoblasts, followed by RT-PCR (Fig. 2B). The wild-type reporter includes the Capzb exon at ∼8% in myoblasts (Fig. 2B, lane 3). Deletion of the conserved region (D1), replacing the conserved region with a nonspecific sequence (R1), or deleting just the ACUAA elements (M-Czb) results in loss of exon inclusion (Fig. 2B, lanes 4–6). These data indicate that the ACUAA motifs downstream from the Capzb exon are required for its proper inclusion in myoblasts, as suggested by their enrichment near a large class of regulated exons (Sugnet et al. 2006; Das et al. 2007).
FIGURE 2.

Splicing reporters based on Capzb exon 9. (A) Cartoon of Capzb gene structure from exons 7 through 10. The region marked by the black bar below the endogenous gene was amplified by PCR and cloned into pDUP51 to create the reporter construct shown. The sequence below the reporter diagram represents the downstream intronic region containing the ACUAA elements (boxed). Mutations tested are (D1) deletion of the conserved region; (R1) replacement of the conserved region with a nonspecific sequence; (M-Czb) deletion of the ACUAA elements. (B) RNA isolated from proliferating C2C12 cells transiently transfected with the constructs in A were analyzed by RT-PCR using primers specific for the flanking β-globin exons; spliced products are indicated to the right of the gel. Exon percent inclusion for each lane is graphed, far right. Mock-transfected and globin-only controls are shown (M, Gl, lanes 1,2); the band in the Gl lane is nonspecific.
Quaking and SF1 bind specifically to RNA containing ACUAA motifs
To identify proteins that bind ACUAA containing RNA, we used RNA affinity chromatography, followed by multidimensional protein identification technology (MudPIT) (Washburn et al. 2001). RNAs containing either the wild-type Capzb sequence from 32 to 72 nucleotides downstream from exon 9 (Fig. 3A, WT) or with mutated ACUAA motifs (Fig. 3A, M) were coupled to agarose beads and incubated with myoblast nuclear extracts. Proteins bound at low salt were released with increasing salt concentrations (Fig. 3B), and the WT and M fractions were compared on a silver-stained gel. The pattern of proteins eluted remains complex; however, specific proteins can be seen in the WT- but not the M-RNA-binding fractions (Fig. 3B, arrow). We chose to analyze the 0.4 M NaCl eluted fraction (Fig. 3B, asterisk) by MudPIT (Yates et al. 2009).
FIGURE 3.

Identification of ACUAA-binding proteins in proliferating C2C12 cells. (A) Sequences of in vitro-transcribed RNAs used. ACUAA motifs or mutant versions are italicized. (B) Silver-stained denaturing gel of proteins bound to the wild-type (WT) or mutant (M) RNAs. Lanes are as follows: NE, nuclear extract; FT, flow through; triangles indicate increasing NaCl elution (0.1, 0.2, 0.4, and 1 M). Arrow points to enriched proteins visible in the WT- but not the M-RNA-binding fractions. Asterisks mark fractions subjected to MudPIT analysis. (C) Western blot of hnRNP K, QK, Ddx5, SF1, and PTB in the eluted fractions. Lanes correlate with those in B.
We expected a complex set of proteins to bind to RNA under these conditions; indeed, numerous cytoskeletal proteins, ribosomal proteins, and DNA-binding proteins are found in both fractions (data not shown). To examine the relative association of RNA processing factors with the WT- and M-RNAs, we considered five classes of proteins: splicing factors, hnRNP proteins, spliceosomal snRNP proteins, DEAD/H RNA helicase family proteins, and other processing enzymes (Table 1; Supplemental Table 1). Among proteins detected in the WT-RNA fraction, but absent from the M-RNA fraction, are the known ACUAA-binding factors QK (Ryder and Williamson 2004; Galarneau and Richard 2005) and Splicing Factor 1 (SF1) (Arning et al. 1996; Liu et al. 2001; Corioni et al. 2011). The peptides observed for the QK protein do not distinguish which of the three major QK isoforms (QK-5, QK-6, and QK-7) are bound, since these forms differ only in their C-terminal tails (Ebersole et al. 1996; Chénard and Richard 2008).
TABLE 1.
Detection of RNA processing factors in WT- and M-RNA affinity chromatography fractions

We validated several proteins from the MudPIT analysis by Western blot using fractions from the WT- and M-RNA columns. QK and SF1 were well retained by the WT- but not the M-RNA affinity matrix (Fig. 3C). Thus, it appears that QK, SF1, and several other proteins depend on the intact ACUAA elements for their efficient association with the WT-RNA. Whether these bind directly to RNA or indirectly to other proteins bound to RNA is not known. hnRNP K and Ddx5 were retained by both of the matrices, although it appears the M-RNA matrix bound less protein overall. Furthermore, the known splicing regulator PTB is bound by the Capzb RNA sequence but is not strongly dependent on the ACUAA sequences (Fig. 3C). We conclude that QK, SF1, PTB, as well as other proteins, bind pre-mRNA sequences downstream from Capzb exon 9 and are candidates for factors that regulate its splicing.
Depletion of QK, but not SF1, inhibits Capzb exon 9 inclusion in myoblasts
To test the hypothesis that QK activates Capzb exon 9 we used siRNA against QK (all isoforms) to deplete the protein in myoblasts. A Western blot using the pan-QK antibody shows substantial reduction in QK protein (Supplemental Fig. 1A), and RT-PCR analysis of the endogenous Capzb transcripts reveals that inclusion of exon 9 is greatly inhibited (Fig. 4A, lane 3). In contrast, when we use siRNA to deplete SF1 in myoblasts, endogenous Capzb exon inclusion is unaffected (Fig. 4A, lane 4; Supplemental Fig. 1A), suggesting that SF1 does not activate exon inclusion through downstream ACUAA sequences. Thus, QK, not SF1, is the ACUAA-binding protein that contributes to Capzb exon 9 inclusion in myoblasts.
FIGURE 4.

QK and PTB control inclusion of Capzb exon 9. (A) Analysis of RT-PCR products from C2C12 cells mock-transfected (M) or transiently transfected with a nonspecific siRNA (NS si), a QK-specific siRNA (QK si), or an SF1-specific siRNA (SF1 si). Spliced products corresponding to exon 9-included or excluded endogenous Capzb mRNAs are indicated to the right of the gel. Percent inclusion was quantified and graphed, far right. (B) Overexpression of QK leads to Capzb exon 9 inclusion provided ACUAA elements are intact. Proliferating C2C12 cells were mock transfected (M) or transiently cotransfected with a QK-5 expression construct (myc-QK-5) or a control GFP expression construct (GFP), along with either the wild-type Capzb reporter (Czb) or the mutant reporter with deletion of the ACUAA elements (M-Czb) from Figure 2. A globin-only control is also shown (Gl, lanes 2,3); the band in the Gl lanes is a nonspecific RT-PCR product. Inclusion of the WT Capzb exon increased with expression of QK by approximately fivefold (cf. lanes 4 and 5, P < 0.001, n = 4). Spliced products are indicated and percent inclusion is graphed, far right. QK depletion significantly reduces exon inclusion (asterisk, P < 0.05, cf. lanes 2 and 3). (C) Western blot of whole-cell extract from cells mock-transfected (lane 1) or transiently cotransfected with the Capzb reporter and either a GFP expression construct (GFP), a QK-5 expression construct (myc-QK-5), a nonspecific siRNA (NS siRNA), or a PTB-specific siRNA (PTB siRNA) as indicated at top. Blots were probed with antibodies to GFP, pan-QK, PTB, and GAPDH as a loading control (as indicated to the left of the blots). (D) Analysis of RT-PCR products from RNA isolated from cells transfected in C. Spliced products are indicated to the right of the gel. Percent inclusion was quantified and graphed, far right. (*) P < 0.002. (E) Model for regulation of Capzb exon 9 by QK and PTB. QK activates inclusion through intronic ACUAA elements downstream, and PTB represses inclusion, possibly through sequences similar to PTB-binding sites upstream and/or through binding downstream, as detected by RNA affinity chromatography (Fig. 3).
Quaking isoform 5 activates Capzb exon 9 through the ACUAA motif
Given that reduction in QK protein levels decreases Capzb exon 9 inclusion, we asked whether increasing QK protein levels would promote inclusion. We focused on Quaking isoform 5 (QK-5) because it is predominantly nuclear, whereas isoforms 6 and 7 (QK-6, QK-7) are mostly cytoplasmic in cell types that have been tested (Chen and Richard 1998; Wu et al. 1999). We transfected a myc-tagged QK-5 construct (generous gift from Sean Ryder) along with wild-type and mutant Capzb reporter into myobloasts (Fig. 4B). Expression of myc-QK-5 results in a nearly eightfold increase in exon 9 inclusion (Fig. 4B, cf. lanes 4 and 5, P < 0.001, n = 4), while the M-Czb reporter lacking the three ACUAA motifs was not affected (lanes 6 and 7, see Fig. 2 for a description of the M-Czb mutant). Thus, increased expression of QK-5 activates inclusion of Capzb exon 9 in a fashion dependent on the downstream intronic ACUAA sequences.
Repression of PTB enhances QK activation of Capzb exon 9 inclusion
RNA affinity chromatography detects PTB binding to the intronic sequence downstream from Capzb exon 9 (Fig. 3). PTB represses exons in C2C12 myoblasts (Boutz et al. 2007a), and PTB repression can be mediated through downstream sites in combination with upstream binding sites (Chan and Black 1997; Southby et al. 1999). To test whether Capzb exon 9 is under the control of PTB, we depleted PTB from myoblasts with or without concomitant overexpression of QK-5 (Fig. 4C,D). After confirming the effects on protein levels by Western blot (Fig. 4C, lanes 4–7), we analyzed splicing of the Capzb exon 9 reporter construct by RT-PCR (Fig. 4D, lanes 4–7). When PTB expression is reduced, inclusion levels increase approximately fourfold (Fig. 4D, cf. lanes 4 and 5, P < 0.0001, n = 4). As discussed above, increased QK-5 expression activates the Capzb exon 9 reporter on its own (Fig. 4D, cf. lanes 4 and 6). However, in combination with reduced PTB expression, increased QK-5 expression leads to greater exon inclusion (Fig. 4D, cf. lanes 3 and 7, P < 0.002, n = 4). Similar results are obtained for the endogenous Capzb exon 9 (Supplemental Fig. 1B). This shows that PTB represses Capzb exon 9 antagonistically to QK. A model describing these relationships is shown in Figure 4E.
QK depletion in myoblasts results in widespread alternative splicing changes
To determine the extent of QK function in splicing, we depleted QK in proliferating C2C12 cells and captured changes in alternative splicing using splicing-sensitive microarrays. Western blots revealed that levels of all three isoforms are reduced by a QK-specific siRNA directed at a common region of their mRNAs (Fig. 5A). We detected 406 cassette exons whose inclusion changes in QK-depleted myoblasts as compared with the mock-depleted controls (q ≤ 0.05 and |Sepscore| ≥ 0.3; Supplemental Table 2). We validated a number of these by RT-PCR (Fig. 5B). Among the 162 exons with the most significant changes (q = 0, |Sepscore| ≥ 0.5), about half show a decrease in inclusion upon QK depletion, indicating that QK promotes their inclusion in myoblasts. In contrast, half show an increase in exon inclusion, indicating that QK represses them. We conclude that QK has a major role in regulating splicing in muscle cells.
FIGURE 5.
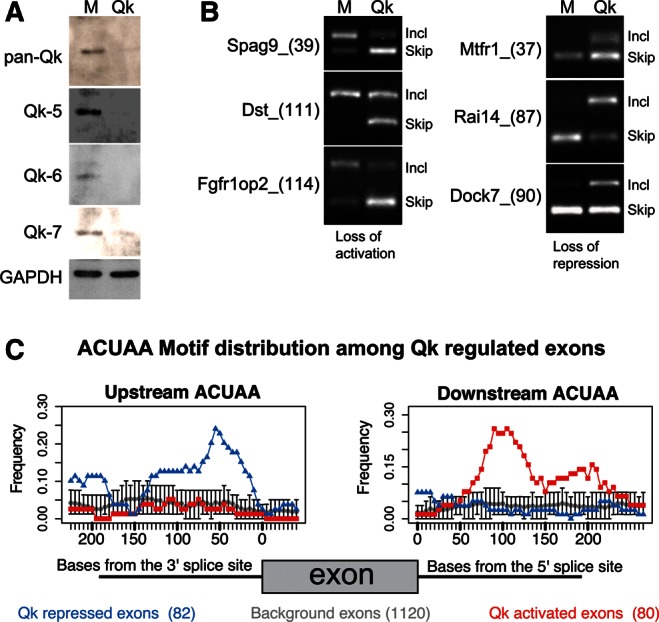
The QK-splicing regulatory network in proliferating myoblasts. (A) QK siRNA depletes all three forms of QK in myoblasts. Western blot of proteins from proliferating C2C12 cells mock-transfected (M) or transfected with a QK-specific siRNA (QK). Blots were probed with antibodies to pan-QK, QK-5, QK-6, QK-7, and GAPDH as a loading control. (B) RT-PCR validation of splicing changes detected by array analysis. Agarose gel analysis of RT-PCR products for alternative cassette exons using RNA from mock-transfected or QK-depleted cells. Exon-included product is always the upper band, exon-skipped product always the lower. The splicing event is labeled by gene name and exon number and size (nucleotides, in parenthesis). (C) The frequency of ACUAA elements upstream of (left) and downstream from (right) the top 162 QK-regulated cassette exons is mapped. Exons up-regulated by QK depletion are in blue and inferred to be repressed by QK; down-regulated exons are in red and inferred to be activated by QK. Control exons are in gray. Error bars indicate the 95% confidence limit for ACUAA frequency in the control exons.
Enrichment of intronic ACUAA sequences near QK-regulated exons
If QK functions in muscle cells like other globally active splicing factors such as the MBNL (Du et al. 2010; Charizanis et al. 2012; Wang et al. 2012), RBFOX (Zhang et al. 2008; Yeo et al. 2009), PTB (Xue et al. 2009), and Nova proteins (Ule et al. 2006), we would expect to find QK-binding sites near the exons it controls. We searched for 5-mers enriched in the regions 150 nucleotides upstream of and downstream from the top 162 QK-regulated exons relative to the corresponding regions of 5166 cassette exons with probesets on the microarray. Significant (FDR ≤ 0.05) enrichment for CUAAC and ACUAA (and no other 5-mers) is found upstream of exons repressed by QK and downstream from exons activated by QK (Supplemental Table 3). We mapped the positional occurrence of the ACUAA 5-mer across the entire set of QK-regulated exons relative to cassette exons whose splicing did not change upon QK depletion (Fig. 5C; Supplemental Table 3). In general, when QK binds upstream of an exon it acts as a repressor, and when bound downstream, it acts as an activator (Fig. 5C). This is comparable to other splicing factors that bind intron sequences, such as Nova, MBNL1, and others (Ule et al. 2006; Zhang et al. 2008; Xue et al. 2009; Yeo et al. 2009; Du et al. 2010; Charizanis et al. 2012; Wang et al. 2012). The distribution of ACUAA both upstream at QK silencers and downstream at QK enhancers is notably bimodal. This is likely due to the fact that QK is a dimer with a bipartit-binding site (Zorn and Krieg 1997; Chen and Richard 1998; Galarneau and Richard 2005; Carmel et al. 2010; Beuck et al. 2012).
The PTB and QK splicing regulatory networks overlap in myoblasts
Capzb exon 9 inclusion is controlled in myoblasts by a balance between QK activation and PTB repression and is induced during myoblast differentiation to myotubes. In addition, PTB down-regulation and several PTB-regulated splicing changes take place during myoblast differentiation (Boutz et al. 2007a). To explore the extent of PTB-splicing regulation in myoblasts, we used splicing-sensitive microarrays to analyze splicing changes after PTB depletion by siRNA (Supplemental Fig. 2A). Depletion of PTB results in splicing changes for 485 cassette exons (q = 0, |Sepscore| ≥ 0.5; Supplemental Table 4), of which 268 show increased inclusion (repressed by PTB), and 217 show decreased inclusion (activated by PTB). A number of 5-mers including UCUCU are significantly enriched in regions upstream of PTB repressed exons (FDR < 0.01; Supplemental Table 5), and a map of the UCUCU frequency over the intronic regions adjacent to PTB repressed exons shows a broad region from ∼100 nucleotides upstream and up to the 3′ splice site where this signal is located (Supplemental Fig. 2B; Supplemental Table 5). Our results for myoblasts are similar to analyses of PTB in other cell types and tissues (Boutz et al. 2007b; Xue et al. 2009; Llorian et al. 2010).
When we intersect the QK-splicing network with the PTB-splicing network (Supplemental Table 6), we find 172 exons whose inclusion is significantly affected (q ≤ 0.05, |Sepscore| ≥ 0.3) by either QK or PTB depletion, including Capzb exon 9. As expected (Fig. 4), Capzb exon 9 is activated by QK and repressed by PTB. The 172 jointly regulated exons represent 42% of the 406 QK-regulated events, but only 17% of the 1012 PTB-regulated events, indicating that while the PTB network detected in this experiment is more than twice as large as the QK network, QK regulation is more strongly influenced by PTB than the other way around. Interestingly, these exons sort out about equally into the four regulatory classes that could be created by the combination of QK and PTB (40 exons are activated by both, 40 are repressed by both, 48 are activated by QK and repressed by PTB, 44 are repressed by QK and activated by PTB). Thus, Capzb is not unique; there are substantial numbers of exons controlled by QK and PTB in all possible combinatorial classes.
Contributions of QK and PTB to alternative splicing during myoblast differentiation
The analysis above details QK- and PTB-controlled splicing events in myoblasts. To assess their roles in alternative splicing during muscle cell differentiation, we asked which exons under QK and PTB control in myoblasts are also regulated during myogenesis. Regulation of PTB levels has been implicated in controlling several alternative splicing events in myogenesis (Boutz et al. 2007a). We compared splicing in RNA from myoblasts to that from 72-h differentiated myotubes using the same array platform as for the depletion experiments and detected many robust splicing changes, as also reported by groups using other platforms (Bland et al. 2010; Trapnell et al. 2010). Data for cassette exons showing changes in inclusion in myoblasts depleted of either QK or PTB or after differentiation into myotubes are shown in Supplemental Table 6.
We followed changes in QK protein expression during myogenesis by Western blot using the pan-QK antibody on proteins from undifferentiated myoblasts and 72 h myotube cultures (Fig. 6A). The upper band represents both QK-5 and QK-7, while the lower band represents QK-6. Relative to GAPDH, QK levels increase two- to threefold during differentiation. This increase might be expected to influence inclusion of the many exons controlled by QK in myoblasts in the opposite direction as observed in QK-depleted myoblasts. Conversely, since PTB is down-regulated during myogenesis (Fig. 6A; Boutz et al. 2007a), the splicing of exons controlled by PTB in myoblasts may change in the same direction as observed in PTB-depleted myoblasts. For exons where QK and PTB appear to act similarly, exon inclusion may not change, as the increase in QK might offset the loss of PTB. Finally, the behavior of some exons may be influenced by other factors in addition to QK or PTB during differentiation.
FIGURE 6.
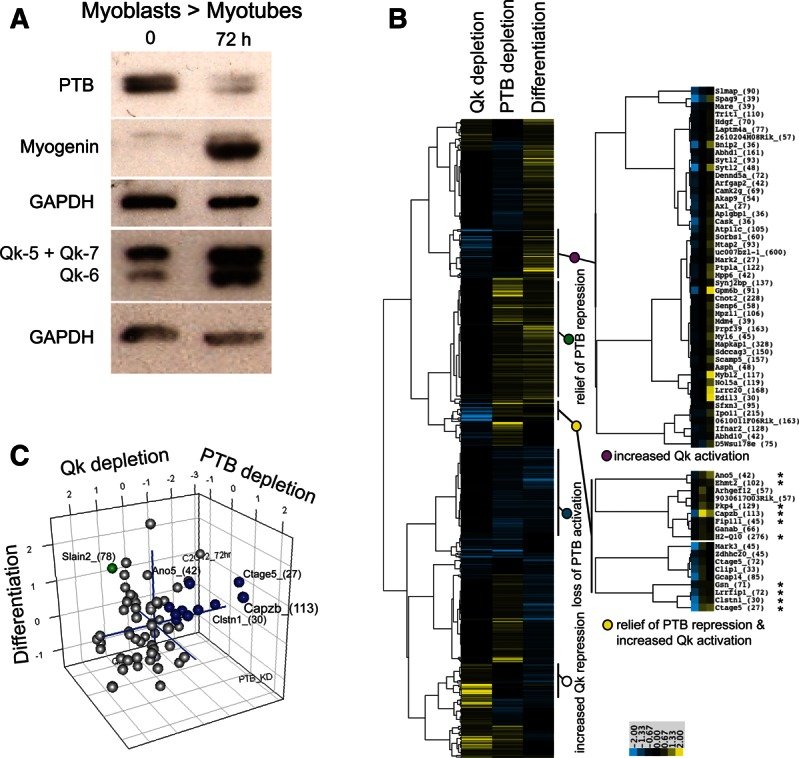
Regulation of QK protein level, PTB protein level, and alternative splicing during myogenic differentiation. (A) Western blot of proteins during differentiation. Proteins from undifferentiated C2C12 Myoblasts (0) or cells after 72 h of differentiation (Myotubes, 72 h) were detected using antibodies against proteins listed at left: PTB, myogenin, pan-QK (co-migration of QK-5 and QK-7 results in one band, QK-6 is the lower band), and GAPDH. (B) Clustering of splicing events based on their response to QK or PTB depletion in myoblasts relative to their change in splicing during differentiation. Yellow means that inclusion increased, whereas blue indicates reduction in exon inclusion. Asterisks mark exons colored blue in C. A total of 612 cassette exon events with significant changes (q-value ≤ 0.05 and |Sepscore| ≥ 0.3) in at least two data sets are shown; Sepscores that did not meet the significance criteria were replaced with zeros for clustering (Supplemental Table 7). (C) Graph of exons with splicing changes in all three data sets. The Sepscores of 66 exons with q-value ≤ 0.01 and |Sepscore| ≥ 0.3 in all three data sets were plotted using R. Exons activated by QK and repressed by PTB in myoblasts that are activated during differentiation are blue. The Slain2_(78) exon is green.
We classified exons with respect to their dependence on these two splicing factors in myoblasts and their changes during muscle cell differentiation by clustering the Sepscores of exons whose inclusion changed in at least two of the three comparisons (Fig. 6B; Supplemental Table 7). Several expected classes are visible. For example, a subset of the exons activated by QK (decreased inclusion after depletion, blue), but not significantly affected by PTB (black) in myoblasts, is also activated during myotube differentiation (yellow), in part due to increasing QK protein (Fig. 6B, purple circle). A large group of exons is not significantly controlled by QK, but repressed by PTB in myoblasts (increased after depletion, yellow) and activated during differentiation (yellow), likely in part by the decrease in PTB protein (Fig. 6B, green circle; Boutz et al. 2007a). Other expected classes such as would be generated by increased QK repression or loss of PTB activation during differentiation can also be observed (Fig. 6B, white and blue circles). The combined regulatory effect of both factors explains another set of events (e.g., Fig. 6B, yellow circle). In total, 22% of the splicing changes observed during myoblast differentiation occur in exons controlled by QK or PTB and are consistent with the changes in QK and PTB protein levels during differentiation. The observed 72-h splicing changes of another 17% of exons that are also regulated by QK or PTB in myoblasts cannot be explained by the simple expectation that the increase in QK and the reduction in PTB relative to the myoblast state will predict the differentiated cell state; these exons may be influenced by other splicing factors also expressed in C2C12 cells such as RBFOX and MBNL family members.
To identify exons whose inclusion during differentiation is likely altered by changes in activity of both QK and PTB, we made a three-dimensional plot of Sepscores for the 66 exons whose splicing changed in all three experiments (Fig. 6C; Supplemental Table 8), allowing the diversity of exon responses to be visualized. There are 10 exons (blue spheres) whose inclusion decreases upon QK depletion and increases upon PTB depletion, which are activated during differentiation, including Capzb exon 9 (Capzb_(113)). The behavior of these exons are consonant with the changes in protein level observed during differentiation or depletion in undifferentiated cells. As with the clustering (Fig. 6B; same exons are marked by asterisks), we observe many combinations of splicing changes within the three experimental conditions, many of which are not consonant with the changes in protein level during differentiation. For example, the Slain2_(78) exon (colored in green) is repressed by QK (exon inclusion increases upon QK depletion) and activated by PTB (exon inclusion decreases upon PTB depletion), but is activated during differentiation. Thus the robust activation of Slain2_(78) must be due to other factors that overcome strong repression due to increasing levels of QK and loss of activation with decreasing levels of PTB. This highlights the incomplete nature of our understanding of splicing regulation during muscle cell differentiation and underscores its complexity. Nonetheless, in this more select set of exons, we again observe that the differentiation behavior of a large number of exons (29%) can be explained by changes in QK and PTB protein levels during differentiation (Fig. 6C; Supplemental Table 8).
DISCUSSION
In a previous study we used genome-wide splicing data from mouse tissues to identify motifs associated with heart and skeletal muscle-specific exons, revealing an RNA sequence motif ACUAA similar to those recognized by SF1 and the STAR family of RNA-binding proteins (Sugnet et al. 2006). We have now shown that this motif mediates splicing regulatory control through QK, in particular the nuclear isoform 5 (QK-5). At Capzb exon 9 the conserved downstream ACUAA elements are required for exon inclusion, bind QK in a fashion dependent on ACUAA, and mediate QK-5 activation (Figs. 2–4; Supplemental Fig. 1). Furthermore, QK regulates hundreds of splicing events in muscle cells, as a repressor or an activator, depending on the location of nearby ACUAA motifs (Fig. 5; Supplemental Tables 3, 4). We found that QK protein levels change during differentiation of myoblasts to myotubes (Fig. 6), suggesting a mechanism for a part of the alternative splicing program during myotube differentiation. Finally, we intersected the QK splicing regulatory network with that of PTB, a splicing factor that is down-regulated during myogenesis (Boutz et al. 2007a), and found classes of exons controlled by both proteins (Fig. 6). This work identifies QK as a global splicing factor in muscle cells and reveals that QK plays a larger role beyond its known activity as a translational regulator binding to 3′ UTRs.
QK RNA-binding proteins are ancient regulators of fundamental processes
Mouse Quaking was originally identified as a recessive mutation causing a jittery phenotype attributed to dysmyelination in the central nervous system (Sidman et al. 1964). Quaking has been implicated in a striking variety of processes in the mouse, such as embryogenesis, blood vessel development, glial cell fate determination, apoptosis, and smooth muscle development, while the human homolog, QKI, has been implicated in a number of diseases, including ataxia, glioblastoma development, and schizophrenia (Chénard and Richard 2008). QK homologs are expressed in genomes as divergent as sea urchin and are critically involved in fundamental cell and developmental processes in deeply diverged metazoans from C. elegans (gld-1, asd-2) (Francis et al. 1995; Ohno et al. 2008) to Drosophila (how) (Baehrecke 1997; Fyrberg et al. 1997; Zaffran et al. 1997) and zebrafish (qkA) (Tanaka et al. 1997; Lobbardi et al. 2011). In vertebrates, a larger family of proteins related to QK also exists, including SAM68 and the less-studied SLM-1 and SLM-2 (Lock et al. 1996; Di Fruscio et al. 1999). Together, this evolutionary depth and breadth of functional contribution implies that QK and its relatives have been integrated into the post-transcriptional regulatory structure of eukaryotic cells for hundreds of millions of years. Here we show that in addition to its previously demonstrated role in translational control, that QK is a global regulator of splicing in vertebrate muscle cells during differentiation.
QK is a splicing factor that directly activates Capzb exon 9
The identifying feature of this ancient STAR protein family is a dimeric RNA-binding domain that recognizes bipartite sequences of the form AYUAAY..N2-20..UAAY (Zorn and Krieg 1997; Chen and Richard 1998; Galarneau and Richard 2005; Carmel et al. 2010; Beuck et al. 2012). Sequence elements related to the ACUAA motif are found in many contexts, bound by different proteins, making it difficult to predict the binding partner of the ACUAA motif observed bioinformatically (Sugnet et al. 2006; Das et al. 2007; Bland et al. 2010). For example, the branchpoint sequence URAY (in yeast, UACUAAC), found near exons and utilized in constitutive splicing during the first catalytic step, is bound by SF1 (in yeast, BBP1) to recruit the splicing machinery to the intron (Kramer 1992; Liu et al. 2001). The response element for QK and its C. elegans homolog GLD-1 (Ryder and Williamson 2004; Ryder et al. 2004) has been found in 3′ UTRs, where it regulates mRNA stability, localization, and translation (Saccomanno et al. 1999; Li et al. 2000; Lakiza et al. 2005; Zhao et al. 2010; Zearfoss et al. 2011). We found that both QK and SF1 bound the ACUAA-like sequences downstream from Capzb exon 9 by RNA affinity chromatography (Fig. 3). We then showed that depletion of SF1 has no detectable effect on Capzb exon 9 inclusion, while QK depletion strongly reduced exon inclusion (Fig. 4A,B). Furthermore, overexpression of the QK-5 isoform increased exon inclusion (Fig. 4D; Supplemental Fig. 1A), demonstrating a direct role for QK in regulating alternative splicing of Capzb exon 9.
Direct and indirect modes of QK influence on splicing
Several studies of invertebrate QK homologs have identified roles in splicing at specific exons. C. elegans asd-2 mutations were isolated using a two-color reporter gene carrying mutually exclusive exons from a body wall muscle myosin gene (Ohno et al. 2008). Mutations in an ACUAA-like motif-containing element (two CUAAC repeats) were shown to abrogate the regulation of adult-specific alternative splicing, phenocopying the asd-2 mutant worms. In flies, the QK homolog how promotes inclusion of neurexin IV exon 3, most likely through downstream ACUAA sequences (Rodrigues et al. 2012). It has remained an open question as to whether these single exon studies reflect a larger involvement of QK in splicing at other exons more globally. In vertebrates, QK has been shown to regulate splicing in oligodendrocyte precursors, but not primarily by binding near exons with ACUAA. In these cells, QK stabilizes hnRNPA1 mRNA by binding to its 3′ UTR; thus, a primary effect of depleting QK appears to be down-regulation of hnRNP A1, which causes gene expression and splicing defects (Zearfoss et al. 2011).
In myoblasts, however, introns neighboring the exons most affected by QK depletion are clearly enriched for the QK-binding site (Fig. 5) and exhibit the “upstream repressing, downstream activating” pattern observed for other globally active splicing factors like Nova (Ule et al. 2006), MBNL1 (Du et al. 2010), MBNL2 (Charizanis et al. 2012), RBFOX1 (Jin et al. 2003), and RBFOX2 (Zhang et al. 2008; Yeo et al. 2009). Splicing of Capzb exon 9 is influenced by the well-defined binding sites for QK (Ryder and Williamson 2004; Galarneau and Richard 2005) in the downstream intron and by expression of QK (Figs. 2B, 4; Supplemental Fig. 1). Thus, we infer that QK acts directly in myoblast splicing, rather than indirectly through hnRNP A1, as in both the Quakingviable mouse (Zhao et al. 2010) and oligodendrocyte precursor cells (Zhao et al. 2010; Zearfoss et al. 2011). This does not exclude the possibility that QK regulates other RNA-binding proteins in myoblasts—a view supported by our observation that a subset of exons is regulated by QK but lacks good matches to the QK response element within 150 nucleotides of their splice sites. These exons could be regulated by other factors that are themselves regulated by QK, or have QK-binding sites farther from the exon, as has been observed for RBFOX regulation (Huh and Hynes 1994; Lim and Sharp 1998; Tang et al. 2009). In addition, we note that a 5-mer related to the RBFOX motif UGCAUG is slightly enriched upstream of exons repressed by QK in myoblasts (Supplemental Table 3), suggesting that either these exons are coregulated by a QK and an RBFOX family member, or perhaps that QK might regulate an RBFOX family member. Clearly, the regulation and overlap of complexity of splicing during muscle differentiation is far from resolved.
QK is a dual regulator of alternative splicing and mRNA function
In mammals, RNA-binding proteins come in families (e.g., PTB/nPTB, RBFOX1 through RBFOX3, MBNL1 through MBBNL3); members of the same family share very similar RNA-binding domains and seem to bind indistinguishable motifs. Yet family members have both shared and distinct functions that are governed by sequence context, cell-type, or tissue-specific information. For example, MBNL1 appears to influence alternative pre-mRNA splicing more significantly in muscle tissue, whereas MBNL2 exerts a similarly predominant influence in brain tissue (Du et al. 2010; Charizanis et al. 2012). In addition, numerous in vivo crosslinking studies on splicing factors show that many also bind to 3′ UTRs of mRNAs as well as to introns near regulated exons (Hafner et al. 2010; Charizanis et al. 2012; Masuda et al. 2012; Wang et al. 2012). This implies that the same protein may function in splicing, localization, or translation within the same cell, or may only participate in some functions in certain cell types. In the case of QK, the three isoforms differ in amino acid sequence only at their C-termini, and this is thought to confer, in part, their distinct localization behaviors. QK-5, the predominantly nuclear form, seems more likely to be involved in direct splicing regulation (this work), whereas the cytoplasmic QK-6 and QK-7 forms may mediate translational control or mRNA stability with indirect effects on splicing (Saccomanno et al. 1999; Zhao et al. 2010; Zearfoss et al. 2011). We currently lack an understanding of the information that specifies which family member or isoform should bind where, or carry out which function. One possibility is that different family members or protein isoforms carry out these different functions, perhaps interacting with separate sets of adaptor proteins that confer specificity. But whether and how these distinct isoforms might be partitioned to different RNA target sites is unknown.
Combinatorial control of the myogenic splicing program by QK and PTB
To understand the relationship between the QK and PTB splicing regulatory networks in myoblasts and during differentiation, we defined the PTB global splicing network in undifferentiated myoblasts and differentiated myotubes using splicing sensitive microarrays. The PTB network is about 2.5 times larger than the QK network; while only 17% of PTB regulated exons are also controlled by QK, PTB coregulates nearly 42% of the QK network. The 172 exons coregulated by QK and PTB are split evenly into four classes: one where both repress, another where both activate, one where QK represses while PTB activates, and one where QK activates and PTB represses. Thus, for just over half of exons regulated by both proteins, they oppose each other and reinforce each other at the remaining exons. Analysis of the genes containing these regulated exons indicates that a diversity of both general and muscle-specific functions are under control of these factors.
During differentiation, the two proteins change levels in opposite directions, with QK increasing and PTB decreasing (Fig. 6) due in part to the induction of miR-133 and its repression of PTB mRNA (Boutz et al. 2007a). Exons such as Capzb exon 9 are activated during differentiation by the combined increase in QK activation and decrease of repression by PTB. However, numerous other splicing factors play roles in controlling alternative splicing in muscle, and given the complex interplay of splicing factors at most exons, dissecting a developmental program of alternative splicing will involve understanding not just why exons change inclusion levels, but how those that do not change are made stable (Fig. 6B). Nonetheless, by mapping splicing regulatory networks in myoblasts and following changes in factor level during differentiation, ∼22%–29% of the alternative splicing changes during myogenesis occur in a fashion consistent with primary regulation by QK alone, PTB alone, or the two proteins together. The close physical proximity of mapped PTB and QK motifs (Fig. 5, cf. Supplemental Fig. 2) in the sequence upstream of exons repressed by both proteins is notable and may indicate a mechanism whereby the relative occupancy and competition for binding plays a key role in the bimodal relationship between the two proteins—i.e., as one protein leaves another is able to bind. Future work will involve integrating our understanding of the roles of these and other splicing factors during myoblast differentiation, including MBNL1 (which also activates Capzb exon 9) (Du et al. 2010), RBFOX proteins, and the CELF proteins (Gromak et al. 2003). In addition, the control of PTB levels by miR-133 during myogenic differentiation (Boutz et al. 2007a) and the interactions between QK and microRNAs in other systems (Chen et al. 2012; Ji et al. 2012; van Mil et al. 2012) suggest that a full understanding of the developmental control of alternative splicing will require expanding our appreciation for other control mechanisms at the same time as we dissect the roles of splicing networks.
MATERIALS AND METHODS
Cell culture
Mouse C2C12 myoblast cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Invitrogen). To induce myogenesis, 1.5 × 105 cells were plated in growth medium (DMEM + 10% FBS) in 6-well tissue culture-treated plates 12–18 h before addition of differentiation medium (DMEM supplemented with 2% horse serum, Invitrogen). Media was changed every 24 h, and myotube formation was visible after ∼48 h. Differentiation was verified visually by myotube formation and Western blot of myogenic markers. RNA and protein were isolated under subconfluent conditions in growth media (for myoblast analyses) or 72 h after addition of differentiation medium (for differentiation analyses).
Microscopy
Live cells were photographed on a Zeiss Axiovert 200 microscope using a Zeiss 20×/0.30 LD A-Plan lens. Images were collected using a SPOT Insight Mosaic 3.2.0 color digital camera and SPOT Advanced software (Diagnostic Instruments, Inc).
Transient transfection
Transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were incubated for 18–24 h post-transfection prior to harvesting RNA and/or protein.
RNA isolation
Whole-cell RNA was isolated using Trizol (Invitrogen) according to manufacturer’s instructions. RNA pellets were resuspended in 50 µL of TE (10 mM Tris, pH 8.0; 1 mM EDTA). Cytoplasmic RNA was extracted as described previously (Modafferi and Black 1997).
Protein isolation
Cells were harvested by trypsinization and washed once with 1× PBS, then resuspended in 50 µL of NP-40 lysis buffer (150 mM NaCl, 10 mM tris-HCl at pH 7.8, 0.65% NP-40) with complete protease inhibitor cocktail tablets (Roche), and incubated on ice for 10 min. After spinning for 10 min at 18,000g at 4°C, the supernatant was flash frozen and stored at −80°C.
Splicing microarrays
Targets were prepared from three replicate cultures for each sample with the RiboMinus Kit (Invitrogen) and the GeneChip Whole Transcript Sense Target Labeling Kit (Affymetrix) Labeled target was hybridized to the MJAY Chip (Affymetrix #540092). Chips were washed and scanned using the Fluidics Station 450 and GeneChip scanner (Affymetrix). Data were analyzed as in Sugnet et al. (2006). The Sepscore is log2 (Include/Skip ratio) of the experimental sample over the reference sample. When exon inclusion increases the Sepscore is positive. Hierarchical clustering (Eisen et al. 1998) was done with Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm); clusters were displayed with Java TreeView (Saldanha 2004) (http://jtreeview.sourceforge.net/).
Motif analysis
Counts of all 5-mers in the selected region of an exon set are compared with their counts from a background set of sequences using Fisher’s exact test, with multiple testing correction. Motif mapping is described in the Supplemental Methods.
RNA affinity chromatography
RNA was bound to beads and incubated with nuclear extract as described in the Supplemental Methods.
Multidimensional Protein Identification Technology (MudPIT)
Mass spectrometry was performed by the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory at UC Berkeley. Peptides were eluted using a 14-step MudPIT procedure (Washburn et al. 2001). The programs SEQUEST and DTASELECT were used to identify peptides and proteins from the mouse database (Eng et al. 1994; Tabb et al. 2002). The false-positive rate for protein identification is calculated to be <0.1% (Elias et al. 2005; Sharma et al. 2008). Detailed methodology is in the Supplemental Methods.
RT-PCR
cDNA synthesis and PCR details are in the Supplemental Methods; primer sequences are in Supplemental Table 9. Percent exon inclusion ([inclusion/(inclusion + skipping)] × 100) is graphed in Excel; error bars are ±SD for n ≥ 3 independent experiments.
DATA DEPOSITION
Microarray data are deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE40962.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Doug Black (Dept. of Microbiology, Immunology and Molecular Genetics, UCLA) and Sean Ryder (Dept. of Biochemistry and Molecular Pharmacology, UMass Medical School) for reagents and helpful advice. Dr. Lori Kohlstaedt (NIH grant 1S10RR025622-01) was a great help with MuDPIT. R.N. and M.H. had postdoctoral fellowships from the California Institute of Regenerative Medicine. This work was supported by NIH training grant TG 5T32GM008646-14 (W.S.F.), MDA135140 from the Muscular Dystrophy Association, NIH grant GM084317, and NIH grant GM040478 to M.A.
REFERENCES
- Arning S, Gruter P, Bilbe G, Kramer A 1996. Mammalian splicing factor SF1 is encoded by variant cDNAs and binds to RNA. RNA 2: 794–810 [PMC free article] [PubMed] [Google Scholar]
- Baehrecke EH 1997. who encodes a KH RNA binding protein that functions in muscle development. Development 124: 1323–1332 [DOI] [PubMed] [Google Scholar]
- Beuck C, Qu S, Fagg WS, Ares M Jr, Williamson JR 2012. Structural analysis of the Quaking homodimerization interface. J Mol Biol 423: 766–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, Burge CB, Cooper TA 2010. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res 38: 7651–7664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Chawla G, Stoilov P, Black DL 2007a. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 21: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL 2007b. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21: 1636–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel AB, Wu J, Lehmann-Blount KA, Williamson JR 2010. High-affinity consensus binding of target RNAs by the STAR/GSG proteins GLD-1, STAR-2 and Quaking. BMC Mol Biol 11: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Black DL 1997. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol 17: 4667–4676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. 2012. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75: 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet-B N, Logan P, Singh G, Cooper TA 2002. Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol Cell 9: 649–658 [DOI] [PubMed] [Google Scholar]
- Chen T, Richard S 1998. Structure-function analysis of Qk1: A lethal point mutation in mouse quaking prevents homodimerization. Mol Cell Biol 18: 4863–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AJ, Paik JH, Zhang H, Shukla SA, Mortensen R, Hu J, Ying H, Hu B, Hurt J, Farny N, et al. 2012. STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev 26: 1459–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chénard CA, Richard S 2008. New implications for the QUAKING RNA binding protein in human disease. J Neurosci Res 86: 233–242 [DOI] [PubMed] [Google Scholar]
- Corioni M, Antih N, Tanackovic G, Zavolan M, Kramer A 2011. Analysis of in situ pre-mRNA targets of human splicing factor SF1 reveals a function in alternative splicing. Nucleic Acids Res 39: 1868–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JB, Patton JG 2006. Activation of α-tropomyosin exon 2 is regulated by the SR protein 9G8 and heterogeneous nuclear ribonucleoproteins H and F. Mol Cell Biol 26: 8791–8802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Clark TA, Schweitzer A, Yamamoto M, Marr H, Arribere J, Minovitsky S, Poliakov A, Dubchak I, Blume JE, et al. 2007. A correlation with exon expression approach to identify cis-regulatory elements for tissue-specific alternative splicing. Nucleic Acids Res 35: 4845–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fruscio M, Chen T, Richard S 1999. Characterization of Sam68-like mammalian proteins SLM-1 and SLM-2: SLM-1 is a Src substrate during mitosis. Proc Natl Acad Sci 96: 2710–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z, Kole R 1991. Selection of splice sites in pre-mRNAs with short internal exons. Mol Cell Biol 11: 6075–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, et al. 2010. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol 17: 187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole TA, Chen Q, Justice MJ, Artzt K 1996. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet 12: 260–265 [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D 1998. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Haas W, Faherty BK, Gygi SP 2005. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2: 667–675 [DOI] [PubMed] [Google Scholar]
- Eng J, McCormack AL, Yates JR 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Am Soc Mass Spectrom 5: 976–989 [DOI] [PubMed] [Google Scholar]
- Francis R, Barton MK, Kimble J, Schedl T 1995. gld-1, a tumor suppressor gene required for oocyte development in Caenorhabditis elegans. Genetics 139: 579–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyrberg C, Becker J, Barthmaier P, Mahaffey J, Fyrberg E 1997. A Drosophila muscle-specific gene related to the mouse quaking locus. Gene 197: 315–323 [DOI] [PubMed] [Google Scholar]
- Galarneau A, Richard S 2005. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12: 691–698 [DOI] [PubMed] [Google Scholar]
- Goo Y-H, Cooper TA 2009. CUGBP2 directly interacts with U2 17S snRNP components and promotes U2 snRNA binding to cardiac troponin T pre-mRNA. Nucleic Acids Res 37: 4275–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding C, Roberts GC, Smith CW 1998. Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated α-tropomyosin exon. RNA 4: 85–100 [PMC free article] [PubMed] [Google Scholar]
- Gromak N, Matlin AJ, Cooper TA, Smith CWJ 2003. Antagonistic regulation of α-actinin alternative splicing by CELF proteins and polypyrimidine tract binding protein. RNA 9: 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Hynes RO 1994. Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes Dev 8: 1561–1574 [DOI] [PubMed] [Google Scholar]
- Ji S, Ye G, Zhang J, Wang L, Wang T, Wang Z, Zhang T, Wang G, Guo Z, Luo Y, et al. 2012. miR-574-5p negatively regulates Qki6/7 to impact β-catenin/Wnt signalling and the development of colorectal cancer. Gut 10.1136/gutjnl-2011-301083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, Yasuda K, Inoue K 2003. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J 22: 905–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Cooper TA 2011. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet 12: 715–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A 1992. Purification of splicing factor SF1, a heat-stable protein that functions in the assembly of a presplicing complex. Mol Cell Biol 12: 4545–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakiza O, Frater L, Yoo Y, Villavicencio E, Walterhouse D, Goodwin EB, Iannaccone P 2005. STAR proteins quaking-6 and GLD-1 regulate translation of the homologues GLI1 and tra-1 through a conserved RNA 3′UTR-based mechanism. Dev Biol 287: 98–110 [DOI] [PubMed] [Google Scholar]
- Lejeune F, Maquat LE 2005. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol 17: 309–315 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Green RE, Brenner SE 2003. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci 100: 189–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Li D, Feng Y 2000. Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J Neurosci 20: 4944–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Sharp PA 1998. Alternative splicing of the fibronectin EIIIB exon depends on specific TGCATG repeats. Mol Cell Biol 18: 3900–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Luyten I, Bottomley MJ, Messias AC, Houngninou-Molango S, Sprangers R, Zanier K, Kramer A, Sattler M 2001. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science 294: 1098–1102 [DOI] [PubMed] [Google Scholar]
- Llorian M, Schwartz S, Clark TA, Hollander D, Tan LY, Spellman R, Gordon A, Schweitzer AC, de la Grange P, Ast G, et al. 2010. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol 17: 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobbardi R, Lambert G, Zhao J, Geisler R, Kim HR, Rosa FM 2011. Fine-tuning of Hh signaling by the RNA-binding protein Quaking to control muscle development. Development 138: 1783–1794 [DOI] [PubMed] [Google Scholar]
- Lock P, Fumagalli S, Polakis P, McCormick F, Courtneidge SA 1996. The human p62 cDNA encodes Sam68 and not the RasGAP-associated p62 protein. Cell 84: 23–24 [DOI] [PubMed] [Google Scholar]
- Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K 2012. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci Rep 2: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modafferi EF, Black DL 1997. A complex intronic splicing enhancer from the c-src pre-mRNA activates inclusion of a heterologous exon. Mol Cell Biol 17: 6537–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno G, Hagiwara M, Kuroyanagi H 2008. STAR family RNA-binding protein ASD-2 regulates developmental switching of mutually exclusive alternative splicing in vivo. Genes Dev 22: 360–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F, Thuma L, Klambt C 2012. The regulation of glial-specific splicing of Neurexin IV requires HOW and Cdk12 activity. Development 139: 1765–1776 [DOI] [PubMed] [Google Scholar]
- Ryder SP, Williamson JR 2004. Specificity of the STAR/GSG domain protein Qk1: Implications for the regulation of myelination. RNA 10: 1449–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder SP, Frater L, Abramovitz DL, Goodwin EB, Williamson JR 2004. RNA target specificity of the STAR/GSG domain post-transcriptional regulatory protein GLD-1. Nat Struct Mol Biol 11: 20–28 [DOI] [PubMed] [Google Scholar]
- Saccomanno L, Loushin C, Jan E, Punkay E, Artzt K, Goodwin EB 1999. The STAR protein QKI-6 is a translational repressor. Proc Natl Acad Sci 96: 12605–12610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248 [DOI] [PubMed] [Google Scholar]
- Sanford JR, Ellis J, Caceres JF 2005. Multiple roles of arginine/serine-rich splicing factors in RNA processing. Biochem Soc Trans 33: 443–446 [DOI] [PubMed] [Google Scholar]
- Schafer DA, Korshunova YO, Schroer TA, Cooper JA 1994. Differential localization and sequence analysis of capping protein β-subunit isoforms of vertebrates. J Cell Biol 127: 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL 2008. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol 15: 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman R, Dickie M, Appel S 1964. Mutant mice (quaking and jimpy) with deficient myelination in the central nervous system. Science 144: 309–311 [DOI] [PubMed] [Google Scholar]
- Southby J, Gooding C, Smith CWJ 1999. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of α-actinin mutally exclusive exons. Mol Cell Biol 19: 2699–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugnet CW, Srinivasan K, Clark TA, O’Brien G, Cline MS, Wang H, Williams A, Kulp D, Blume JE, Haussler D, et al. 2006. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput Biol 2: e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb D, McDonald W, Yates JR 2002. DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1: 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Abe K, Kim CH 1997. Cloning and expression of the quaking gene in the zebrafish embryo. Mech Dev 69: 209–213 [DOI] [PubMed] [Google Scholar]
- Tang ZZ, Zheng S, Nikolic J, Black DL 2009. Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol Cell Biol 29: 4757–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB 2006. An RNA map predicting Nova-dependent splicing regulation. Nature 444: 580–586 [DOI] [PubMed] [Google Scholar]
- van Mil A, Grundmann S, Goumans MJ, Lei Z, Oerlemans MI, Jaksani S, Doevendans PA, Sluijter JP 2012. MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc Res 93: 655–665 [DOI] [PubMed] [Google Scholar]
- Vernet C, Artzt K 1997. STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet 13: 479–484 [DOI] [PubMed] [Google Scholar]
- Voelker RB, Berglund JA 2007. A comprehensive computational characterization of conserved mammalian intronic sequences reveals conserved motifs associated with constitutive and alternative splicing. Genome Res 17: 1023–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. 2012. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150: 710–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Diegel JV, von Hippel PH, Berglund JA 2009. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. Proc Natl Acad Sci 106: 9203–9208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19: 242–247 [DOI] [PubMed] [Google Scholar]
- Wu J, Zhou L, Tonissen K, Tee R, Artzt K 1999. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem 274: 29202–29210 [DOI] [PubMed] [Google Scholar]
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, et al. 2009. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 36: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J, Ruse CI, Nakorchevsky A 2009. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu Rev Biomed Eng 11: 49–79 [DOI] [PubMed] [Google Scholar]
- Yeo GW, Nostrand ELV, Liang TY 2007. Discovery and analysis of evolutionarily conserved intronic splicing regulatory elements. PLoS Genet 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH 2009. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16: 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffran S, Astier M, Gratecos D, Semeriva M 1997. The held out wings (how) Drosophila gene encodes a putative RNA-binding protein involved in the control of muscular and cardiac activity. Development 124: 2087–2098 [DOI] [PubMed] [Google Scholar]
- Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP 2011. Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet 7: e1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, Zhang MQ 2008. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 22: 2550–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Mandler MD, Yi H, Feng Y 2010. Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin-associated glycoprotein. Proc Natl Acad Sci 107: 19061–19066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM, Krieg PA 1997. The KH domain protein encoded by quaking functions as a dimer and is essential for notochord development in Xenopus embryos. Genes Dev 11: 2176–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]