

Alfonzo and colleagues show that in the parasitic protist Trypanosoma brucei, a tRNA anticodon-loop guanosine base methylation affects translation differentially in mitochondria versus the cytoplasm. Mitochondrial protein synthesis is particularly sensitized. The anticodon-loop guanosine methylation was not required for tRNA import to mitochondria. Instead, the methyltransferase TRM5 is imported. Inhibition of expression of TRM5 and the resulting loss of anticodon-loop guanosine methylation greatly affected mitochondrially encoded protein synthesis and protist growth.
Keywords: Trypanosoma, tRNA, methylation, tRNA import, mitochondrion
Abstract
All tRNAs undergo post-transcriptional chemical modifications as part of their natural maturation pathway. Some modifications, especially those in the anticodon loop, play important functions in translational efficiency and fidelity. Among these, 1-methylguanosine, at position 37 (m1G37) of the anticodon loop in several tRNAs, is evolutionarily conserved and participates in translational reading frame maintenance. In eukaryotes, the tRNA methyltransferase TRM5 is responsible for m1G formation in nucleus-encoded as well as mitochondria-encoded tRNAs, reflecting the universal importance of this modification for protein synthesis. However, it is not clear what role, if any, mitochondrial TRM5 serves in organisms that do not encode tRNAs in their mitochondrial genomes. These organisms may easily satisfy the m1G37 requirement through their robust mitochondrial tRNA import mechanisms. We have explored this possibility in the parasitic protist Trypanosoma brucei and show that down-regulation of TRM5 by RNAi leads to the expected disappearance of m1G37, but with surprisingly little effect on cytoplasmic translation. On the contrary, lack of TRM5 causes a marked growth phenotype and a significant decrease in mitochondrial functions, including protein synthesis. These results suggest mitochondrial TRM5 may be needed to mature unmethylated tRNAs that reach the mitochondria and that could pose a problem for translational fidelity. This study also reveals an unexpected lack of import specificity between some fully matured and potentially defective tRNA species.
INTRODUCTION
Most nucleic acids in cells undergo naturally occurring post-replicative and post-transcriptional chemical modifications. These include a myriad of chemical groups that impart distinct local effects at the site of modification and can also globally affect the structure, and thus the function, of the particular nucleic acid they target (Dunin-Horkawicz et al. 2006). By far, tRNAs are the recipients of the most numerous and diverse post-transcriptional chemical modifications, which influence the stability and abundance of tRNAs, as well as their recognition by cognate synthetases, consequently affecting translation (Agris et al. 2007; Juhling et al. 2009). In addition, modifications can have direct impact on protein synthesis by contributing to the strength and specificity of codon–anticodon interactions during decoding (Gustilo et al. 2008).
In most tRNAs, position 37 of the anticodon loop is an encoded purine that is always, with rare exceptions, post-transcriptionally modified (Juhling et al. 2009). In cases where this position is a guanosine (G37), it is methylated at the base to form 1-methylguanosine (m1G37). Because of its important role in translational frame maintenance, m1G is proposed as one of the “primordial” modifications already used in decoding by the last universal common ancestor (Björk et al. 2001). In all organisms studied so far, lack of m1G37 leads to increased +1 frameshifting, triggering growth phenotypes from global defects in the fidelity, but not the efficiency, of protein synthesis (Björk et al. 1989).
In eukaryotes, TRM5, the enzyme responsible for m1G37 formation in a number of tRNAs, has dual localization to the nucleus and the mitochondrion (Lee et al. 2007; Ohira and Suzuki 2012). Most, if not all, nucleus-encoded tRNAs that contain m1G are presumably methylated in the nucleus, following transcription and prior to export to the cytoplasm. Likewise, mitochondria-encoded tRNAs are methylated at G37 by the same nucleus-encoded TRM5 enzyme, which is imported into the organelle from the cytoplasm (Lee et al. 2007). The function of TRM5 in mitochondria has being investigated in yeast, where lack of m1G37 in the mitochondria-encoded initiator tRNAMet causes a reduction in mitochondrial respiration, but the reasons for this decrease remain unknown (Lee et al. 2007). However, given the degree of conservation of this modification, similar functions in translational fidelity are expected for TRM5 and m1G37 in mitochondria.
The parasitic protist Trypanosoma brucei has two genome-containing compartments, the nucleus and mitochondrion. Because of this intracellular compartmentalization, cells also contain two separate protein-synthesizing machineries: one of eukaryotic origin in the cytoplasm and one of bacterial origin in the mitochondria. What sets T. brucei and related kinetoplastid flagellates apart from most other eukaryotes is the complete absence of tRNA genes in the mitochondrial genome (Alfonzo and Soll 2009). Thus, following nuclear transcription, a full complement of tRNAs transits through the cytoplasm and is subsequently imported into the mitochondrion, where they participate in organellar translation. The mitochondrial genome of these protists, termed kinetoplast DNA (kDNA), still encodes a number of subunits of the electron transport chain, but the majority of organellar proteins are also imported from the cytoplasm to supplement all that is needed for mitochondrial biogenesis and function. These include additional subunits of the respiratory complexes, various metabolic enzymes, as well as all proteins involved in the synthesis and processing of mitochondrial nucleic acids, including tRNA modification enzymes. Hence, tRNA and protein import are essential for mitochondrial translation (Sieber et al. 2011).
Little is known about the function of the majority of tRNA modifications in kinetoplastid protists, let alone their roles in kinetoplast mitochondria. The lack of tRNA genes in T. brucei kDNA raises an interesting question about the importance of m1G formation in the organelle. It is possible that due to the robust mitochondrial tRNA import system in kinetoplastids, including T. brucei, tRNAs are imported into the mitochondria fully methylated at G37. Cytosolic methylation would therefore satisfy all requirements for m1G37 in both cellular compartments. This scenario supposes that the tRNA import system is able to discriminate between fully G37-methylated and unmethylated tRNAs, where the latter are presumably not functional in translation. Alternatively, there is no discrimination between the two tRNA species, and the mitochondrial translation system evolved counter-measures to deal with the unmethylated species and their potential effects on translational fidelity. This is especially relevant given numerous suggestions of possible roles of tRNA modifications in mitochondrial tRNA import. For example, in plants, three nucleus-encoded tRNALeu contain a ribose methylated guanosine at position 18 (Gm18) in the mitochondrion, whereas this position is not methylated in their cytosolic counterparts (Marechal-Drouard et al. 1988). A similar observation was made in T. brucei using enzymatic sequencing of tRNALys, tRNALeu, and tRNATyr, each of which bears a mitochondrion-specific ribose methylated cytidine residue at position 32 (Cm32) of the anticodon (Schneider et al. 1994a,b). Likewise, thiolation of tRNAs has been implicated as a negative determinant for tRNA import in the parasitic flagellate Leishmania tarentolae (Kaneko et al. 2003), but the same is not true in T. brucei (Paris et al. 2009).
Because of the potential role of modifications in tRNA distribution in T. brucei, we have investigated the nature of m1G37 synthesis in this organism. Our interest was prompted by the observation of significant amounts of m1G37-lacking tRNAs in the mitochondrion. As shown here, the T. brucei import system does not appear to discriminate between tRNAs fully methylated and unmethylated at G37, raising the question of how the mitochondrial translational system copes with the potentially harmful accumulation of undermodified tRNAs, which may cause increased frameshifting. We show that down-regulation of the T. brucei TRM5 (TbTRM5) enzyme leads to a marked growth phenotype. Despite only having minor effects on the synthesis of some proteins in the cytoplasm, the lack of m1G37 leads to a decrease in mitochondrial protein synthesis, cytochrome C oxidase activity, an associated reduction in respiration, and accumulation of reactive oxygen species (ROS). Combined, these phenotypes partly explain the growth defects due to ablation of TbTRM5. These experiments suggest that in T. brucei, the potential translational problem posed by accumulation of m1G37-lacking tRNAs in the mitochondrion is solved by the import of the TRM5 enzyme. TRM5 may then methylate any m1G37-lacking tRNA that may find its way into the organelle due to the relaxed selectivity of the import system for methylated and unmethylated species.
RESULTS
T. brucei mitochondria contains a significant amount of tRNA lacking m1G37
In organisms that import all tRNA from the cytoplasm, we have questioned whether or not mitochondrial m1G formation is still needed. Total mitochondrial and extra-mitochondrial RNA fractions were probed in Northern blot hybridization experiments using an oligonucleotide specific for tRNAIle UAU (one of seven tRNAs predicted to contain m1G37). This probe distinguishes between m1G37-containing and unmethylated tRNAIle UAU, since m1G disrupts base-pairing and prevents probe hybridization (Fig. 1A). We found negligible hybridization signal with the extra-mitochondrial RNA fraction, consistent with the presence of a modification that blocks probe hybridization and supporting the fact that under steady-state growth conditions, cytosolic tRNAIle UAU is essentially fully modified at G37 (Fig. 1B). On the contrary, a significant level of unmodified tRNAIle UAU was observed in the mitochondrial RNA fraction (a 6.8-fold increase over the cytoplasmic signal) (Fig. 1C). We also probe the same membrane with a new oligonucleotide that hybridizes downstream from position 37. This oligonucleotide does not overlap the m1G37 position (Fig. 1A, dashed arrow), and it does not discriminate between unmethylated and methylated tRNAILe UAU. This probe could easily detect a signal in the cytoplasmic fraction for tRNAIle UAU. The relative levels of this tRNA are slightly higher in the mitochondria that seen with the control tRNAGln and tRNAGlu. These differences are likely due to variations in import efficiency between different tRNAs (Kapushoc et al. 2002). To assess fraction purity and rule out the possibility of fraction cross-contamination, the same membrane was hybridized with a probe specific for the spliced leader RNA (SL RNA, a cytoplasmic marker) and mitochondrial ribosomal RNA (12S rRNA, a mitochondrial marker) (Fig. 1B). This experiment showed the absence of SL RNA in the mitochondrial fraction, verifying its high purity and negligible cytoplasmic contamination. A small amount of 12S rRNA signal was detected in the extra-mitochondrial fraction, suggesting that the faint signal in this fraction with the tRNAIle probe may be due to mitochondrial breakage during purification. Probes specific for two tRNAs that are known to lack m1G37 (tRNAGln UUG and tRNAGlu UUC) were used as positive controls for tRNA hybridization and showed that these tRNAs could be easily detected in both RNA fractions (Fig. 1B). Taken together, these experiments suggest that a significant portion of the mitochondrial tRNAIle UAU is unmethylated compared with its fully methylated cytosolic counterpart (Fig. 1C).
FIGURE 1.

Presence of m1G37-lacking tRNAIle UAU in mitochondria. (A) Cloverleaf structure of tRNAIle UAU. U34 corresponds to the first position of the anticodon, and G37 is the site of m1G formation. The solid arrow represents the G37-overlapping oligonucleotide primer specific for tRNAIle UAU used as a probe in Northern hybridizations. The presence of m1G prevents hybridization of this probe. The dashed arrow represents a m1G37-nonoverlapping probe, which should not discriminate between m1G37-containing and unmethylated tRNAs. (B) Northern blots of cytosolic (C, extra-mitochondrial) and mitochondrial (M) RNA fractions. The panel shows a significant signal with the m1G37-overlapping probe, which detects nonmethylated tRNAIle UAU (IleUAU*). The probe that does not cover the methylated position shows a signal for cytoplasmic and mitochondrial fractions (IleUAU). Probes for 12S rRNA (12S, a mitochondrial marker) and spliced leader RNA (SL, cytoplasmic marker) were used as controls for fraction purity. Probes specific for tRNAGln UUG (GlnUUG) and tRNAGlu UUC (GluUUC) were used as hybridization controls for tRNAs that do not contain m1G. These probes have similar melting temperatures for hybridization and hybridize to a similar region of these tRNAs as the tRNAIle probe. (C) The relative levels of unmethylated tRNA, in either cellular fraction, were calculated by normalizing the signals for the unmethylated (IleUAU*) and the nondiscriminating probe (IleUAU) from each compartment to the tRNAGln UUG and tRNAGlu UUC signals. The normalized unmethylated value for each compartment was then divided by the total signal (from IleUAU) and the cytoplasmic level set at one. The normalized unmethylated signal for the mitochondrial fraction was divided by the normalized unmethylated cytoplasmic signal.
Lack of m1G37 leads to a growth phenotype
To corroborate that the lack of hybridization with the cytosolic fraction is indeed due to the presence of m1G37, we searched the T. brucei genomic databases (TriTrypDB) with the yeast TRM5 sequence as a query and identified its potential homolog (accession no. Tb927.8.5720). The putative T. brucei enzyme (TbTRM5) contains all the conserved residues important for TRM5 function in other eukaryotes, including the S-adenosyl methionine (SAM)–binding domain. The putative TbTRM5 coding sequence was cloned into a T. brucei RNAi vector, which allows inducible synthesis of double-stranded RNA upon the addition of tetracycline to the growth media. Growth curves with the RNAi-induced cells (+TET) were compared with uninduced cells (−TET) and wild-type culture controls (Fig. 2A). A strong growth phenotype was observed after 8 d of RNAi induction, which persisted until day 16, at which point the growth curve was stopped. To confirm the down-regulation of TbTRM5 levels, a Western blot was performed with polyclonal antibodies raised against the recombinant TbTRM5 protein and total cell extracts from the 8 d of induction time point (Fig. 2A, inset). This experiment showed a significant decrease in TbTRM5 levels compared with wild-type or uninduced controls.
FIGURE 2.

TRM5 is important for growth in T. brucei. (A) Growth curve of wild-type (WT), uninduced (−TET), and induced (+TET) cell lines. The uninduced and induced lines are T. brucei 29-13 cells transformed with a dual T7 plasmid containing a portion of the putative TRM5 m1G methyltransferase coding sequence. RNAi was induced by addition of tetracycline to the media. Total protein extracts were prepared at the onset of the growth phenotype (indicated by the arrow). Western blots with WT, −TET, and +TET protein samples were performed using rabbit polyclonal antibodies against recombinant TbTRM5. Shown in the inset is a representative Western blot; “anti-TRM5” refers to a membrane probed with the anti-TRM5 polyclonal antibody; the signal with anti-enolase antibodies was used as a loading control. (B) Total (T), cytoplasmic (C), and mitochondrial (M) protein fractions were used in Western blots to establish the intracellular localization of TRM5. “Anti-enolase” refers to Western blots performed with antibodies against enolase, a known cytoplasmic marker. The Isd-11 protein was used as a mitochondrial marker. These protein markers were also used as controls for localization and to determine fraction purity. (C) Immunofluorescence to determine TRM5 localization using the same anti-TRM5 antibodies as above. In addition, the cells were stained with DAPI (DNA) and MitoTracker Red as a mitochondrial marker. The anti-TRM5 antibodies are shown in green. Merged image refers to the superposition of the DAPI (blue), TRM5 (green), and MitoTracker (red). Yellow fluorescence results from the colocalization of MitoTracker and TRM5, indicative of mitochondrial localization. Nomarski refers to a phase-contrast image. N and K in the DAPI panel refer to nuclear and mitochondrial (kinetoplast, kDNA), respectively.
TbTRM5 localizes to nucleus and mitochondrion
In the yeast system, TRM5 localizes to the nucleus and the mitochondrion, with a single enzyme being responsible for m1G37 formation in both cellular compartments. We performed localization experiments with the α-TbTRM5 antibodies. Western blots with subcellular T. brucei fractions (total, cytoplasmic, and mitochondrial) showed that a significant portion of the TRM5 protein localizes to the organelle compared with enolase and Isd11, the cytoplasmic and mitochondrial markers, respectively (Fig. 2B). Immunofluorescence experiments with the α-TbTRM5 antibodies, mitochondria-specific MitoTracker dye, as well as DAPI—which labels both nuclear (N) and mitochondrial (K) DNAs—also revealed a significant amount of TbTRM5 in the mitochondrion (Fig. 2C, yellow-orange color in the merged image), although a majority of the protein localizes within the nuclear periphery. These experiments are again consistent with the nuclear and mitochondrial localization of the putative TbTRM5 protein.
TbTRM5 is responsible for m1G37 formation in several tRNAs
We then tested whether this putative enzyme is indeed specific for G37 methylation of tRNAs. To this end, we performed a primer extension assay with an oligonucleotide primer specific for tRNAIle UAU. Because m1G prevents Watson-Crick base-pairing, the presence of this modification leads to a strong stop 1 nucleotide (nt) short of the modified position (Fig. 3A). Similar results were obtained with tRNAArg CCG, tRNACys GCA, tRNAHis GUG, tRNALeu CAG, and tRNAPro AGG, all predicted to have m1G37 (data not shown). We performed primer extension assays with extra-mitochondrial and mitochondrial RNA fractions derived from the RNAi-induced and uninduced cell lines. We found that down-regulation of TbTRM5 leads to the disappearance of the strong stop in both RNA fractions (Fig. 3B, left panel). This experiment shows that the target enzyme is clearly required for m1G formation at position 37 of tRNAIle UAU. This finding was further confirmed by incubation of an in vitro tRNAIle UAU transcript with recombinant TbTRM5 in the presence of SAM, followed by primer extension reaction, which again led to the appearance of the strong stop characteristic of the presence of m1G37 (Fig. 3B, right panel). We thus conclude that we have identified the TbTRM5 enzyme, which localizes to the nucleus and mitochondrion and is responsible for m1G37 formation in cytosolic and mitochondrial tRNAIle UAU.
FIGURE 3.

TRM5 is responsible for m1G37 formation. (A) Schematic of the primer extension assay used for detection of m1G37. The radioactive oligonucleotide probe anneals 8 nt away from the methylated position. In the presence of m1G, a “strong” stop 1 nt short of the methylated position (primer +7) is expected, while lack of m1G leads to read-through G37. (B) Primer extension assay with extra-mitochondrial (nuclear/cytoplasmic [cyto]) and mitochondrial (mito) RNA fractions isolated from TRM5 RNAi-induced (+) and uninduced cells (−; left panel). The right panel shows an in vitro transcript corresponding to tRNAIle UAU incubated with recombinant TbTRM5 purified from E. coli (+). A similar reaction but in the absence of enzyme (−) was used as a negative control for methylation. The arrows point at the position of the primer used for the assay and the positions of the primer +7 product indicative of m1G37. Primer alone refers to a reaction where no template RNA was added to the primer extension reaction.
To further test this proposal, we also performed Northern blots with subcellular RNA fractions derived from the TbTRM5-induced and uninduced cells and probes specific for tRNAIle UAU. A strong hybridization signal appeared in the cytosolic fraction obtained from the induced cell line. This is consistent with the idea that the lack of hybridization in Figure 1, with the same probe, was due to the presence of m1G37. As an additional control, we also probed the same membrane for the other tRNAIle UAA isoacceptor (Fig. 4B). Notably, this isoacceptor does not contain a G37 and therefore can be detected with a probe covering a comparable region of the tRNA and overlapping A37 in this case. We also probed the same membrane with the m1G nonoverlapping oligonucleotide (see Fig. 1A, dashed arrow as reference). This probe shows similar hybridization signals between the induced and uninduced samples (Fig. 4A,B). These observations strongly support the view that the reduced hybridization signal with tRNAIle UAU in the uninduced cells is due to the presence of the methylated nucleotide. In these experiments, the level of unmethylated tRNA in the uninduced samples from mitochondria was slightly lower than that seen with samples from wild-type cells (cf. Figs. 1 and 4A); this may reflect the fact that RNAi is sometimes leaky in T. brucei. We also showed, by hybridizing the same membrane with probes specific for other G37-containing tRNAs, that the levels of unmethylated species varies depending on the given tRNA species (Fig. 4C,D). In most cases, however, the bulk of the tested tRNA was mostly methylated in both compartments with the exception of tRNALeu CAG, which showed a significant signal in the cytosol of uninduced cells. Although we do not know if these relative differences in methylation levels are of biological significance, it is clear that in all cases the hybridization signal increases when TbTRM5 is down-regulated by RNAi, strongly suggesting its involvement in m1G37 formation in these additional isoacceptors.
FIGURE 4.

m1G37 levels vary between compartments and different tRNAs. (A) Northern analysis with subcellular fractions from the TRM5 RNAi cell line comparing induced (+) and uninduced (−) samples (as before). IleUAU* refers to a probe that overlaps the methylated position, which only yields a signal if the tRNA is unmethyalted (as in Fig. 1). IleUAU refers to the dashed probe from Figure 1, which does not discriminate between methylated and unmethylated tRNA. (B) IleUAA refers to a probe specific for the other tRNAIIe isoacceptor. This tRNA has an A at position 37, and it is therefore not a substrate for TRM5 methylation. GlnUUG, SL, and 12S are controls (as in Fig. 1). (C,D) Same membrane as in panel A, which was stripped and rehybridized with probes specific for other G37-containing tRNAs (as indicated), expected to be substrates for TRM5. Cyto and mito refer to extra-mitochondrial and mitochondrial fractions as before. In all cases, the asterisk denotes the oligonucleotide probe specific for the unmethylated species. The relative level of unmethylated tRNA between compartments was calculated as before and provided as a fold difference over the uninduced cytoplasmic signal set as 1 (umt, fold diff.). − and + refer to the TRM5-uninduced and TRM5-RNAi–induced RNA fractions.
Lack of m1G37 does not globally affect cytosolic translation
The previous experiments raise the question of what leads to the strong growth phenotype seen with the TbTRM5-depleted cells. In other systems, similar phenotypes have been ascribed to a decrease in translation fidelity due to the absence of m1G37 in cytosolic tRNAs (Urbonavicius et al. 2001, 2003; Waas et al. 2007). Moreover, in yeast lack of m1G37 in mitochondrial initiator tRNAMet led to a respiratory defect (Lee et al. 2007). It was suggested that this defect was the result of a decrease in mitochondrial protein synthesis, but this hypothesis was not formally tested. To delineate the specific contribution of m1G37 to cytosolic protein synthesis, we generated a transgenic T. brucei cell line constitutively expressing a chloramphenicol acetyltransferase (CAT) construct integrated as a single copy in the genome. This cell line was then transformed with the TbTRM5 RNAi plasmid described above, and cell growth was monitored after the addition of tetracycline to induce RNAi. Again, a strong growth phenotype was observed after 8 d of induction, and the lack of m1G37 was confirmed as before (data not shown). At this point, cells were collected from the RNAi-induced and uninduced cultures, and the specific activity of CAT was measured (see the Materials and Methods). Comparable levels of CAT activity were observed in the presence or absence of tetracycline (Fig. 5), leading to the conclusion that lack of m1G37 does not significantly affect synthesis of the CAT reporter. Importantly, the CAT construct contains several codons that are decoded by m1G37-containing tRNAs in addition to tRNAIle (38 codons, or 17.35% of the coding sequence). We also performed a series of Western blots with antibodies to various nucleus-encoded proteins (that likewise contain codons that would require m1G-containing tRNAs), these showed no significant difference between the induced and uninduced samples (Fig. 5C). In this experiment, TRM5 was used as positive control for down-regulation of expression and behaved as expected (Fig. 5C). Notably, some of these proteins (e.g., Isd11 or MRP1) are imported into the mitochondria following synthesis in the cytoplasm. This leads to the conclusion that lack of m1G in the cytoplasm does not affect the levels of a number of nucleus-encoded proteins that are synthesized in the cytoplasm regardless of their final intracellular localization.
FIGURE 5.

Down-regulation of TRM5 does not affect cytoplasmic translation. (A) A chloramphenicol acetyl transferase (CAT) assay with the same cell lines as above comparing the levels of a constitutive CAT translational reporter with TRM5 RNAi-induced (+) and uninduced (−) cell lines. “Substrate” refers to the input fluorescently labeled chloramphenicol and product refers to its acetylated form. A vector alone control refers to the same cell line transformed with the constitutive expression plasmid lacking the CAT open reading frame. This control was used as a negative control and as a background control in these experiments. Substrate and product were separated by thin-layer chromatography (Materials and Methods). (B) Quantitation of similar experiments as in A. The table shows five independent assays, following five different RNAi inductions. All samples come from day 8 after induction. The levels of CAT did not change in either earlier or later time points (data not shown). CAT activity is expressed in units (U) per milligram, where 1 unit equals 1 pmol of acetylated chloramphenicol produced per minute. All measurements were performed in the linear phase of the enzyme activity curve. (C) Western blots with antibodies specific for various nucleus-encoded proteins, including TRM5 (used as positive control for down-regulation) and enolase (a cytoplasmic marker). Isd-11, MRP1, and Phb1 are all nucleus-encoded proteins that are synthesized in the cytoplasm and then imported into the mitochondria. WT refers to total protein from wild-type cells; TET − and + refer to total protein sample from uninduced and RNAi-induced cells, as before.
TbTRM5 is important for mitochondrial protein synthesis and biogenesis
In light of the previous results, we decided to explore the possibility that the bulk of the growth defect following RNAi may be due to the lack of m1G37 in mitochondrial tRNAs. We performed in vivo labeling experiments with 35S methionine. In this experiment, RNAi was induced as before, except that cells were washed and incubated with cycloheximide prior to the addition of the labeled amino acid mixture. Under these conditions, cytosolic translation is inhibited but mitochondrial translation remains unaffected. Total protein samples were collected at 6, 7, and 8 d (6 d, 7 d, 8 d) following RNAi induction and compared with an uninduced control (UND) upon separation by two-dimensional (2D) SDS-PAGE as described previously (Horváth et al. 2000, 2002). Partly due to the very hydrophobic nature of the T. brucei mitochondria-encoded proteins, only two proteins are discernible in this type of gel, namely, cytochrome c oxidase subunit 1 (CO1) and apocytochrome b (Cyb), with their identity corroborated by protein sequencing (Horváth et al. 2000). We observed a great reduction in the levels of both mitochondria-encoded proteins (Fig. 6). To ensure equal sample loading, these gels were also stained with Coomassie Blue dye prior to autoradiography (Fig. 6, insets). Since tetracycline does not affect mitochondrial translation in T. brucei (Richterová et al. 2011), this observation leads to the conclusion that the lack of m1G37 specifically affects global mitochondrial protein synthesis.
FIGURE 6.
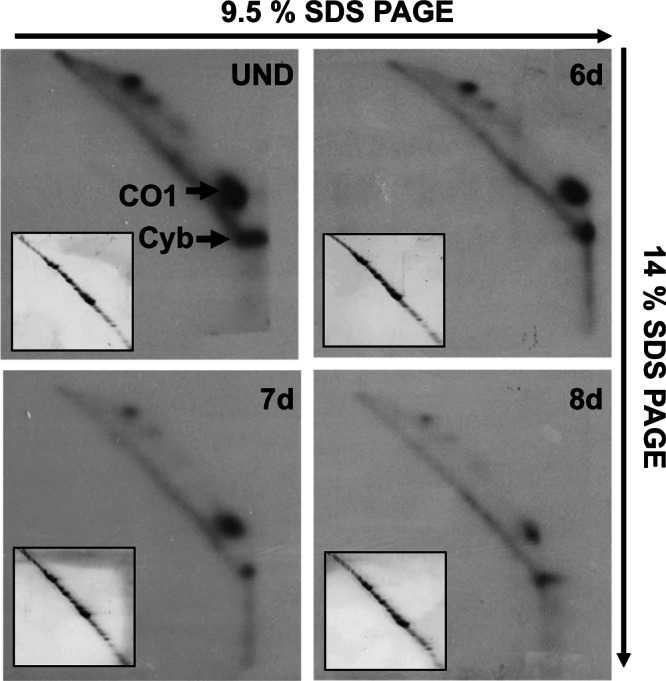
Lack of TRM5 leads to a decrease in mitochondrial protein synthesis. Uninduced (−) and induced (+) samples were separated in two-dimensional gels as described previously (Neboháčová et al. 2004). The samples were radioactively labeled as described in the Materials and Methods. The arrows mark the position of the two identifiable mitochondria-encoded proteins, cytochrome oxydase subunit 1 (CO1) and cytochrome oxydase subunit b (Cyb), used as reporters for the steady-state levels of protein synthesis following RNAi induction for 6 (6 d), 7 (7 d), or 8 d (8 d). The insets show Coomassie Blue–stained versions of the same gels, prior to exposure to X-ray film, used to ensure equal loading with the different samples. Arrows indicate the direction of electrophoresis, where the first dimension was on 9.5% SDS-PAGE and the second on a 14% SDS gel.
To determine the biological impact of this down-regulation, we also measured respiration as well as mitochondrial membrane potential in the TbTRM5 RNAi-induced cells. While a significant decrease in respiration was observed upon the ablation of TbTRM5, respiration remained unaltered in the uninduced cell line and the wild-type control (Fig. 7A). The same was true of cytochrome c oxidase, which showed a decrease in activity concomitant with the RNAi growth phenotype (Fig. 8). Contrary to our expectations, the mitochondrial membrane potential increased (Fig. 7C). Such increases may be the result of increase in mitochondrial mass/volume. We tested this possibility by staining the cells with the membrane potential insensitive, mitochondria-specific dye MitoTracker Green FM, where fluorescence was determined as before. A slight but significant effect in volume as judged by an increase in fluorescence was seen after 10 d of RNAi induction, while the significant increase in membrane potential is already obvious on day 6 after induction (Fig. 7D). We conclude that although there is slight increase in mitochondrial volume, such an increase cannot account for the much earlier increase in membrane potential. Therefore the volume increase at day 10 is likely an effect of RNAi but not a cause for the change in membrane potential. Although we do not fully understand the meaning of the observed increased in potential, it explains why protein import does not seem to be impaired; for example, the levels of the Isd11 mitochondrial marker did not change after TRM5 RNAi (Fig. 5C). We also assayed for markers of mitochondrial defects, such as DNA fragmentation and mitochondrial iron accumulation, to see how this respiratory phenotype could be correlated to the observed growth phenotype. In these aspects, no significant differences were observed between the various cell lines (data not shown), with one notable exception, namely an increase in ROS (Fig. 7B), which could provide a level of toxicity related to the observed growth phenotype. However, given the pleiotropic effects due to lack of m1G37 in other systems additional factors are likely at play.
FIGURE 7.

m1G37 formation in mitochondria is important for respiration. (A) Mitochondrial membrane potential determination comparing uninduced (−) and 6, 8, and 10 d of TRM5-RNAi induction as before. A wild-type (WT) sample was used as a positive control, and the same sample incubated with the CCCP (an uncoupler inhibitor of oxidative phosphorylation) was used as a negative control. (B) Similar samples as in A were incubated with a fluorescent indicator of reactive oxygen species (ROS) and compared with uninduced, WT, and a WT control treated with paraquat, a known inducer of ROS production. (C) The rate of oxygen consumption was determined as a measure of respiration. (D) Relative fluorescence determined by flow-cytometry following incubation of induced an uninduced cells with the membrane-potential independent but mitochondria-specific MitoTracker Green FM dye (molecular probes). The graph compares WT values (set to 100%) to uninduced and RNAi-induced cell lines. The mean and the SD of three independent experiments are shown. A Tukey-Kramer analysis determined that the results are significant in comparison to wild-type levels, with P-values <0.05 (denoted by asterisks in the different graphs).
FIGURE 8.

Cytochrome c oxidase assay. (A) A qualitative gradient gel showing and in-gel staining for cytochrome oxidase activity. Equal amounts of total protein fractions obtained from mitochondria purified from wild-type cells, uninduced cells, and TRM-RNAi–induced cells (for 8 and 9 d) were loaded in each lane. (B) A quantitative cytochrome c oxidase assay using similar samples as in A comparing the levels in the wild-type to that of the uninduced and RNAi-induced fractions. Relative COX activity refers to the levels in each sample compared with the wild type, which was set as 100% activity. The assays are representative of three independent measurements for each sample. (C) Comparison of the induced activity to that of the uninduced sample, where the uninduced was set to 100%. P-values are <0.05. Numbers above each bar in the graph show the percentage of activity, including the standard deviation between the different replicates.
DISCUSSION
The dynamic nature of intracellular tRNA distribution in T. brucei has led us to the question of how tRNAs are matured in the different cellular compartments. It may well be that the tRNA import system is so efficient that it alone suffices in providing fully modified (mature) tRNAs needed for mitochondrial translation. However, a number of reports have shown that, following import, tRNAs are still further modified in the mitochondrion (Schneider 2011). Consequently, some, if not all, imported tRNAs have two sets of modifications. A typical tRNA destined for the mitochondrion receives some modifications in the nucleus after transcription, additional modifications in the cytosol, and then more modifications following import into the mitochondrion (Paris et al. 2009; Phizicky and Alfonzo 2010). This then implies that in common with organisms that encode most or all of their mitochondrial tRNAs in the organellar genome, T. brucei still imports a subset of nucleus-encoded modification enzymes. We have suggested that some modifications are necessary to readapt tRNAs intended for eukaryotic translation in the cytosol to the nuances of the prokaryote-type translation system in the mitochondrion (Rubio and Alfonzo 2005). In this respect then, a universally conserved modification like m1G37, which plays similar and important roles in archaea, bacteria, and eukarya, could be easily provided by the robust import system and need not to be synthesized in the mitochondria.
Presented here are studies prompted by two primary observations: (1) a significant amount of the m1G37-containing tRNAs are found in the mitochondria in their unmethylated form, and (2) the enzyme responsible for m1G formation is imported to the mitochondrion in addition to its nucleo/cytosolic localization. These findings led to the question of the role of m1G formation in the organelle. We hypothesized that because of the important role of m1G37 in reading frame maintenance in all organisms (including mitochondria), TbTRM5 import was necessary to deal with unmethylated tRNAs, which could potentially affect mitochondrial translational fidelity. In support of this hypothesis, ablation of TbTRM5 leads to a growth defect and a dramatic decrease of mitochondrial protein synthesis. Surprisingly, lack of m1G37 had negligible effects on the levels of the CAT reporter and/or several nucleus-encoded proteins (including some that are imported into the mitochondria). However, lack of m1G greatly impacted mitochondrial protein synthesis. Although these results suggest that the growth phenotype may be the result of the mitochondrial defect, we cannot formally rule out the possibility that the expression of some cytosolic proteins is also affected.
In the case of tRNAIle UAU, the bulk of the cytosolic tRNAs is fully methylated, but a significant portion of it remains unmethylated in the mitochondrion. This reveals a new facet of mitochondrial tRNA import: The import machinery cannot clearly differentiate between tRNAs lacking or containing m1G37. Yet if this postulate is correct, the import system must be sufficiently robust as to efficiently import; however, little unmethylated tRNAs escape TbTRM5 in the nucleo/cytosolic compartments. Alternatively, it is possible that the unmethylated tRNA, as has been suggested for other imported tRNAs, is the preferred substrate for import and only then is methylated to become functional. With the data presented, we cannot distinguish between these scenarios. However, in either case, it is clear that TbTRM5 in the mitochondrion ensures methylation of G37-containing tRNAs, which in turn are essential for mitochondrial function. We emphasize that the observed defects cannot be ascribed solely to tRNAIle UAU, given that seven other tRNAs contain m1G37. However, tRNAIle UAU is highlighted here because it shows the highest levels of unmethylated species in the organelle.
In a broader context, a number of mitochondrial DNA mutations are associated with human diseases (Florentz et al. 2003; Putz et al. 2007; Yarham et al. 2011). In the specific case of AISA (acquired idiopathic sideroblastic anemia), onset of the disease is correlated with a G-to-A mutation that targets position 37 of tRNAs, including the methylated G37 position of tRNALeu CUN (Gattermann et al. 1996). Although the reasons for this correlation are not well understood, our data suggest that this defect may well be due to a reduced efficiency of mitochondrial translation, as shown here in the T. brucei system. The results described here also show a striking similarity to the “threshold” effect observed with many human mitochondrial mutations that lead to malfunction, where a certain level of unmodified or mutated tRNA may be tolerated, but a clear threshold exists that, when surpassed, allows phenotypes to manifest themselves in the form of physiological defects. By analogy in the T. brucei case the (wild-type) mitochondrial protein synthesis can handle a significant amount of G37-unmethylated tRNA, but in the absence of TRM5, a threshold for this unmethylated species may be exceeded. Since mitochondrial genetics in the T. brucei system is not currently possible, we cannot determine if increased translational frameshifting in mitochondria is at the root of the problem.
MATERIALS AND METHODS
Plasmid constructs, transfections, RNAi induction, and cell cultivation
A 1001-nt-long portion of the TRM5 gene (Tb927.8.5720) was PCR amplified using primers TRM5-F (5′-ACTAGTCTTCAAGTTGCACTTACCACACA-3′) and TRM5-R (5′-CTCGAGTCTTTTCTCAGCTCGAAAGAAAG-3′; added SpeI and XhoI restriction sites are underlined) from total genomic DNA of T. brucei strain 29-13. The amplicon was cloned into the p2T7-177 vector, which was linearized with NotI (for genome integration) and then used to transform procyclic T. brucei 29-13 cells and selected as described elsewhere (Wickstead et al. 2002). RNAi was triggered by the addition of 1 μg/mL of tetracycline to the growth medium (SDM-79). Cell density was measured every 24 h using the Beckman Z2 Coulter counter over a period of 16 d after the induction of double-stranded RNA synthesis.
For endogenous expression of TRM5 in T. brucei, the C-terminal tagged TRM5 protein was created by PCR amplification of the full-size TRM5 gene and inserted into the pLew-79-MHTAP plasmid containing His and Myc epitope tags (Jensen et al. 2007). This construct was tranformed into the 29-13 cell line for inducible expresion following tetracycline addition. For expression in Escherichia coli, the full-length TRM5 open reading frame was amplified with TRM5 specific primers by PCR. The amplicon was gel-purified and cloned into the pET100 expression vector (Invitrogen). The resulting expression plasmid encoding 6xHis-tagged TRM5 was transformed into the E. coli strain BL21(DE3) (Novagen). Soluble protein was obtained by IPTG induction of recombinant protein expression in bacterial cells (incubation for 20 h at 25°C) and purified under native conditions using Ni2+-chelate chromatography according to the method described previously (Spears et al. 2011).
Preparation of antibodies and Western blot analysis
Polyclonal antibodies were prepared by immunizing rabbits at 2-wk intervals with four subcutaneous injections of 0.5 mg purified recombinant TRM5 protein emulsified with complete (first injection) and incomplete (following injections) Freund’s adjuvant. The specific TRM5 antibody was affinity purified from rabbit serum by using immobilized recombinant TRM5 antigen and was used for further experiments. Cell lysates corresponding to 5 × 106 cells/lane were separated on 15% SDS–polyacrylamide gel, blotted, and probed. The polyclonal rabbit antibodies against the T. brucei TRM5, Isd11 (Paris et al. 2011), Phb1 (Týč et al. 2010), MRP1 (Vondrušková et al. 2005), and enolase (kindly provided by P.A.M. Michels) were used at 1:100, 1:1.000, 1:1.000, 1:1.000, and 1:1.000 dilutions, respectively. Secondary anti-rabbit IgG antibodies (1:2000) coupled to horseradish peroxidase (GE Healthcare) were visualized according to the manufacturer’s protocol using the ECL kit (Pierce).
Membrane potential, mitochondrial volume, ROS, and oxygen consumption measurement
Exponentially growing procyclic cells were resuspended in 1 mL of fresh SDM-79 medium (at a concentration 5 × 106 cells/mL), and mitochondrial inner membrane potential/mitochondrial volume was measured by the uptake of 50 nM MitoTracker Red CMX ROS/500 nM MitoTracker Green FM (Molecular Probes) for 30 min at 27°C. After staining, the cells were resuspended in 1× PBS and instantly measured by flow cytometry using a Facs Canto II (BD). For the measurement of free ROS, cells were incubated for 30 min at 27°C with the dye dihydrorhodamine 123 at a final concentration of 10 μg/mL and subsequently analyzed using a Facs Canto II (BD). Oxygen consumption was determined with a Clark electrode (Strathkelvin) at 4-min intervals according to the method described previously (Horváth et al. 2005).
Northern blot analysis and primer extension assay
RNA was isolated using the guanidinium thiocyanate/phenol/chloroform extraction method (Chomczynski and Sacchi 1987). Ten micrograms of extramitochondrial (nuclear and cytosolic) or 2.5 μg of mitochondrial RNA was separated on denaturating 8% polyacrylamide gel with 8 M urea and electroblotted to Zeta-probe (Bio-Rad) membranes, which were subsequently probed with [32P]-5′ end-labeled oligonucleotides specific for each RNA. Hybridization procedures were carried out according to the manufacturer’s instructions (Bio-Rad). Images were taken with a Storm PhosphorImager (Molecular Dynamics). Primer extension analysis was carried out according to the method described previously (Alfonzo et al. 1999).
Oligonucleotide probes
The following oligonucleotides were used for Northern analysis and primer extension of the indicated RNAs: tRNAIle UAU*, 5′-CCCGCGATATTCGGTTCATA-3′; tRNAIle UAU, 5′-GGGGTTCGAACCCGCGATATTCGGT-3′; tRNAIle UAU pe:, 5′-CCGGCGGGGTTCGAACCCGCATAT-3′; tRNAIle AAU, 5′-CCAACAGGGGTCGAACCTGTGACC-3′; tRNAGlu UUC, 5′-TTCCGGTACCGGGAATCGAAC-3′; tRNAGln UUG, 5′-CAGGATTCGAACCTGGGTTTTCG-3′; 12S rRNA, 5′-AGGAGAGTAGGACTTGCCCT-3′; SL RNA, 5′-GCTGCTACTGGGAGCTTCTCATAC-3′; tRNALeu CAG *, 5′-CGGAGAGATGACGACCTG-3′; tRNALeu CAG, 5′-AACCCACGCCTCCGGAGAG-3′; tRNAPhe GAA*, 5′-TTCAGATCTTCAGTCTGACGCT-3′; tRNAPhe GAA, 5′-GCGACCCGGGATCGAACCAGGGACC-3′; tRNAHis GUG*, 5′-CCGGGTATTCAGAGCCAC-3′; and tRNAHis GUG: 5′-GGAATCGAACCCGGGTATT-3′.
Chloramphenicol acetyl transferase assay and in vivo mitochondrial protein synthesis
Chloramphenicol acetyl transferase (CAT) assays were performed using total protein extracts from uninduced and RNAi-induced cells constitutively expressing CAT from a single-copy of the gene stably integrated in the genome. CAT levels were determined with the FAST CAT (deoxy) CAT assay kit (Invitrogen), as recommended by the manufacturer. The amount of substrate converted into product during the CAT reactions was quantified using a Storm 860 blue fluorescence scanner and the ImageQuant software (Molecular Dynamics). Analysis of de novo mitochondrial translation followed procedures described previously (Horváth et al. 2002; Neboháčová et al. 2004). In brief, 1 × 107 exponentially growing cells were pelleted by centrifugation at 2000g for 10 min, washed twice, and resuspended in 100 μL of SoTE buffer (0.6 M sorbitol, 20 mM Tris–HCl at pH 7.5, 2 mM EDTA). Mitochondrial translation products were labeled by incubating the cells for 2 h at 27°C in the presence of EasyTag EXPRESS35S protein labeling mix (100 μCi per 100 μL reaction) with the concurrent inhibition of cytosolic translation by addition of 100 μg/mL cycloheximide. The labeled cells were analyzed in denaturing 2D (9.5% vs. 14%) polyacrylamide Tris–glycine SDS gels as described previously (Horváth et al. 2002).
Cytochrome c oxidase assays
Mitochondria were purified from 5 × 108 T. brucei cells by hypotonic cell lysis, as described previously (Horváth et al. 2005), and stored as pellets at −70°C. These mitochondria were then lysed with 2% dodecyl maltoside, and the cytochrome c oxidase activity was determined in vitro by measuring the change in absorbance of cytochrome c as it becomes oxidized after donating its electrons to complex IV. In parallel, the same dodecyl maltoside lysed mitochondrial samples were resolved (100 μg of protein per lane) on a 3%–12% deep blue native (BN) PAGE gel, and the cytochrome c oxidase activity was detected by an in-gel assay. The enzymatic activity of complex IV causes the native complex to be stained a dark blue as the electron acceptor 3,3′-diaminobenzidine is precipitated when it becomes reduced (Horváth et al. 2005).
ACKNOWLEDGMENTS
We thank all members of the Alfonzo and Lukeš laboratories for helpful discussions. This work was supported by a GACR P305/11/2179, Czech Ministry of Education (AMVIS LH12104), and the Praemium Academiae award to J.L., and a GM084065-05 grant from NIH to J.D.A.
REFERENCES
- Agris PF, Vendeix FA, Graham WD 2007. tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol 366: 1–13 [DOI] [PubMed] [Google Scholar]
- Alfonzo JD, Soll D 2009. Mitochondrial tRNA import: The challenge to understand has just begun. Biol Chem 390: 717–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonzo JD, Blanc V, Estevez AM, Rubio MA, Simpson L 1999. C to U editing of the anticodon of imported mitochondrial tRNATrp allows decoding of the UGA stop codon in Leishmania tarentolae. EMBO J 18: 7056–7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk GR, Wikstrom PM, Bystrom AS 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244: 986–989 [DOI] [PubMed] [Google Scholar]
- Björk GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Dunin-Horkawicz S, Czerwoniec A, Gajda MJ, Feder M, Grosjean H, Bujnicki JM 2006. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res 34: D145–D149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florentz C, Sohm B, Tryoen-Toth P, Putz J, Sissler M 2003. Human mitochondrial tRNAs in health and disease. Cell Mol Life Sci 60: 1356–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattermann N, Retzlaff S, Wang YL, Berneburg M, Heinisch J, Wlaschek M, Aul C, Schneider W 1996. A heteroplasmic point mutation of mitochondrial tRNALeu(CUN) in non-lymphoid haemopoietic cell lineages from a patient with acquired idiopathic sideroblastic anaemia. Br J Haematol 93: 845–855 [DOI] [PubMed] [Google Scholar]
- Gustilo EM, Vendeix FA, Agris PF 2008. tRNA’s modifications bring order to gene expression. Curr Opin Microbiol 11: 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth A, Berry EA, Maslov DA 2000. Translation of the edited mRNA for cytochrome b in trypanosome mitochondria. Science 287: 1639–1640 [DOI] [PubMed] [Google Scholar]
- Horváth A, Neboháčová M, Lukeš J, Maslov DA 2002. Unusual polypeptide synthesis in the kinetoplast-mitochondria from Leishmania tarentolae. Identification of individual de novo translation products. J Biol Chem 277: 7222–7230 [DOI] [PubMed] [Google Scholar]
- Horváth A, Horáková E, Dunajcikova P, Verner Z, Pravdova E, Šlapetová I, Cuninkova L, Lukeš J 2005. Downregulation of the nuclear-encoded subunits of the complexes III and IV disrupts their respective complexes but not complex I in procyclic Trypanosoma brucei. Mol Microbiol 58: 116–130 [DOI] [PubMed] [Google Scholar]
- Jensen BC, Kifer CT, Brekken DL, Randall AC, Wang Q, Drees BL, Parsons M 2007. Characterization of protein kinase CK2 from Trypanosoma brucei. Mol Biochem Parasitol 151: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J 2009. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37: D159–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Suzuki T, Kapushoc ST, Rubio MA, Ghazvini J, Watanabe K, Simpson L 2003. Wobble modification differences and subcellular localization of tRNAs in Leishmania tarentolae: Implication for tRNA sorting mechanism. EMBO J 22: 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapushoc ST, Alfonzo JD, Simpson L 2002. Differential localization of nuclear-encoded tRNAs between the cytosol and mitochondrion in Leishmania tarentolae. RNA 8: 57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kramer G, Graham DE, Appling DR 2007. Yeast mitochondrial initiator tRNA is methylated at guanosine 37 by the Trm5-encoded tRNA (guanine-N1-)-methyltransferase. J Biol Chem 282: 27744–27753 [DOI] [PubMed] [Google Scholar]
- Marechal-Drouard L, Weil JH, Guillemaut P 1988. Import of several tRNAs from the cytoplasm into the mitochondria in bean Phaseolus vulgaris. Nucleic Acids Res 16: 4777–4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neboháčová M, Maslov DA, Falick AM, Simpson L 2004. The effect of RNA interference down-regulation of RNA editing 3′-terminal uridylyl transferase (TUTase) 1 on mitochondrial de novo protein synthesis and stability of respiratory complexes in Trypanosoma brucei. J Biol Chem 279: 7819–7825 [DOI] [PubMed] [Google Scholar]
- Ohira T, Suzuki T 2012. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc Natl Acad Sci 108: 10502–10507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris Z, Rubio MA, Lukeš J, Alfonzo JD 2009. Mitochondrial tRNA import in Trypanosoma brucei is independent of thiolation and the Rieske protein. RNA 15: 1398–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris Z, Changmai P, Rubio MA, Zíková A, Stuart KD, Alfonzo JD, Lukeš J 2011. The Fe/S cluster assembly protein Isd11 is essential for tRNA thiolation in Trypanosoma brucei. J Biol Chem 285: 22394–22402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Alfonzo JD 2010. Do all modifications benefit all tRNAs? FEBS Lett 584: 265–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz J, Dupuis B, Sissler M, Florentz C 2007. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA 13: 1184–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richterová L, Vávrová Z, Lukeš J 2011. DEAD-box RNA helicase is dispensable for mitochondrial translation in Trypanosoma brucei. Exp Parasitol 127: 300–303 [DOI] [PubMed] [Google Scholar]
- Rubio MAT, Alfonzo JD 2005. Editing and modification in trypanosomatids: The reshaping of non-coding RNAs. In Topics in current genetics, Vol. 12 (ed. Grosjean H), pp. 71–86 Springer-Verlag, Berlin/Heidelberg [Google Scholar]
- Schneider A 2011. Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu Rev Biochem 80: 1033–1053 [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin J, Agabian N 1994a. A nuclear encoded tRNA of Trypanosoma brucei is imported into mitochondria. Mol Cell Biol 14: 2317–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, McNally KP, Agabian N 1994b. Nuclear-encoded mitochondrial tRNAs of Trypanosoma brucei have a modified cytidine in the anticodon loop. Nucleic Acids Res 22: 3699–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber F, Duchene AM, Marechal-Drouard L 2011. Mitochondrial RNA import: From diversity of natural mechanisms to potential applications. Int Rev Cell Mol Biol 287: 145–190 [DOI] [PubMed] [Google Scholar]
- Spears JL, Rubio MA, Gaston KW, Wywial E, Strikoudis A, Bujnicki JM, Papavasiliou FN, Alfonzo JD 2011. A single zinc ion is sufficient for an active Trypanosoma brucei tRNA editing deaminase. J Biol Chem 286: 20366–20374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Týč J, Faktorová D, Kriegová E, Jirků M, Vávrová Z, Maslov DA, Lukeš J 2010. Probing for primary functions of prohibitin in Trypanosoma brucei. Int J Parasitol 40: 73–83 [DOI] [PubMed] [Google Scholar]
- Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Björk GR 2001. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20: 4863–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavicius J, Stahl G, Durand JM, Ben Salem SN, Qian Q, Farabaugh PJ, Björk GR 2003. Transfer RNA modifications that alter +1 frameshifting in general fail to affect −1 frameshifting. RNA 9: 760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondrušková E, van den Burg J, Zíková A, Ernst NL, Stuart K, Benne R, Lukeš J 2005. RNA interference analyses suggest a transcript-specific regulatory role for mitochondrial RNA-binding proteins MRP1 and MRP2 in RNA editing and other RNA processing in Trypanosoma brucei. J Biol Chem 280: 2429–2438 [DOI] [PubMed] [Google Scholar]
- Waas WF, Druzina Z, Hanan M, Schimmel P 2007. Role of a tRNA base modification and its precursors in frameshifting in eukaryotes. J Biol Chem 282: 26026–26034 [DOI] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K 2002. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol 125: 211–216 [DOI] [PubMed] [Google Scholar]
- Yarham JW, Elson JL, Blakely EL, McFarland R, Taylor RW 2011. Mitochondrial tRNA mutations and disease. Wiley Interdiscip Rev RNA 1: 304–324 [DOI] [PubMed] [Google Scholar]